
The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives



Abstract
1. Introduction
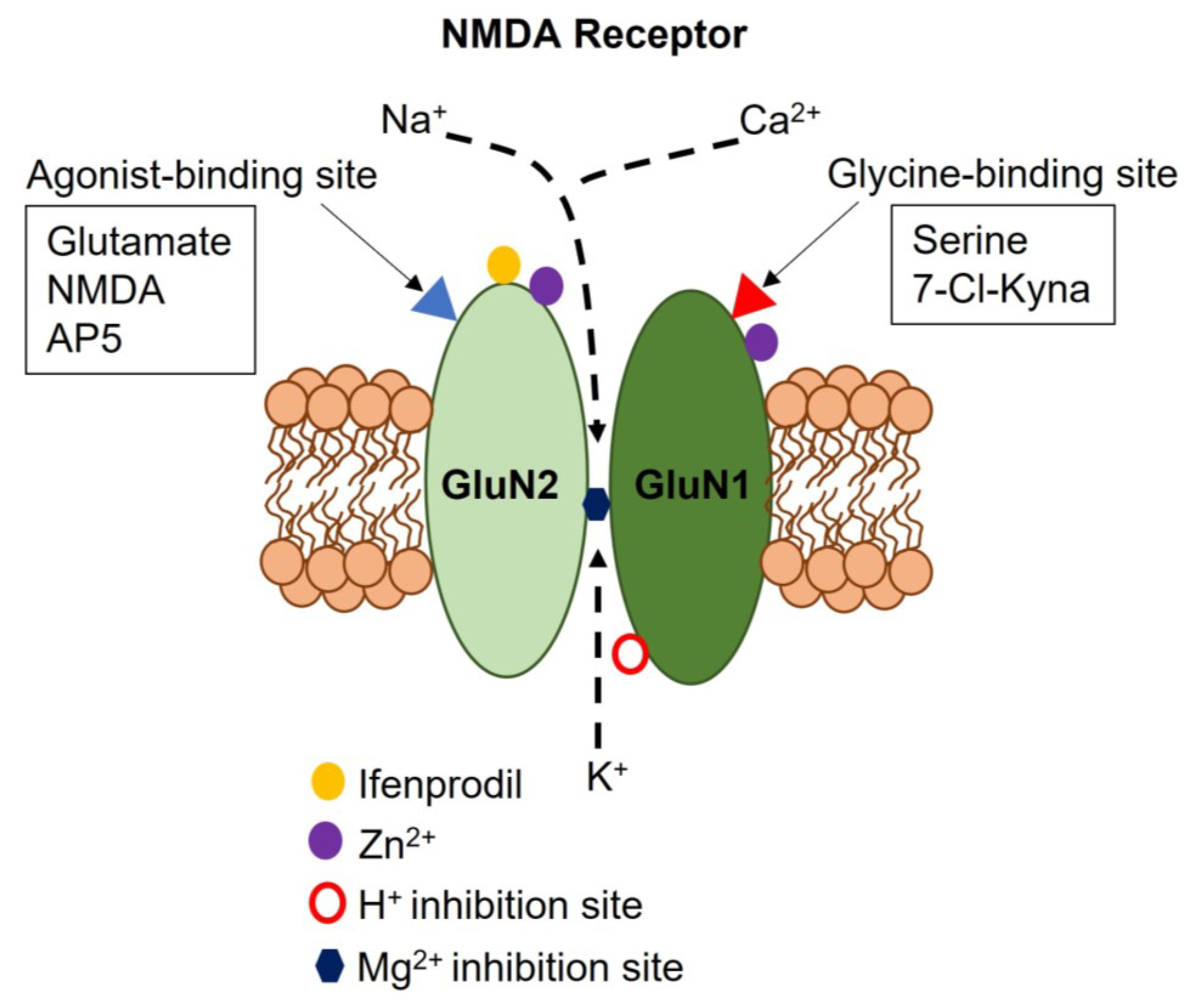
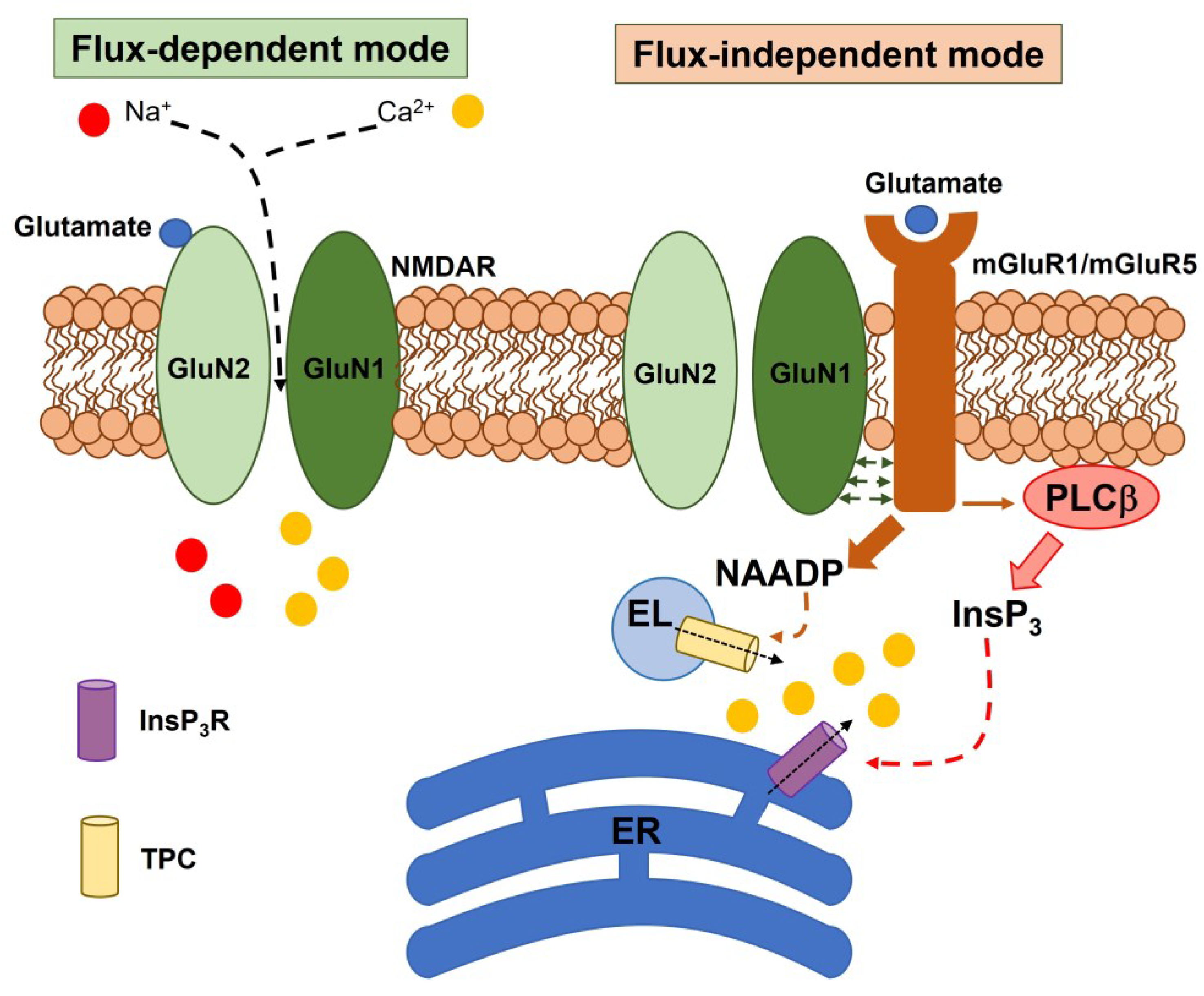
2. Subunit Composition, Regulatory Mechanisms, and Signaling Modes of Neuronal NMDARs
3. NMDAR Expression, Signaling, and Function in the Heart
3.1. The Role of NMDAR Activation in Ventricular Arrhythmias and Acute Myocardial Infarction (AMI)
3.2. The Role of Extracellular Ca2+ Entry in NMDA-Induced Ventricular Arrhythmia and Cardiac Remodeling
3.3. The Role of NMDARs in Atrial Excitability and Conductivity and Their Involvement in Atrial Fibrillation
3.4. The Endogenous Agonist of Cardiac NMDARs: Is There A Role for Homocysteine (Hcy)?
4. The Role of NMDARs in VSMCs and Vascular Endothelial Cells
4.1. The Hemodynamic Effect of NMDAR Activation in Peripheral Vasculature
4.2. The Role of Peripheral NMDARs in Vascular Remodelling during HHcy-Induced Atherosclerosis and in PAH
5. The Role of NMDARs in Cerebro-Microvascular Endothelial Cells (CMVECs)
5.1. Endothelial NMDARs Control BBB Permeability
5.2. Endothelial NMDARs Control NVC
5.3. The Pathogenic Role of Endothelial NMDARs in the CNS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Soda, T.; Mapelli, L.; Locatelli, F.; Botta, L.; Goldfarb, M.; Prestori, F.; D’Angelo, E. Hyperexcitability and Hyperplasticity Disrupt Cerebellar Signal Transfer in the IB2 KO Mouse Model of Autism. J. Neurosci. 2019, 39, 2383–2397. [Google Scholar]
- Locatelli, F.; Soda, T.; Montagna, I.; Tritto, S.; Botta, L.; Prestori, F.; D’Angelo, E. Calcium Channel-Dependent Induction of Long-Term Synaptic Plasticity at Excitatory Golgi Cell Synapses of Cerebellum. J. Neurosci. 2021, 41, 3307–3319. [Google Scholar] [CrossRef]
- Loftis, J.M.; Janowsky, A. The N-methyl-d-aspartate receptor subunit NR2B: Localization, functional properties, regulation, and clinical implications. Pharmacol. Ther. 2003, 97, 55–85. [Google Scholar] [CrossRef]
- Pagano, J.; Giona, F.; Beretta, S.; Verpelli, C.; Sala, C. N-methyl-d-aspartate receptor function in neuronal and synaptic development and signaling. Curr. Opin. Pharmacol. 2021, 56, 93–101. [Google Scholar] [CrossRef]
- Mapelli, L.; Soda, T.; D’Angelo, E.; Prestori, F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. Int. J. Mol. Sci. 2022, 23, 3894. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Chvatal, A. NMDA Receptors in Astrocytes. Neurochem. Res. 2020, 45, 122–133. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef]
- Bozic, M.; Valdivielso, J.M. The potential of targeting NMDA receptors outside the CNS. Expert Opin. Ther. Targets 2015, 19, 399–413. [Google Scholar] [CrossRef]
- McGee, M.A.; Abdel-Rahman, A.A. N-Methyl-d-Aspartate Receptor Signaling and Function in Cardiovascular Tissues. J. Cardiovasc. Pharmacol. 2016, 68, 97–105. [Google Scholar] [CrossRef]
- Kalev-Zylinska, M.L.; Hearn, J.I.; Makhro, A.; Bogdanova, A. N-Methyl-d-Aspartate Receptors in Hematopoietic Cells: What Have We Learned? Front. Physiol. 2020, 11, 577. [Google Scholar] [CrossRef]
- Kalev-Zylinska, M.L.; Morel-Kopp, M.C.; Ward, C.M.; Hearn, J.I.; Hamilton, J.R.; Bogdanova, A.Y. Ionotropic glutamate receptors in platelets: Opposing effects and a unifying hypothesis. Platelets 2021, 32, 998–1008. [Google Scholar] [CrossRef]
- Alexander, S.P.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The concise guide to pharmacology 2021/22: Ion channels. Br. J. Pharmacol. 2021, 178 (Suppl. 1), S157–S245. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Pachernegg, S.; Strutz-Seebohm, N.; Hollmann, M. GluN3 subunit-containing NMDA receptors: Not just one-trick ponies. Trends Neurosci. 2012, 35, 240–249. [Google Scholar] [CrossRef]
- Perez-Otano, I.; Larsen, R.S.; Wesseling, J.F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat. Rev. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef]
- Burzomato, V.; Frugier, G.; Perez-Otano, I.; Kittler, J.T.; Attwell, D. The receptor subunits generating NMDA receptor mediated currents in oligodendrocytes. J. Physiol. 2010, 588 Pt 18, 3403–3414. [Google Scholar] [CrossRef]
- Pina-Crespo, J.C.; Talantova, M.; Micu, I.; States, B.; Chen, H.S.; Tu, S.; Nakanishi, N.; Tong, G.; Zhang, D.; Heinemann, S.F.; et al. Excitatory glycine responses of CNS myelin mediated by NR1/NR3 “NMDA” receptor subunits. J. Neurosci. 2010, 30, 11501–11505. [Google Scholar] [CrossRef]
- Dore, K.; Stein, I.S.; Brock, J.A.; Castillo, P.E.; Zito, K.; Sjostrom, P.J. Unconventional NMDA Receptor Signaling. J. Neurosci. 2017, 37, 10800–10807. [Google Scholar] [CrossRef]
- Park, D.K.; Stein, I.S.; Zito, K. Ion flux-independent NMDA receptor signaling. Neuropharmacology 2022, 210, 109019. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P. Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. Int. J. Mol. Sci. 2018, 19, 3800. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef]
- Stein, I.S.; Gray, J.A.; Zito, K. Non-Ionotropic NMDA Receptor Signaling Drives Activity-Induced Dendritic Spine Shrinkage. J. Neurosci. 2015, 35, 12303–12308. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P.; Aguilera, P. A Metabotropic-Like Flux-Independent NMDA Receptor Regulates Ca2+ Exit from Endoplasmic Reticulum and Mitochondrial Membrane Potential in Cultured Astrocytes. PLoS ONE 2015, 10, e0126314. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P.; Matus Nunez, M.; Picones, A.; Hernandez-Cruz, A. NMDAR in cultured astrocytes: Flux-independent pH sensor and flux-dependent regulator of mitochondria and plasma membrane-mitochondria bridging. FASEB J. 2020, 34, 16622–16644. [Google Scholar] [CrossRef]
- Birnbaum, J.H.; Bali, J.; Rajendran, L.; Nitsch, R.M.; Tackenberg, C. Calcium flux-independent NMDA receptor activity is required for Abeta oligomer-induced synaptic loss. Cell. Death Dis. 2015, 6, e1791. [Google Scholar] [CrossRef]
- Park, D.K.; Petshow, S.; Anisimova, M.; Barragan, E.V.; Gray, J.A.; Stein, I.S.; Zito, K. Reduced d-serine levels drive enhanced non-ionotropic NMDA receptor signaling and destabilization of dendritic spines in a mouse model for studying schizophrenia. Neurobiol. Dis. 2022, 170, 105772. [Google Scholar] [CrossRef]
- Ashton, J.L.; Burton, R.A.B.; Bub, G.; Smaill, B.H.; Montgomery, J.M. Synaptic Plasticity in Cardiac Innervation and Its Potential Role in Atrial Fibrillation. Front. Physiol. 2018, 9, 240. [Google Scholar] [CrossRef]
- Reilly, S.; Nattel, S. Finding a new job: Glutamate signaling acts in atrial cardiomyocytes. Cell Res. 2021, 31, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.W.; Gill, S.S.; Pulido, O.M. The monkey (Macaca fascicularis) heart neural structures and conducting system: An immunochemical study of selected neural biomarkers and glutamate receptors. Toxicol. Pathol. 2003, 31, 227–234. [Google Scholar] [PubMed]
- Gill, S.S.; Pulido, O.M.; Mueller, R.W.; McGuire, P.F. Molecular and immunochemical characterization of the ionotropic glutamate receptors in the rat heart. Brain Res. Bull. 1998, 46, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Veinot, J.; Kavanagh, M.; Pulido, O. Human heart glutamate receptors—Implications for toxicology, food safety, and drug discovery. Toxicol. Pathol. 2007, 35, 411–417. [Google Scholar] [CrossRef]
- Lin, Y.J.; Bovetto, S.; Carver, J.M.; Giordano, T. Cloning of the cDNA for the human NMDA receptor NR2C subunit and its expression in the central nervous system and periphery. Brain Res. Mol. Brain Res. 1996, 43, 57–64. [Google Scholar] [CrossRef]
- Xie, D.; Xiong, K.; Su, X.; Wang, G.; Ji, Q.; Zou, Q.; Wang, L.; Liu, Y.; Liang, D.; Xue, J.; et al. Identification of an endogenous glutamatergic transmitter system controlling excitability and conductivity of atrial cardiomyocytes. Cell Res. 2021, 31, 951–964. [Google Scholar] [CrossRef]
- Shi, S.; Liu, T.; Wang, D.; Zhang, Y.; Liang, J.; Yang, B.; Hu, D. Activation of N-methyl-d-aspartate receptors reduces heart rate variability and facilitates atrial fibrillation in rats. Europace 2017, 19, 1237–1243. [Google Scholar] [CrossRef]
- Nesterov, S.V.; Skorobogatova, Y.A.; Panteleeva, A.A.; Pavlik, L.L.; Mikheeva, I.B.; Yaguzhinsky, L.S.; Nartsissov, Y.R. NMDA and GABA receptor presence in rat heart mitochondria. Chem. Biol. Interact. 2018, 291, 40–46. [Google Scholar] [CrossRef]
- Seeber, S.; Humeny, A.; Herkert, M.; Rau, T.; Eschenhagen, T.; Becker, C.M. Formation of molecular complexes by N-methyl-d-aspartate receptor subunit NR2B and ryanodine receptor 2 in neonatal rat myocard. J. Biol. Chem. 2004, 279, 21062–21068. [Google Scholar] [CrossRef]
- Shi, S.; Liu, T.; Li, Y.; Qin, M.; Tang, Y.; Shen, J.Y.; Liang, J.; Yang, B.; Huang, C. Chronic N-methyl-d-aspartate receptor activation induces cardiac electrical remodeling and increases susceptibility to ventricular arrhythmias. Pacing Clin. Electrophysiol. 2014, 37, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Di Filippo, C.; Rossi, F.; Rossi, F. Arrhythmias induced by myocardial ischaemia-reperfusion are sensitive to ionotropic excitatory amino acid receptor antagonists. Eur. J. Pharmacol. 1999, 366, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhong, J.; Wang, D.; Xu, J.; Su, H.; An, C.; Zhu, H.; Yan, J. Increasing glutamate promotes ischemia-reperfusion-induced ventricular arrhythmias in rats in vivo. Pharmacology 2014, 93, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Y.; Hu, S.; Zhong, Q.W.; Tian, C.N.; Ma, H.M.; Yu, J.J. N-Methyl-d-Aspartate Receptor-Driven Calcium Influx Potentiates the Adverse Effects of Myocardial Ischemia-Reperfusion Injury Ex Vivo. J. Cardiovasc. Pharmacol. 2017, 70, 329–338. [Google Scholar] [CrossRef]
- Lu, J.; Gao, X.; Gu, J.; Zhou, L.; Guo, S.; Hao, W.; Zhou, Z.; Cao, J.M. Nerve sprouting contributes to increased severity of ventricular tachyarrhythmias by upregulating iGluRs in rats with healed myocardial necrotic injury. J. Mol. Neurosci. 2012, 48, 448–455. [Google Scholar] [CrossRef]
- Gao, X.; Xu, X.; Pang, J.; Zhang, C.; Ding, J.M.; Peng, X.; Liu, Y.; Cao, J.M. NMDA receptor activation induces mitochondrial dysfunction, oxidative stress and apoptosis in cultured neonatal rat cardiomyocytes. Physiol. Res. 2007, 56, 559–569. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, Z.; Liao, Z.; Wang, M.; Zhou, Y.; Luo, S.; Ding, Y.; Liu, T.; Cao, C.; Yue, S. Effects of Excessive Activation of N-methyl-D-aspartic Acid Receptors in Neonatal Cardiac Mitochondrial Dysfunction Induced by Intrauterine Hypoxia. Front. Cardiovasc. Med 2022, 9, 837142. [Google Scholar] [CrossRef]
- McGee, M.A.; Abdel-Rahman, A.A. Enhanced vascular neuronal nitric-oxide synthase-derived nitric-oxide production underlies the pressor response caused by peripheral N-methyl-d-aspartate receptor activation in conscious rats. J. Pharmacol. Exp. Ther. 2012, 342, 461–471. [Google Scholar] [CrossRef]
- Roepke, T.K.; Kontogeorgis, A.; Ovanez, C.; Xu, X.; Young, J.B.; Purtell, K.; Goldstein, P.A.; Christini, D.J.; Peters, N.S.; Akar, F.G.; et al. Targeted deletion of kcne2 impairs ventricular repolarization via disruption of I(K,slow1) and I(to,f). FASEB J. 2008, 22, 3648–3660. [Google Scholar] [CrossRef]
- Schmitt, N.; Grunnet, M.; Olesen, S.P. Cardiac potassium channel subtypes: New roles in repolarization and arrhythmia. Physiol. Rev. 2014, 94, 609–653. [Google Scholar] [CrossRef]
- Liu, Z.; Vuohelainen, V.; Tarkka, M.; Tenhunen, J.; Lappalainen, R.S.; Narkilahti, S.; Paavonen, T.; Oksala, N.; Wu, Z.; Mennander, A. Glutamate release predicts ongoing myocardial ischemia of rat hearts. Scand. J. Clin. Lab. Investig. 2010, 70, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Jannesar, K.; Abbaszadeh, S.; Malekinejad, H.; Soraya, H. Cardioprotective effects of memantine in myocardial ischemia: Ex vivo and in vivo studies. Eur. J. Pharmacol. 2020, 882, 173277. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadeh, S.; Javidmehr, A.; Askari, B.; Janssen, P.M.L.; Soraya, H. Memantine, an NMDA receptor antagonist, attenuates cardiac remodeling, lipid peroxidation and neutrophil recruitment in heart failure: A cardioprotective agent? Biomed. Pharmacother. 2018, 108, 1237–1243. [Google Scholar] [CrossRef]
- Maldonado, C.; Soni, C.V.; Todnem, N.D.; Pushpakumar, S.; Rosenberger, D.; Givvimani, S.; Villafane, J.; Tyagi, S.C. Hyperhomocysteinemia and sudden cardiac death: Potential arrhythmogenic mechanisms. Curr. Vasc. Pharmacol. 2010, 8, 64–74. [Google Scholar] [CrossRef]
- Winter, C.R.; Baker, R.C. L-glutamate-induced changes in intracellular calcium oscillation frequency through non-classical glutamate receptor binding in cultured rat myocardial cells. Life Sci. 1995, 57, 1925–1934. [Google Scholar] [CrossRef]
- Gilbert, G.; Demydenko, K.; Dries, E.; Puertas, R.D.; Jin, X.; Sipido, K.; Roderick, H.L. Calcium Signaling in Cardiomyocyte Function. Cold Spring Harb. Perspect. Biol. 2020, 12, a004242. [Google Scholar] [CrossRef]
- Moccia, F.; Lodola, F.; Stadiotti, I.; Pilato, C.A.; Bellin, M.; Carugo, S.; Pompilio, G.; Sommariva, E.; Maione, A.S. Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration? Int. J. Mol. Sci. 2019, 20, 3986. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafa, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca2+ oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell. Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef]
- Panama, B.K.; Korogyi, A.S.; Aschar-Sobbi, R.; Oh, Y.; Gray, C.B.; Gang, H.; Brown, J.H.; Kirshenbaum, L.A.; Backx, P.H. Reductions in the Cardiac Transient Outward K+ Current Ito Caused by Chronic beta-Adrenergic Receptor Stimulation Are Partly Rescued by Inhibition of Nuclear Factor kappaB. J. Biol. Chem. 2016, 291, 4156–4165. [Google Scholar] [CrossRef]
- Zarain-Herzberg, A.; Fragoso-Medina, J.; Estrada-Aviles, R. Calcium-regulated transcriptional pathways in the normal and pathologic heart. IUBMB Life 2011, 63, 847–855. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Zhong, Q.W.; Tian, C.N.; Ma, H.M.; Yu, J.J.; Hu, S. NMDA receptor-driven calcium influx promotes ischemic human cardiomyocyte apoptosis through a p38 MAPK-mediated mechanism. J. Cell. Biochem. 2019, 120, 4872–4882. [Google Scholar] [CrossRef]
- Govoruskina, N.; Jakovljevic, V.; Zivkovic, V.; Milosavljevic, I.; Jeremic, J.; Bradic, J.; Bolevich, S.; Omarov, I.A.; Djuric, D.; Radonjic, K.; et al. The Role of Cardiac N-Methyl-d-Aspartate Receptors in Heart Conditioning-Effects on Heart Function and Oxidative Stress. Biomolecules 2020, 10, 1065. [Google Scholar] [CrossRef]
- Stojic, I.; Srejovic, I.; Zivkovic, V.; Jeremic, N.; Djuric, M.; Stevanovic, A.; Milanovic, T.; Djuric, D.; Jakovljevic, V. The effects of verapamil and its combinations with glutamate and glycine on cardiodynamics, coronary flow and oxidative stress in isolated rat heart. J. Physiol. Biochem 2017, 73, 141–153. [Google Scholar] [CrossRef]
- Li, Q.; Qin, M.; Tan, Q.; Li, T.; Gu, Z.; Huang, P.; Ren, L. MicroRNA-129-1-3p protects cardiomyocytes from pirarubicin-induced apoptosis by down-regulating the GRIN2D-mediated Ca2+ signalling pathway. J. Cell. Mol. Med. 2020, 24, 2260–2271. [Google Scholar] [CrossRef]
- Lai, S.; Hua, X.; Gao, R.; Zeng, L.; Song, J.; Liu, J.; Zhang, J. Combinational Biomarkers for Atrial Fibrillation Derived from Atrial Appendage and Plasma Metabolomics Analysis. Sci. Rep. 2018, 8, 16930. [Google Scholar] [CrossRef]
- Leblanc, F.J.A.; Hassani, F.V.; Liesinger, L.; Qi, X.; Naud, P.; Birner-Gruenberger, R.; Lettre, G.; Nattel, S. Transcriptomic Profiling of Canine Atrial Fibrillation Models After One Week of Sustained Arrhythmia. Circ. Arrhythm. Electrophysiol. 2021, 14, e009887. [Google Scholar] [CrossRef]
- Buccelletti, E.; Gilardi, E.; Scaini, E.; Galiuto, L.; Persiani, R.; Biondi, A.; Basile, F.; Silveri, N.G. Heart rate variability and myocardial infarction: Systematic literature review and metanalysis. Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 299–307. [Google Scholar] [PubMed]
- Schultz, J.; Uddin, Z.; Singh, G.; Howlader, M.M.R. Glutamate sensing in biofluids: Recent advances and research challenges of electrochemical sensors. Analyst 2020, 145, 321–347. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic Glutamatergic Transmission and Its Implications in Neurodegenerative Disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef]
- Herrmann, W.; Herrmann, M. The Controversial Role of HCY and Vitamin B Deficiency in Cardiovascular Diseases. Nutrients 2022, 14, 1412. [Google Scholar] [CrossRef] [PubMed]
- Moshal, K.S.; Tipparaju, S.M.; Vacek, T.P.; Kumar, M.; Singh, M.; Frank, I.E.; Patibandla, P.K.; Tyagi, N.; Rai, J.; Metreveli, N.; et al. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H890–H897. [Google Scholar] [CrossRef]
- Moshal, K.S.; Kumar, M.; Tyagi, N.; Mishra, P.K.; Metreveli, N.; Rodriguez, W.E.; Tyagi, S.C. Restoration of contractility in hyperhomocysteinemia by cardiac-specific deletion of NMDA-R1. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H887–H892. [Google Scholar] [CrossRef]
- Tyagi, N.; Vacek, J.C.; Givvimani, S.; Sen, U.; Tyagi, S.C. Cardiac specific deletion of N-methyl-d-aspartate receptor 1 ameliorates mtMMP-9 mediated autophagy/mitophagy in hyperhomocysteinemia. J. Recept. Signal Transduct. Res. 2010, 30, 78–87. [Google Scholar] [CrossRef]
- Pedriali, G.; Ramaccini, D.; Bouhamida, E.; Wieckowski, M.R.; Giorgi, C.; Tremoli, E.; Pinton, P. Perspectives on mitochondrial relevance in cardiac ischemia/reperfusion injury. Front. Cell Dev. Biol. 2022, 10, 1082095. [Google Scholar] [CrossRef]
- Srejovic, I.; Zivkovic, V.; Nikolic, T.; Jeremic, N.; Stojic, I.; Jeremic, J.; Djuric, D.; Jakovljevic, V. Modulation of N-methyl-d-aspartate receptors in isolated rat heart. Can. J. Physiol. Pharmacol. 2017, 95, 1327–1334. [Google Scholar] [CrossRef]
- Srejovic, I.; Jakovljevic, V.; Zivkovic, V.; Barudzic, N.; Radovanovic, A.; Stanojlovic, O.; Djuric, D.M. The effects of the modulation of NMDA receptors by homocysteine thiolactone and dizocilpine on cardiodynamics and oxidative stress in isolated rat heart. Mol. Cell. Biochem. 2015, 401, 97–105. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Earley, S.; England, S.K.; Filosa, J.A.; Goulopoulou, S.; Gutterman, D.D.; Isakson, B.E.; Kanagy, N.L.; Martinez-Lemus, L.A.; et al. Guidelines for the measurement of vascular function and structure in isolated arteries and veins. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H77–H111. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Chen, H.; Fitzgerald, R.; Brown, A.T.; Qureshi, I.; Breckenridge, J.; Kazi, R.; Wang, Y.; Wu, Y.; Zhang, X.; Mukunyadzi, P.; et al. Identification of a homocysteine receptor in the peripheral endothelium and its role in proliferation. J. Vasc. Surg. 2005, 41, 853–860. [Google Scholar] [CrossRef]
- Qureshi, I.; Chen, H.; Brown, A.T.; Fitzgerald, R.; Zhang, X.; Breckenridge, J.; Kazi, R.; Crocker, A.J.; Stuhlinger, M.C.; Lin, K.; et al. Homocysteine-induced vascular dysregulation is mediated by the NMDA receptor. Vasc. Med. 2005, 10, 215–223. [Google Scholar] [CrossRef]
- Dumas, S.J.; Bru-Mercier, G.; Courboulin, A.; Quatredeniers, M.; Rucker-Martin, C.; Antigny, F.; Nakhleh, M.K.; Ranchoux, B.; Gouadon, E.; Vinhas, M.C.; et al. NMDA-Type Glutamate Receptor Activation Promotes Vascular Remodeling and Pulmonary Arterial Hypertension. Circulation 2018, 137, 2371–2389. [Google Scholar] [CrossRef]
- Dong, Y.N.; Hsu, F.C.; Koziol-White, C.J.; Stepanova, V.; Jude, J.; Gritsiuta, A.; Rue, R.; Mott, R.; Coulter, D.A.; Panettieri, R.A., Jr.; et al. Functional NMDA receptors are expressed by human pulmonary artery smooth muscle cells. Sci. Rep. 2021, 11, 8205. [Google Scholar] [CrossRef]
- Peters, E.C.; Gee, M.T.; Pawlowski, L.N.; Kath, A.M.; Polk, F.D.; Vance, C.J.; Sacoman, J.L.; Pires, P.W. Amyloid-beta disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries. J. Cereb. Blood Flow Metab. 2021, 42, 145–161. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vascul. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef]
- Mehra, A.; Guerit, S.; Macrez, R.; Gosselet, F.; Sevin, E.; Lebas, H.; Maubert, E.; De Vries, H.E.; Bardou, I.; Vivien, D.; et al. Non-ionotropic action of endothelial NMDA receptors on blood-brain barrier permeability via Rho/ROCK mediated phosphorylation of myosin. J. Neurosci. 2020, 40, 1778–1787. [Google Scholar] [CrossRef]
- Wang, J.; Swanson, R.A. Superoxide and Non-ionotropic Signaling in Neuronal Excitotoxicity. Front. Neurosci. 2020, 4, 861. [Google Scholar] [CrossRef] [PubMed]
- McGee, M.A.; Abdel-Rahman, A.A. Ethanol attenuates peripheral NMDAR-mediated vascular oxidative stress and pressor response. Alcohol 2015, 49, 499–506. [Google Scholar] [CrossRef]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- LeMaistre, J.L.; Sanders, S.A.; Stobart, M.J.; Lu, L.; Knox, J.D.; Anderson, H.D.; Anderson, C.M. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J. Cereb. Blood Flow Metab. 2012, 32, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Lu, L.; Anderson, H.D.; Mori, H.; Anderson, C.M. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2013, 110, 3149–3154. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial dysfunction: The link between homocysteine and hydrogen sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef]
- Doronzo, G.; Russo, I.; Del Mese, P.; Viretto, M.; Mattiello, L.; Trovati, M.; Anfossi, G. Role of NMDA receptor in homocysteine-induced activation of mitogen-activated protein kinase and phosphatidyl inositol 3-kinase pathways in cultured human vascular smooth muscle cells. Thromb. Res. 2010, 125, e23–e32. [Google Scholar] [CrossRef]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L.; Han, C.; Li, M.; Wang, S.; Wu, D. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-kappaB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef]
- Nassar, T.; Bdeir, K.; Yarovoi, S.; Fanne, R.A.; Murciano, J.C.; Idell, S.; Allen, T.C.; Cines, D.B.; Higazi, A.A. tPA regulates pulmonary vascular activity through NMDA receptors. Am J. Physiol.-Lung Cell. Mol. Physiol. 2011, 301, L307–L314. [Google Scholar] [CrossRef]
- Nassar, T.; Yarovoi, S.; Fanne, R.A.; Waked, O.; Allen, T.C.; Idell, S.; Cines, D.B.; Higazi, A.A. Urokinase plasminogen activator regulates pulmonary arterial contractility and vascular permeability in mice. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1015–1021. [Google Scholar] [CrossRef]
- White, R.J.; Wilkins, M.R. New Therapeutic Approaches in Pulmonary Arterial Hypertension: The Pantheon Is Getting Crowded. Circulation 2018, 137, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.B.; Blanpied, T.A.; Swanson, G.T.; Zhang, C.; Ehlers, M.D. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J. Neurosci. 2001, 21, 3063–3072. [Google Scholar] [CrossRef] [PubMed]
- Campos-Sandoval, J.A.; Martin-Rufian, M.; Cardona, C.; Lobo, C.; Penalver, A.; Marquez, J. Glutaminases in brain: Multiple isoforms for many purposes. Neurochem. Int. 2015, 88, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Quatredeniers, M.; Nakhleh, M.K.; Dumas, S.J.; Courboulin, A.; Vinhas, M.C.; Antigny, F.; Phan, C.; Guignabert, C.; Bendifallah, I.; Vocelle, M.; et al. Functional interaction between PDGFbeta and GluN2B-containing NMDA receptors in smooth muscle cell proliferation and migration in pulmonary arterial hypertension. Am J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L445–L455. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Naik, J.S.; Resta, T.C. Acid-sensing ion channel 1 contributes to pulmonary arterial smooth muscle cell depolarization following hypoxic pulmonary hypertension. J. Physiol. 2021, 599, 4749–4762. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Sladojevic, N.; Yu, B.; Liao, J.K. Regulator of G-Protein Signaling 5 Maintains Brain Endothelial Cell Function in Focal Cerebral Ischemia. J. Am. Heart Assoc. 2020, 9, e017533. [Google Scholar] [CrossRef]
- Lv, J.M.; Guo, X.M.; Chen, B.; Lei, Q.; Pan, Y.J.; Yang, Q. The Noncompetitive AMPAR Antagonist Perampanel Abrogates Brain Endothelial Cell Permeability in Response to Ischemia: Involvement of Claudin-5. Cell. Mol. Neurobiol. 2016, 36, 745–753. [Google Scholar] [CrossRef]
- Lim, D.; Mapelli, L.; Canonico, P.L.; Moccia, F.; Genazzani, A.A. Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. Int. J. Mol. Sci. 2018, 19, 2997. [Google Scholar] [CrossRef]
- Andras, I.E.; Deli, M.A.; Veszelka, S.; Hayashi, K.; Hennig, B.; Toborek, M. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J. Cereb. Blood Flow Metab. 2007, 27, 1431–1443. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Hogan-Cann, A.D.; Lu, P.; Anderson, C.M. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10229–10231. [Google Scholar] [CrossRef]
- Kim, K.S.; Jeon, M.T.; Kim, E.S.; Lee, C.H.; Kim, D.G. Activation of NMDA receptors in brain endothelial cells increases transcellular permeability. Fluids Barriers CNS 2022, 19, 70. [Google Scholar] [CrossRef]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2019, 39, 481–496. [Google Scholar] [CrossRef]
- Avemary, J.; Salvamoser, J.D.; Peraud, A.; Remi, J.; Noachtar, S.; Fricker, G.; Potschka, H. Dynamic regulation of P-glycoprotein in human brain capillaries. Mol. Pharm. 2013, 10, 3333–3341. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef]
- Epping, L.; Schroeter, C.B.; Nelke, C.; Bock, S.; Gola, L.; Ritter, N.; Herrmann, A.M.; Rauber, S.; Henes, A.; Wasser, B.; et al. Activation of non-classical NMDA receptors by glycine impairs barrier function of brain endothelial cells. Cell. Mol. Life Sci. 2022, 79, 479. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef]
- Reijerkerk, A.; Kooij, G.; van der Pol, S.M.; Leyen, T.; Lakeman, K.; van Het Hof, B.; Vivien, D.; de Vries, H.E. The NR1 subunit of NMDA receptor regulates monocyte transmigration through the brain endothelial cell barrier. J. Neurochem. 2010, 113, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Leger, C.; Dupre, N.; Aligny, C.; Benard, M.; Lebon, A.; Henry, V.; Hauchecorne, M.; Galas, L.; Frebourg, T.; Leroux, P.; et al. Glutamate controls vessel-associated migration of GABA interneurons from the pial migratory route via NMDA receptors and endothelial protease activation. Cell. Mol. Life Sci. 2020, 77, 1959–1986. [Google Scholar] [CrossRef]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson, T.H., IV; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2592–H2598. [Google Scholar] [CrossRef]
- Neuhaus, W.; Freidl, M.; Szkokan, P.; Berger, M.; Wirth, M.; Winkler, J.; Gabor, F.; Pifl, C.; Noe, C.R. Effects of NMDA receptor modulators on a blood-brain barrier in vitro model. Brain Res. 2011, 1394, 49–61. [Google Scholar] [CrossRef]
- Kuhlmann, C.R.; Gerigk, M.; Bender, B.; Closhen, D.; Lessmann, V.; Luhmann, H.J. Fluvastatin prevents glutamate-induced blood-brain-barrier disruption in vitro. Life Sci. 2008, 82, 1281–1287. [Google Scholar] [CrossRef]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef]
- Legros, H.; Launay, S.; Roussel, B.D.; Marcou-Labarre, A.; Calbo, S.; Catteau, J.; Leroux, P.; Boyer, O.; Ali, C.; Marret, S.; et al. Newborn- and adult-derived brain microvascular endothelial cells show age-related differences in phenotype and glutamate-evoked protease release. J. Cereb. Blood Flow Metab. 2009, 29, 1146–1158. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Tyagi, S.C.; Tyagi, N. Hydrogen Sulfide Epigenetically Attenuates Homocysteine-Induced Mitochondrial Toxicity Mediated Through NMDA Receptor in Mouse Brain Endothelial (bEnd3) Cells. J. Cell. Physiol. 2015, 230, 378–394. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient Receptor Potential Channel Ankyrin 1: A Unique Regulator of Vascular Function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Chow, B.W.; Nunez, V.; Kaplan, L.; Granger, A.J.; Bistrong, K.; Zucker, H.L.; Kumar, P.; Sabatini, B.L.; Gu, C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 2020, 579, 106–110. [Google Scholar] [CrossRef]
- Anfray, A.; Drieu, A.; Hingot, V.; Hommet, Y.; Yetim, M.; Rubio, M.; Deffieux, T.; Tanter, M.; Orset, C.; Vivien, D. Circulating tPA contributes to neurovascular coupling by a mechanism involving the endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2020, 40, 2038–2054. [Google Scholar] [CrossRef]
- Hopper, R.A.; Garthwaite, J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J. Neurosci. 2006, 26, 11513–11521. [Google Scholar] [CrossRef]
- Haul, S.; Godecke, A.; Schrader, J.; Haas, H.L.; Luhmann, H.J. Impairment of neocortical long-term potentiation in mice deficient of endothelial nitric oxide synthase. J. Neurophysiol. 1999, 81, 494–497. [Google Scholar] [CrossRef]
- Blackshaw, S.; Eliasson, M.J.; Sawa, A.; Watkins, C.C.; Krug, D.; Gupta, A.; Arai, T.; Ferrante, R.J.; Snyder, S.H. Species, strain and developmental variations in hippocampal neuronal and endothelial nitric oxide synthase clarify discrepancies in nitric oxide-dependent synaptic plasticity. Neuroscience 2003, 119, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Salvamoser, J.D.; Avemary, J.; Luna-Munguia, H.; Pascher, B.; Getzinger, T.; Pieper, T.; Kudernatsch, M.; Kluger, G.; Potschka, H. Glutamate-Mediated Down-Regulation of the Multidrug-Resistance Protein BCRP/ABCG2 in Porcine and Human Brain Capillaries. Mol. Pharm. 2015, 12, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.D.; Houghton, J.; Elrod, J.W.; Warren, A.; Jackson, T.H., IV; Jawahar, A.; Nanda, A.; Minagar, A.; Alexander, J.S. N-methyl-d-aspartate receptor activation in human cerebral endothelium promotes intracellular oxidant stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1893–H1899. [Google Scholar] [CrossRef]
- Scott, G.S.; Bowman, S.R.; Smith, T.; Flower, R.J.; Bolton, C. Glutamate-stimulated peroxynitrite production in a brain-derived endothelial cell line is dependent on N-methyl-d-aspartate (NMDA) receptor activation. Biochem. Pharmacol. 2007, 73, 228–236. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 2016, 1862, 461–471. [Google Scholar] [CrossRef]
- Macrez, R.; Ortega, M.C.; Bardou, I.; Mehra, A.; Fournier, A.; Van der Pol, S.M.; Haelewyn, B.; Maubert, E.; Lesept, F.; Chevilley, A.; et al. Neuroendothelial NMDA receptors as therapeutic targets in experimental autoimmune encephalomyelitis. Brain 2016, 139 Pt 9, 2406–2419. [Google Scholar] [CrossRef]
- Louet, E.R.; Glavan, M.; Orset, C.; Parcq, J.; Hanley, D.F.; Vivien, D. tPA-NMDAR Signaling Blockade Reduces the Incidence of Intracerebral Aneurysms. Transl Stroke Res 2022, 13, 1005–1016. [Google Scholar] [CrossRef]
- Seillier, C.; Lesept, F.; Toutirais, O.; Potzeha, F.; Blanc, M.; Vivien, D. Targeting NMDA Receptors at the Neurovascular Unit: Past and Future Treatments for Central Nervous System Diseases. Int. J. Mol. Sci. 2022, 23, 10336. [Google Scholar] [CrossRef] [PubMed]
- Wagnon, I.; Helie, P.; Bardou, I.; Regnauld, C.; Lesec, L.; Leprince, J.; Naveau, M.; Delaunay, B.; Toutirais, O.; Lemauff, B.; et al. Autoimmune encephalitis mediated by B-Cell Res.ponse against N-methyl-d-aspartate receptor. Brain 2020, 143, 2957–2972. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Wang, C. Anti-NMDA Receptor Autoimmune Encephalitis: Diagnosis and Management Strategies. Int. J. Gen. Med. 2023, 16, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Hammer, C.; Stepniak, B.; Schneider, A.; Papiol, S.; Tantra, M.; Begemann, M.; Siren, A.L.; Pardo, L.A.; Sperling, S.; Mohd Jofrry, S.; et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol. Psychiatry 2014, 19, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Sonkar, V.K.; Katare, P.B.; Kumar, R.; Kruger, W.D.; Arning, E.; Bottiglieri, T.; Lentz, S.R.; Dayal, S. Memantine Protects From Exacerbation of Ischemic Stroke and Blood Brain Barrier Disruption in Mild But Not Severe Hyperhomocysteinemia. J. Am. Heart Assoc. 2020, 9, e013368. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Sonkusare, S.K. The Calcium Signaling Mechanisms in Arterial Smooth Muscle and Endothelial Cells. Compr. Physiol. 2021, 11, 1831–1869. [Google Scholar] [PubMed]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chamber | Species | Subunit Expression | Proposed NMDAR Composition | Role | References |
|---|---|---|---|---|---|
| Atrium | |||||
| Human | GluN1, GluN3 | Not defined | Excitability and conductivity | [35,37] | |
| Rat | GluN1, GluN2A, GluN2B, GluN2C, GluN2D, GluN3A, GluN3B | Not defined | Excitability and conductivity, fibrillation | [34,37,38] | |
| Monkey | GluN1 | Not defined | Not investigated | [33] | |
| Conduction system | |||||
| Human | GluN1 | Not defined | Not investigated | [35] | |
| Rat | GluN1 | Not defined | Not investigated | [34] | |
| Monkey | GluN1 | Not defined | Not investigated | [33] | |
| Ventricle | |||||
| Human | GluN1 | Not defined | [35] | ||
| Rat | GluN1, GluN2B | Not defined | Delayed repolarization, arrhythmias, fibrosis, apoptosis, mitochondrial dysfunction | [34,39,40,41,42,43,44,45,46,47] | |
| Monkey | GluN1 | Not defined | Not investigated | [33] |
| Cell Type | Species | Subunit Expression | Proposed NMDAR Composition | Role | References |
|---|---|---|---|---|---|
| Aortic VSMCs | Rat | GluN1, GluN2A, GluN2B, GluN2C, GluN2D | Not defined | Proliferation, vasoconstriction, MMP-9 and IL-1β expression, reduction in basal NO release | [48,85] |
| hPASMCs | Human | GluN1, GluN2A, GluN2B, GluN2C, GluN2D | Not defined | Proliferation, hypoxia-induced pulmonary hypertension and vascular remodeling | [86,87] |
| Aortic endothelial cells | Rat | GluN1, GluN2A, GluN2B, GluN2C, GluN2D | Not defined | Proliferation | [85] |
| Cell Type | Species | Subunit Expression | Proposed NMDAR Composition | Role | References |
|---|---|---|---|---|---|
| Endothelial cell | Human | GluN1, GluN2C, GluN3A | Not defined | NO release, increase in transcellular and paracellular permeability | [24,90,115] |
| Endothelial cell | Mouse | GluN1, GluN2A, GluN2B, GluN3A | Not defined | NO release, increase in transcellular and paracellular permeability, monocyte transmigration, GABA interneuron migration | [90,94,95,114,115,116,123,124] |
| Disease | Altered Function | Pharmacological Targeting | References |
|---|---|---|---|
| Epilepsy | BBB disruption | AP5 | [128] |
| Stroke | BBB disruption | MK-801 | [108] |
| Intracerebral aneurysm | BBB disruption | Glunomab | [150] |
| Multiple sclerosis | BBB disruption and monocyte infiltration | Glunomab | [148,149] |
| HHyc | BBB disruption | Memantine | [155] |
| AD | Reduced arterial vasodilation | Not assessed | [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soda, T.; Brunetti, V.; Berra-Romani, R.; Moccia, F. The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 3914. https://doi.org/10.3390/ijms24043914
Soda T, Brunetti V, Berra-Romani R, Moccia F. The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. International Journal of Molecular Sciences. 2023; 24(4):3914. https://doi.org/10.3390/ijms24043914
Chicago/Turabian StyleSoda, Teresa, Valentina Brunetti, Roberto Berra-Romani, and Francesco Moccia. 2023. "The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives" International Journal of Molecular Sciences 24, no. 4: 3914. https://doi.org/10.3390/ijms24043914
APA StyleSoda, T., Brunetti, V., Berra-Romani, R., & Moccia, F. (2023). The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. International Journal of Molecular Sciences, 24(4), 3914. https://doi.org/10.3390/ijms24043914