

Genetic Heterogeneity of X-Linked Ichthyosis in the Republic of North Ossetia–Alania, Case Series Report

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Clinical Description
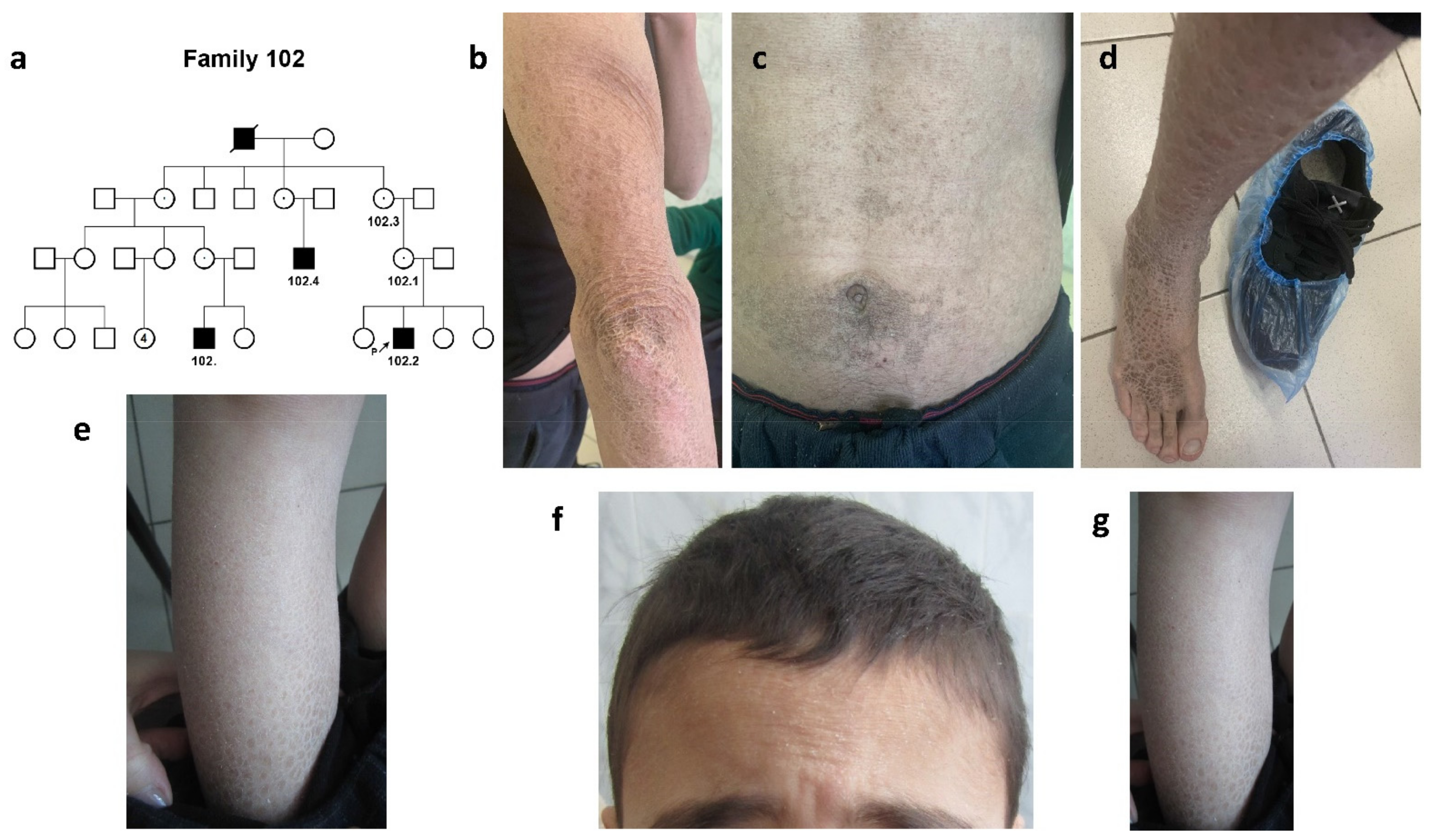
2.1.1. Family #102
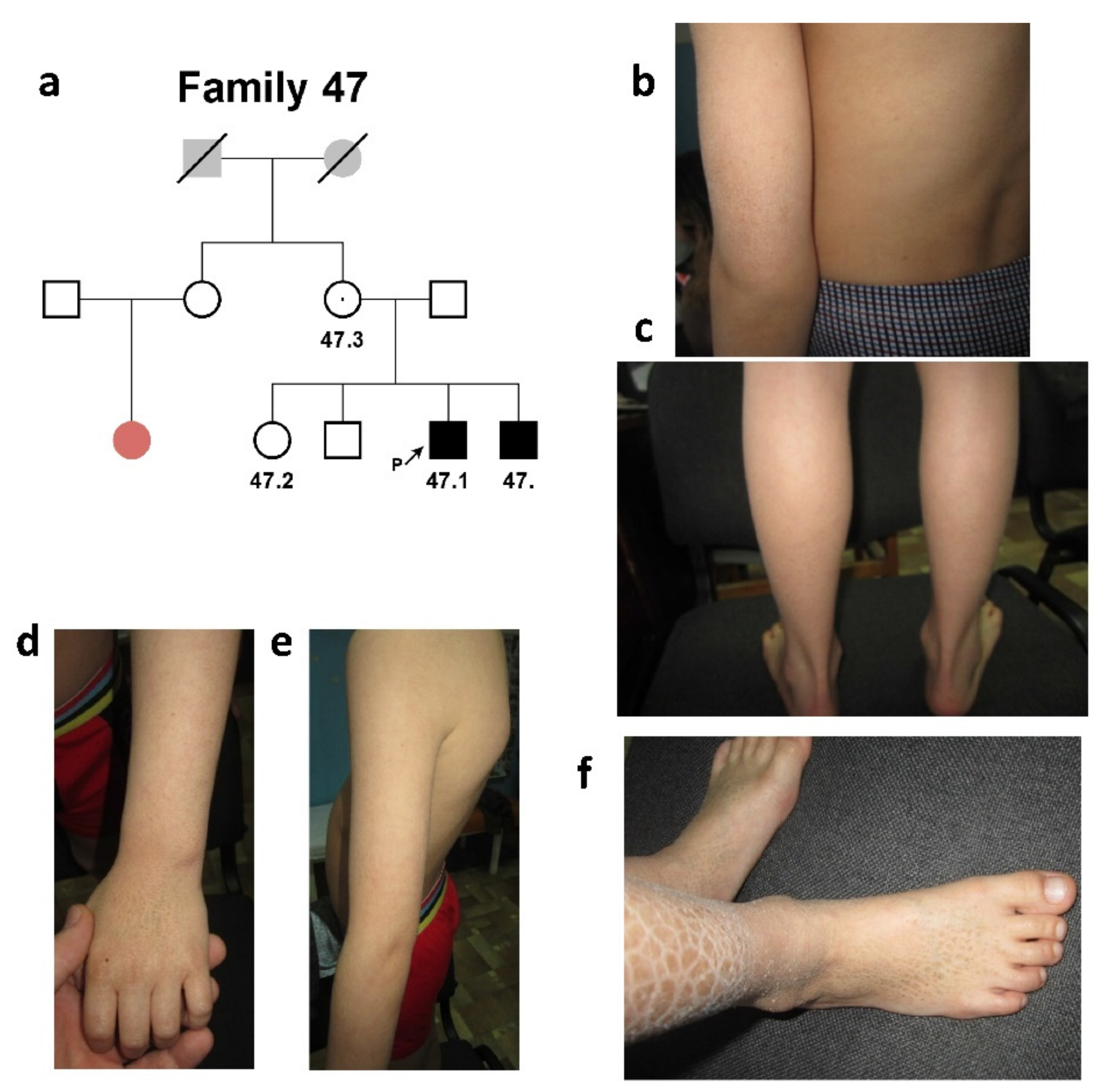
2.1.2. Family #47
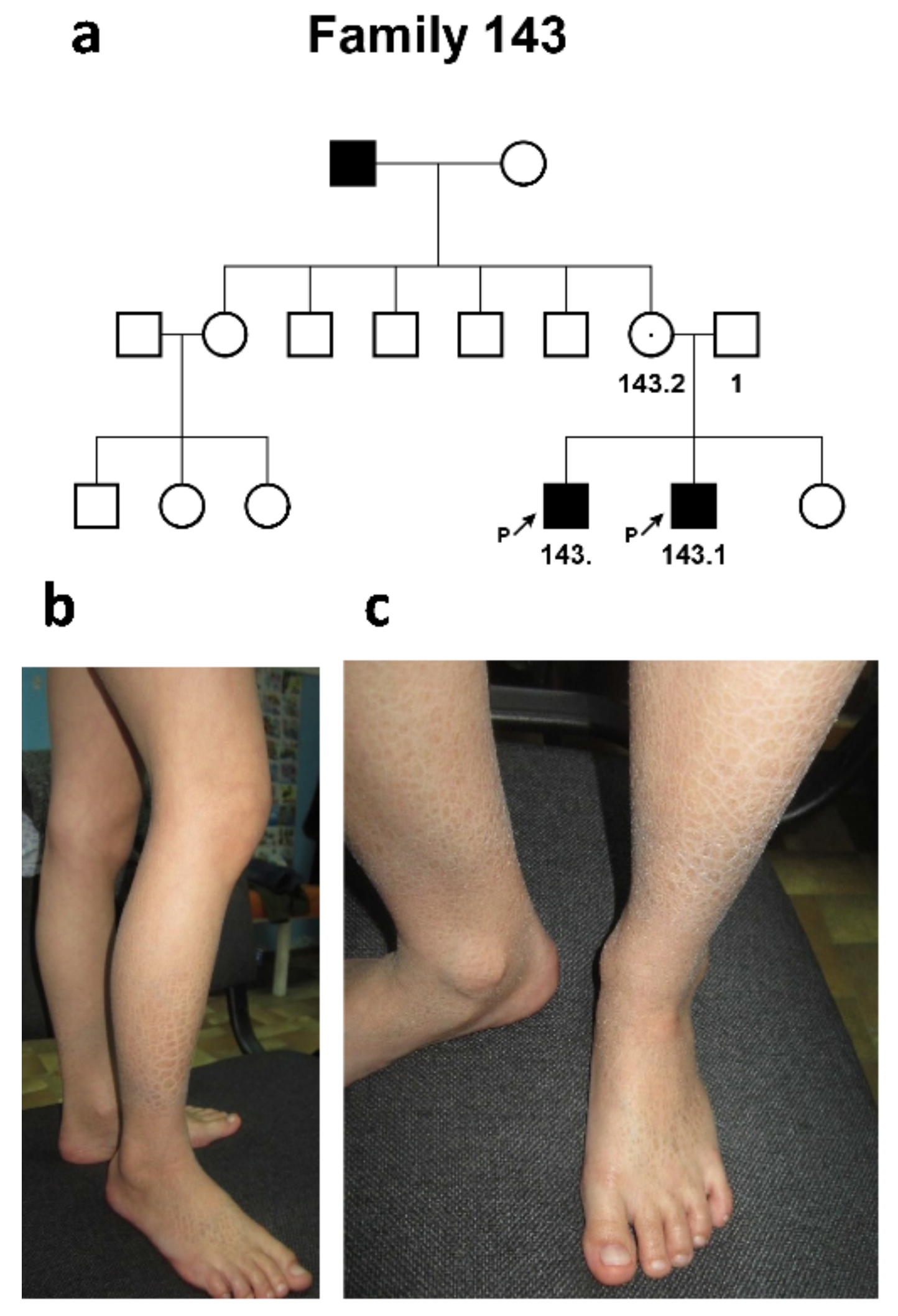
2.1.3. Family #143
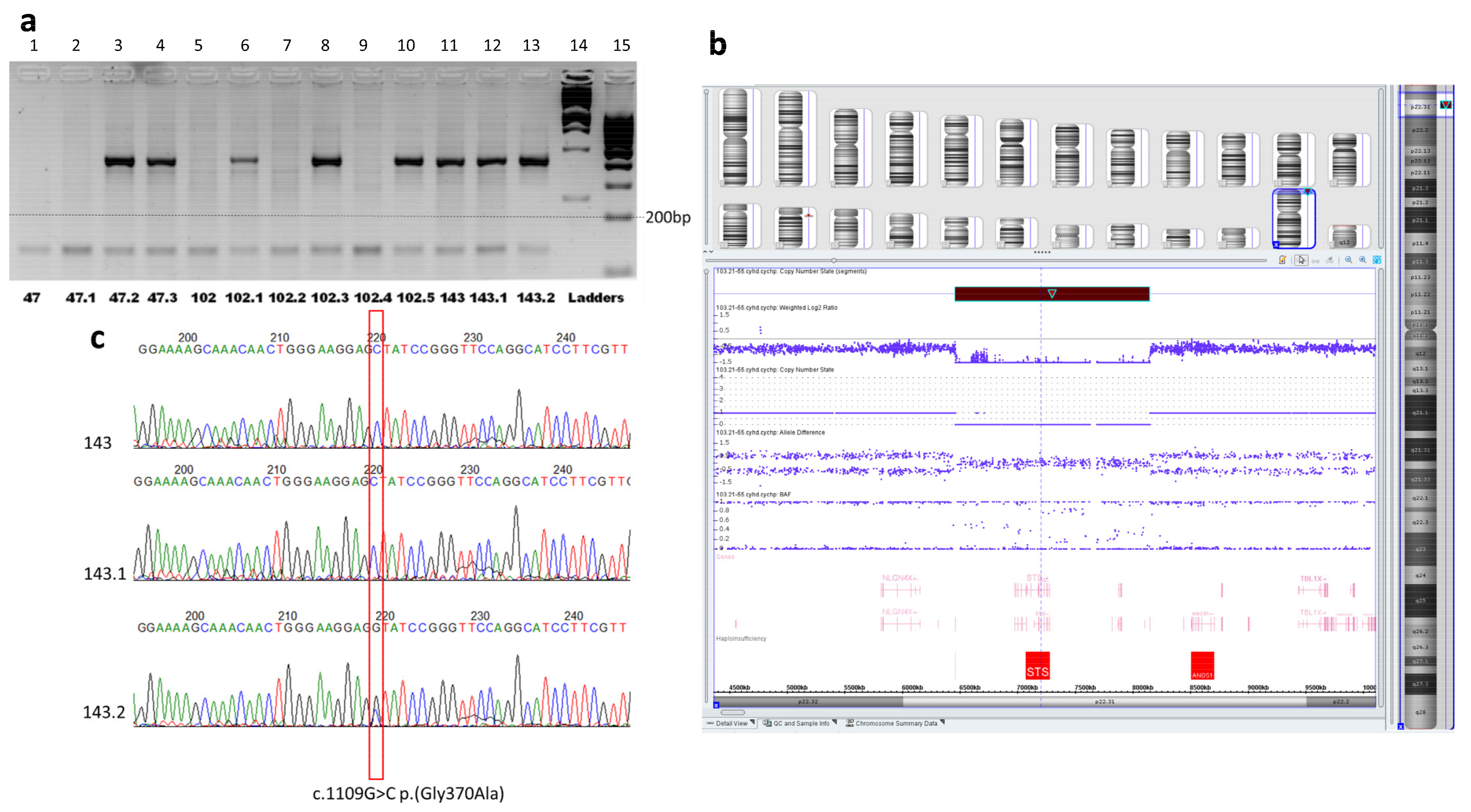
2.2. Molecular Genetic Diagnosis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Informed Consent
4.3. DNA Purification
4.4. High-Throughput Sequencing
4.5. Chromosomal Microarray Analysis
4.6. AFLP-PCR Analysis
4.7. Polymerase Chain Reaction (PCR), Sanger Sequencing
4.8. Amplified Fragment Length Polymorphism of STR Markers Loci Analysis
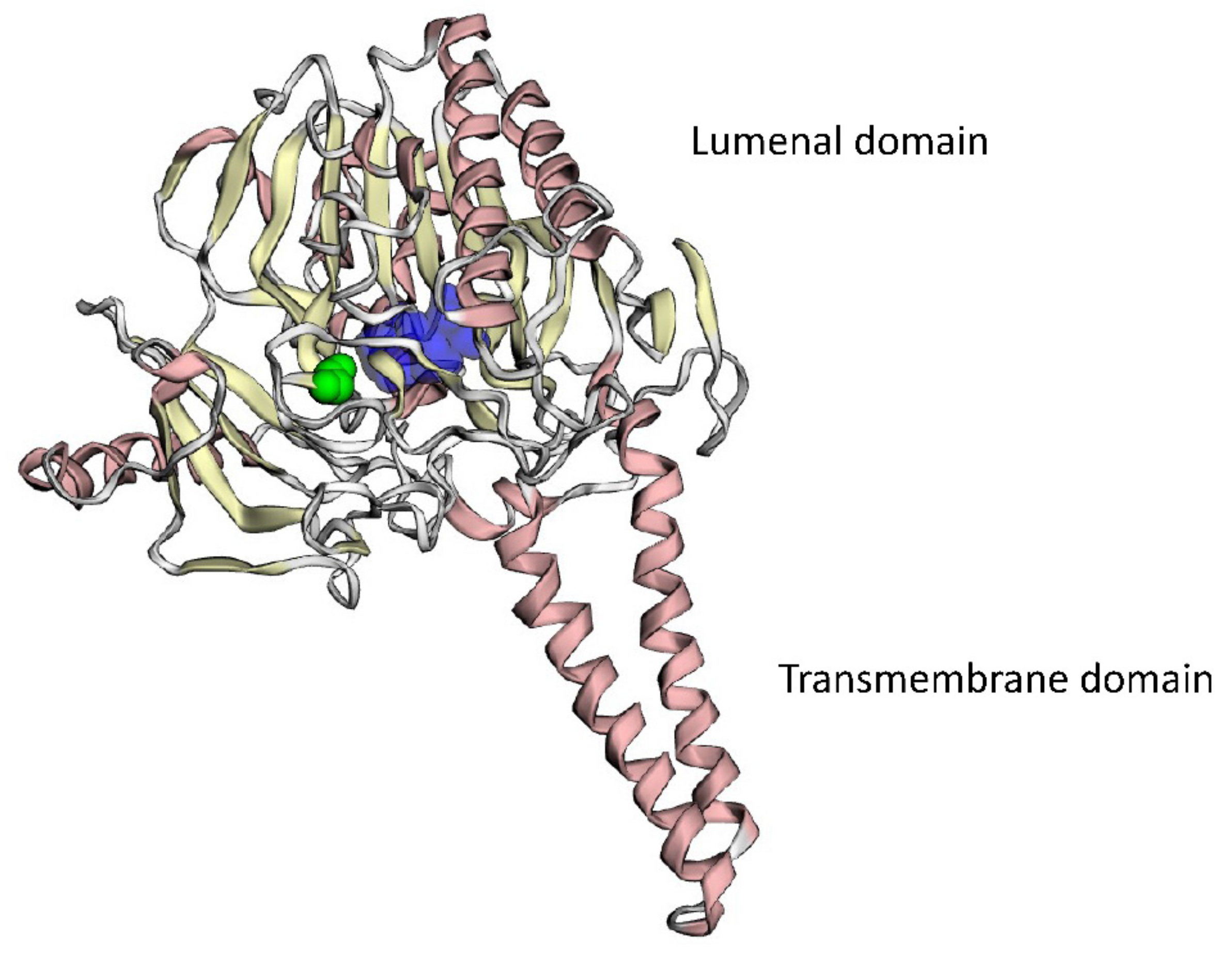
4.9. 3D Modeling of Mutant Protein
4.10. Calculation of the Point Prevalence Rate of the Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernandez-Martin, A.; Gonzalez-Sarmiento, R.; De Unamuno, P. X-linked ichthyosis: An update. Br. J. Dermatol. 1999, 141, 617–627. [Google Scholar] [CrossRef]
- Elias, P.M.; Williams, M.L.; Maloney, M.E.; Bonifas, J.A.; Brown, B.E.; Grayson, S.; Epstein, E.H., Jr. Stratum corneum lipids in disorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis of recessive x-linked ichthyosis. J. Clin. Investig. 1984, 74, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.S.; Kwon, T.U.; Shin, S.; Kwon, Y.J.; Chun, Y.J. Steroid sulfatase deficiency causes cellular senescence and abnormal differentiation by inducing yippee-like 3 expression in human keratinocytes. Sci. Rep. 2021, 11, 20867. [Google Scholar] [CrossRef]
- Crane, J.S.; Paller, A.S. X-linked ichthyosis. In Statpearls; 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448149/ (accessed on 5 September 2022).
- Orphanet Report Series—Prevalence of Rare Diseases: Bibliographic Data. 2022. Available online: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf (accessed on 5 September 2022).
- Miller, A.P.; Gustashaw, K.; Wolff, D.J.; Rider, S.H.; Monaco, A.P.; Eble, B.; Schlessinger, D.; Gorski, J.L.; van Ommen, G.J.; Weissenbach, J.; et al. Three genes that escape x chromosome inactivation are clustered within a 6 mb yac contig and sts map in xp11.21-p11.22. Hum. Mol. Genet. 1995, 4, 731–739. [Google Scholar] [CrossRef]
- Sever, R.J.; Frost, P.; Weinstein, G. Eye changes in ichthyosis. JAMA 1968, 206, 2283–2286. [Google Scholar] [CrossRef]
- France, J.T.; Liggins, G.C. Placental sulfatase deficiency. J. Clin. Endocrinol. Metab. 1969, 29, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, F.E.; Abdulrahman, G.O.; Waring, G.; Hinshaw, K. Placental steroid sulphatase deficiency: An approach to antenatal care and delivery. Ann. Saudi Med. 2018, 38, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, R.A.; Makaov, A.K.; Marakhonov, A.V.; Galkina, V.A.; Kadyshev, V.V.; El’chinova, G.I.; Dadali, E.L.; Mikhailova, L.K.; Petrova, N.V.; Petrina, N.E.; et al. Epidemiology of hereditary diseases in the karachay-cherkess republic. Int. J. Mol. Sci. 2020, 21, 325. [Google Scholar] [CrossRef]
- Bonifas, J.M.; Morley, B.J.; Oakey, R.E.; Kan, Y.W.; Epstein, E.H., Jr. Cloning of a cdna for steroid sulfatase: Frequent occurrence of gene deletions in patients with recessive x chromosome-linked ichthyosis. Proc. Natl. Acad. Sci. USA 1987, 84, 9248–9251. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Alperin, E.S.; Shapiro, L.J. Characterization of point mutations in patients with x-linked ichthyosis. Effects on the structure and function of the steroid sulfatase protein. J. Biol. Chem. 1997, 272, 20756–20763. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Guzman, F.G.; Higashiyama, T.; Pangborn, W.; Osawa, Y.; Ghosh, D. Structure of human estrone sulfatase suggests functional roles of membrane association. J. Biol. Chem. 2003, 278, 22989–22997. [Google Scholar] [CrossRef] [PubMed]
- Brcic, L.; Underwood, J.F.; Kendall, K.M.; Caseras, X.; Kirov, G.; Davies, W. Medical and neurobehavioural phenotypes in carriers of x-linked ichthyosis-associated genetic deletions in the UK biobank. J. Med. Genet. 2020, 57, 692–698. [Google Scholar] [CrossRef]
- Aviram-Goldring, A.; Goldman, B.; Netanelov-Shapira, I.; Chen-Shtoyerman, R.; Zvulunov, A.; Tal, O.; Ilan, T.; Peleg, L. Deletion patterns of the sts gene and flanking sequences in israeli x-linked ichthyosis patients and carriers: Analysis by polymerase chain reaction and fluorescence in situ hybridization techniques. Int. J. Dermatol. 2000, 39, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Kuwata, S.; Nakagawa, H.; Shimada, S.; Tamaki, K.; Ishibashi, Y. Deletion pattern of the steroid sulphatase gene in Japanese patients with x-linked ichthyosis. Br. J. Dermatol. 1998, 139, 96–98. [Google Scholar] [CrossRef]
- Yen, P.H.; Li, X.M.; Tsai, S.P.; Johnson, C.; Mohandas, T.; Shapiro, L.J. Frequent deletions of the human x chromosome distal short arm result from recombination between low copy repetitive elements. Cell 1990, 61, 603–610. [Google Scholar] [CrossRef]
- Hering, S.; Edelmann, J.; Augustin, C.; Kuhlisch, E.; Szibor, R. X chromosomal recombination—A family study analysing 39 str markers in German three-generation pedigrees. Int. J. Leg. Med. 2010, 124, 483–491. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Konovalov, F.A.; Makaov, A.K.; Vasilyeva, T.A.; Kadyshev, V.V.; Galkina, V.A.; Dadali, E.L.; Kutsev, S.I.; Zinchenko, R.A. Primary microcephaly case from the karachay-cherkess republic poses an additional support for microcephaly and seckel syndrome spectrum disorders. BMC Med. Genom. 2018, 11, 8. [Google Scholar] [CrossRef]
- Kohler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Dansault, A.; David, G.; Schwartz, C.; Jaliffa, C.; Vieira, V.; de la Houssaye, G.; Bigot, K.; Catin, F.; Tattu, L.; Chopin, C.; et al. Three new pax6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol. Vis. 2007, 13, 511–523. [Google Scholar] [PubMed]
- Ribeiro Rodrigues, E.M.; Leite, F.P.; Hutz, M.H.; Palha Tde, J.; Ribeiro dos Santos, A.K.; dos Santos, S.E. A multiplex pcr for 11 x chromosome str markers and population data from a Brazilian amazon region. Forensic Sci. Int. Genet. 2008, 2, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Vazna, A.; Musova, Z.; Vlckova, M.; Novotna, D.; Dvorakova, L.; Hrdlicka, M.; Havlovicova, M.; Sedlacek, Z. Fmr1 gene expansion, large deletion of xp, and skewed x-inactivation in a girl with mental retardation and autism. Am. J. Med. Genet. A 2010, 152A, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Matise, T.C.; Chen, F.; Chen, W.; De La Vega, F.M.; Hansen, M.; He, C.; Hyland, F.C.; Kennedy, G.C.; Kong, X.; Murray, S.S.; et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007, 17, 1783–1786. [Google Scholar] [CrossRef] [PubMed]
- Niknafs, N.; Kim, D.; Kim, R.; Diekhans, M.; Ryan, M.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Mupit interactive: Webserver for mapping variant positions to annotated, interactive 3d structures. Hum. Genet. 2013, 132, 1235–1243. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | #102 | #102.2 | #102.4 | #47 | #47.1 | #143 | #143.1 |
|---|---|---|---|---|---|---|---|
| Genotype at chrXp22.31 region or NM_000351.7 (STS) | g.[chrXp22.31del];[0] | g.[chrXp22.31del];[0] | g.[chrXp22.31del];[0] | g.[chrXp22.31del];[0] | g.[chrXp22.31del]; [0] | c.[1109G>C];[0] | c.[1109G>C];[0] |
| Ancestry | Kumyks | Kumyks | Kumyks | Turkish Meskhetians | Turkish Meskhetians | Ossetians | Ossetians |
| Age/gender | 7 y.o./male | 10 y.o./male | 35 y.o./male | 11 y.o./male | 7 y.o./male | 12 y.o./male | 9 y.o./male |
| Dry skin, infantile atopic dermatitis, and mild scaling appeared in the first few weeks of life | + | + | n/d | + | + | + | + |
| Tightly adherent, brown scales at the surface of the extremities | + | + | + | + | + | + | + |
| Scales at the surface of the trunk and neck | + | + | + | + | none | none | none |
| Scales on the forehead | + | + | + | none | none | none | none |
| Scales on the hairy part of the head | + | + | + | none | none | none | none |
| Loci Location and Patients’ ID | delXp22.31 (Encompassing the STS Gene) | DXS10148 | DXS10135 | DXS8378 | Ethnic Origin |
|---|---|---|---|---|---|
| GenBank accession numbers | KC662330.1 | MT607405.1 | MT132938.1 | ||
| Chromosomal location (hg19) (Distance from a deletion in Mb) | chrX:6449753_8135644 (0 Mb) | chrX:9238969_9239205 (1.1 Mb) | chrX:9306118_9306616 (1.2 Mb) | chrX:9370150_9370429 (1.3 Mb) | |
| Female map position in Kosambi cM (Distance from deletion) | 16.2–18.4 cM (0 cM) | 20.2 cM (1.79 cM) | 20.4 cM (1.94 cM) | 20.6 cM (2.15 cM) | |
| #102 | [chrXp22.31del];[0] | 18 | 23.1 | 10 | Kumyks |
| #102.2 | [chrXp22.31del];[0] | 18 | 23.1 | 9 | Kumyks |
| #102.4 | [chrXp22.31del];[0] | 18 | 23.1 | 10 | Kumyks |
| #47 | [chrXp22.31del];[0] | 27.1 | 20 | 9 | Turkish Meskhetians |
| #47.1 | [chrXp22.31del];[0] | 27.1 | 20 | 10 | Turkish Meskhetians |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilyeva, T.A.; Marakhonov, A.V.; Tebieva, I.S.; Kadyshev, V.V.; Borovikov, A.O.; Markova, Z.G.; Chukhrova, A.L.; Ginter, E.K.; Kutsev, S.I.; Zinchenko, R.A. Genetic Heterogeneity of X-Linked Ichthyosis in the Republic of North Ossetia–Alania, Case Series Report. Int. J. Mol. Sci. 2023, 24, 4515. https://doi.org/10.3390/ijms24054515
Vasilyeva TA, Marakhonov AV, Tebieva IS, Kadyshev VV, Borovikov AO, Markova ZG, Chukhrova AL, Ginter EK, Kutsev SI, Zinchenko RA. Genetic Heterogeneity of X-Linked Ichthyosis in the Republic of North Ossetia–Alania, Case Series Report. International Journal of Molecular Sciences. 2023; 24(5):4515. https://doi.org/10.3390/ijms24054515
Chicago/Turabian StyleVasilyeva, Tatyana A., Andrey V. Marakhonov, Inna S. Tebieva, Vitaly V. Kadyshev, Artem O. Borovikov, Zhanna G. Markova, Alyona L. Chukhrova, Evgeny K. Ginter, Sergey I. Kutsev, and Rena A. Zinchenko. 2023. "Genetic Heterogeneity of X-Linked Ichthyosis in the Republic of North Ossetia–Alania, Case Series Report" International Journal of Molecular Sciences 24, no. 5: 4515. https://doi.org/10.3390/ijms24054515
APA StyleVasilyeva, T. A., Marakhonov, A. V., Tebieva, I. S., Kadyshev, V. V., Borovikov, A. O., Markova, Z. G., Chukhrova, A. L., Ginter, E. K., Kutsev, S. I., & Zinchenko, R. A. (2023). Genetic Heterogeneity of X-Linked Ichthyosis in the Republic of North Ossetia–Alania, Case Series Report. International Journal of Molecular Sciences, 24(5), 4515. https://doi.org/10.3390/ijms24054515