
Pathogenicity of Type I Interferons in Mycobacterium tuberculosis
 ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Background
2.1. M. tb: Transmission and Clinical Manifestations
2.2. M. tb: Pathogenesis of Latent Infection
2.3. M. tb: Pathogenesis of Active Infection
2.4. M. tb: Risk Factors and Epidemiology
2.5. M. tb: Prevention
2.6. M. tb: Treatment
3. Interferons
3.1. Type I Interferons: Production and Signaling
3.2. Role of Type II Interferons during M. tb Infection
3.3. Other Key Mediators in Host M. tb Immune Response
4. Role of Type I Interferons in M. tb Infection
4.1. Type I IFN Transcriptional Signature in M. tb Infection
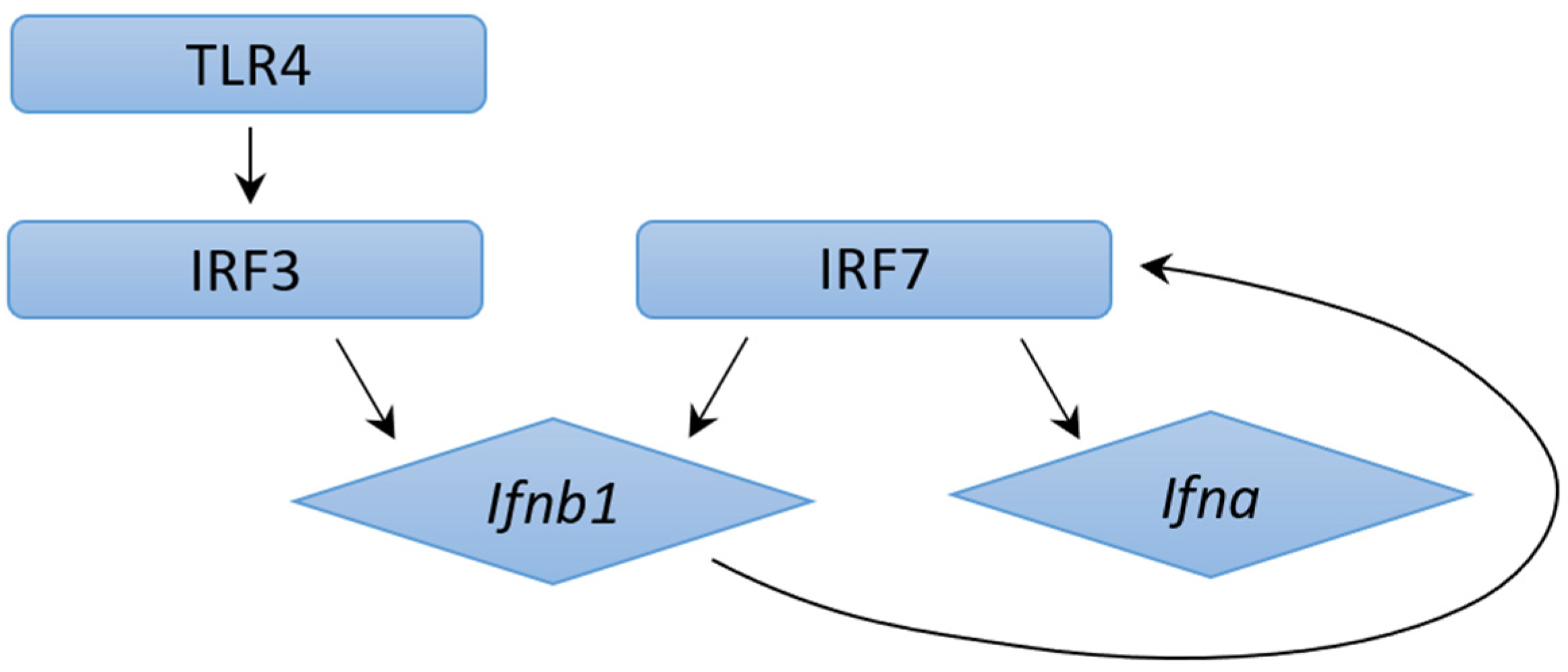
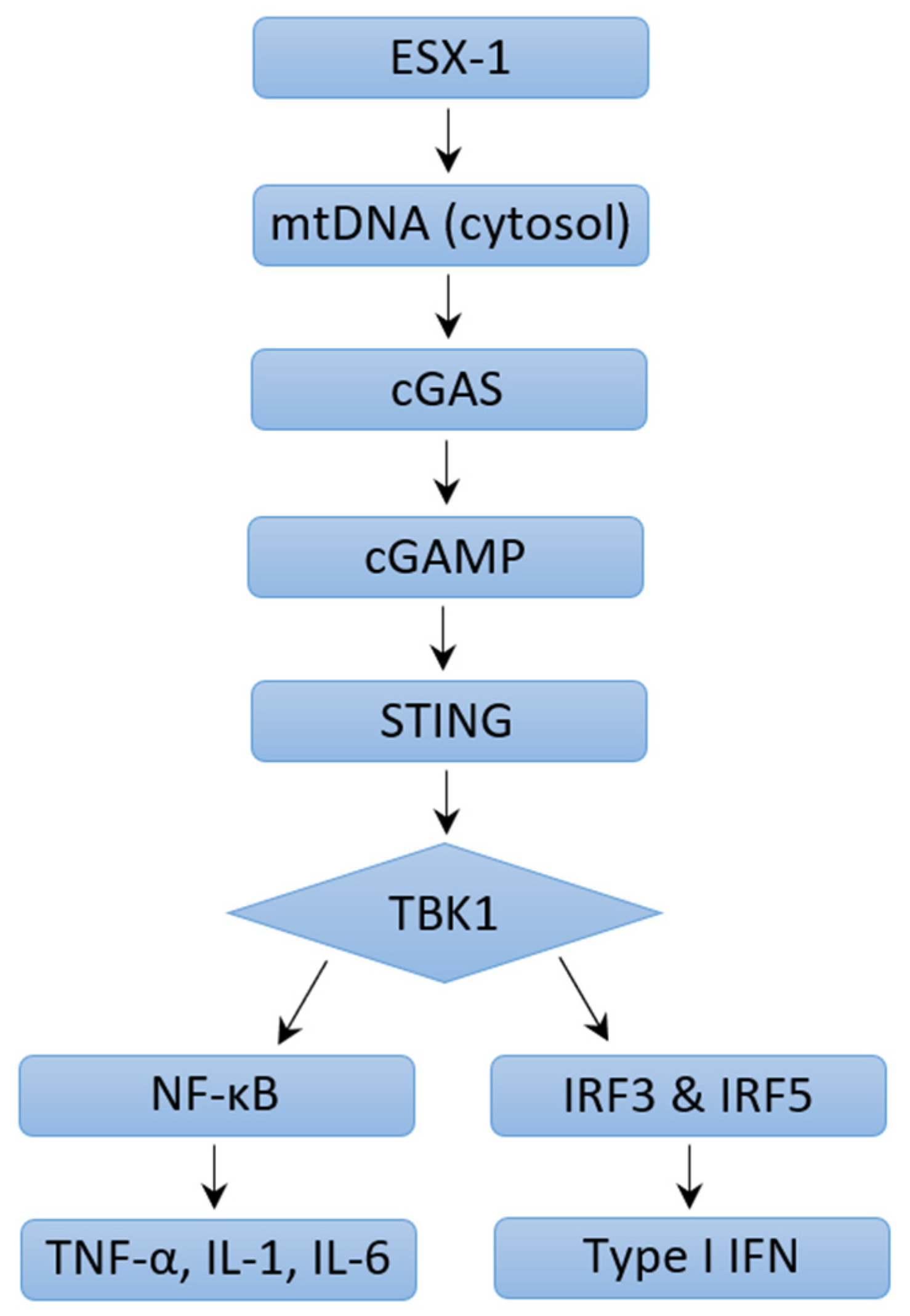
4.2. Mechanisms of Type I IFN Induction in M. tb Infection: ESX-1 Protein Secretion System and Pattern Recognition Receptors
4.3. Mechanisms of Type I IFN Induction in M. tb Infection: ESX-1 Protein Secretion System Independent Mechanisms
4.4. Pathogenic Effects of Type I IFN Signaling during M. tb Infection
4.5. Potential Protective Functions of Type I IFN in M. tb Infection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herzog, B.H. History of Tuberculosis. Respiration 1998, 65, 5–15. [Google Scholar] [CrossRef]
- Koch, A.; Mizrahi, V. Mycobacterium tuberculosis. Trends Microbiol. 2018, 26, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Bañuls, A.-L.; Sanou, A.; van Anh, N.T.; Godreuil, S. Mycobacterium tuberculosis: Ecology and evolution of a human bacterium. J. Med. Microbiol. 2015, 64, 1261–1269. [Google Scholar] [CrossRef]
- Bannon, M.J.; Finn, A. BCG and tuberculosis Commentary. Arch. Dis. Child. 1999, 80, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Frieden, T.R.; Sterling, T.R.; Munsiff, S.S.; Watt, C.J.; Dye, C. Tuberculosis. Lancet 2003, 362, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, A.T. History of Pulmonary Tuberculosis. Thorac. Surg. Clin. 2019, 29, 1–17. [Google Scholar] [CrossRef]
- World Health Organization Global Tuberculosis Report 2022. 2022. Available online: http://apps.who.int/bookorders (accessed on 3 January 2023).
- Low, J.G.H.; Lee, C.C.; Leo, Y.S.; Low, J.G.-H.; Lee, C.-C.; Leo, Y.-S. Severe Acute Respiratory Syndrome and Pulmonary Tuberculosis. Clin. Infect. Dis. 2004, 38, e123–e125. [Google Scholar] [CrossRef]
- Chakaya, J.; Petersen, E.; Nantanda, R.; Mungai, B.; Migliori, G.; Amanullah, F.; Lungu, P.; Ntoumi, F.; Kumarasamy, N.; Maeurer, M.; et al. The WHO Global Tuberculosis 2021 Report—Not so good news and turning the tide back to End TB. Int. J. Infect. Dis. 2022, 124, S26–S29. [Google Scholar] [CrossRef]
- Zimmer, A.J.; Klinton, J.S.; Oga-Omenka, C.; Heitkamp, P.; Nyirenda, C.N.; Furin, J.; Pai, M. Tuberculosis in times of COVID-19. J. Epidemiol. Community Health 2022, 76, 310–316. [Google Scholar] [CrossRef]
- Koegelenberg, C.F.N.; Schoch, O.D.; Lange, C. Tuberculosis: The Past, the Present and the Future. Respiration 2021, 100, 553–556. [Google Scholar] [CrossRef]
- Bloom, B.R.; Atun, R.; Cohen, T.; Dye, C.; Fraser, H.; Gomez, G.B.; Knight, G.; Murray, M.; Nardell, E.; Rubin, E.; et al. Tuberculosis. In Disease Control Priorities, Third Edition (Volume 6): Major Infectious Diseases; The World Bank: Washington, DC, USA, 2017; pp. 233–313. [Google Scholar] [CrossRef]
- Langer, A.J.; Navin, T.R.; Winston, C.A.; LoBue, P. Epidemiology of Tuberculosis in the United States. Clin. Chest Med. 2019, 40, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Churchyard, G.; Kim, P.; Shah, N.S.; Rustomjee, R.; Gandhi, N.; Mathema, B.; Dowdy, D.; Kasmar, A.; Cardenas, V. What We Know About Tuberculosis Transmission: An Overview. J. Infect. Dis. 2017, 216 (Suppl. S6), S629–S635. [Google Scholar] [CrossRef] [PubMed]
- Lawn, S.D.; Zumla, A.I. Tuberculosis. Lancet 2011, 378, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Loddenkemper, R.; Lipman, M.; Zumla, A. Clinical Aspects of Adult Tuberculosis. Cold Spring Harb. Perspect. Med. 2016, 6, a017848. [Google Scholar] [CrossRef]
- Sunnetcioglu, A.; Sunnetcioglu, M.; Binici, I.; Baran, A.I.; Karahocagil, M.K.; Saydan, M.R. Comparative analysis of pulmonary and extrapulmonary tuberculosis of 411 cases. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Song, Y.G.; Park, W.I.; Choi, J.P.; Chang, K.H.; Kim, J.M. Clinical Manifestations and Diagnosis of Extrapulmonary Tuberculosis. Yonsei Med. J. 2004, 45, 453–461. [Google Scholar] [CrossRef]
- Dev, N.; Bhowmick, M.; Chaudhary, S.; Kant, J. Tuberculous encephalopathy without meningitis: A rare manifestation of disseminated tuberculosis. Int. J. Mycobacteriol. 2019, 8, 406. [Google Scholar] [CrossRef]
- Sharma, S.K.; Mohan, A. Miliary Tuberculosis. Microbiol. Spectr. 2017, 5, 10. [Google Scholar] [CrossRef]
- Ahmad, S. Pathogenesis, immunology, and diagnosis of latent mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2011, 2011, 814943. [Google Scholar] [CrossRef]
- Bonavida, V.; Frame, M.; Nguyen, K.H.; Rajurkar, S.; Venketaraman, V. Mycobacterium tuberculosis: Implications of Ageing on Infection and Maintaining Protection in the Elderly. Vaccines 2022, 10, 1892. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.T.; Young, E.F.; Mccann, J.R.; Braunstein, M. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect. Immun. 2012, 80, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, D.; Caws, M.; Marais, B.; Farrar, J. Tuberculosis in Adults and Children. Available online: http://www.springer.com/series/10138 (accessed on 8 February 2023).
- Jilani, T.N.; Avula, A.; Gondal, Z.A.; Siddiqui, A.H. Active Tuberculosis; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Narasimhan, P.; Wood, J.; Macintyre, C.R.; Mathai, D. Risk factors for tuberculosis. In Pulmonary Medicine; Hindawi Publishing Corporation: London, UK, 2013. [Google Scholar] [CrossRef]
- Lewinsohn, D.A.; Gennaro, M.L.; Scholvinck, L.; Lewinsohn, D.M. Tuberculosis immunology in children: Diagnostic and therapeutic challenges and opportunities. Int. J. Tuberc. Lung Dis. 2004, 8, 658–674. [Google Scholar]
- Roy, A.; Eisenhut, M.; Harris, R.J.; Rodrigues, L.C.; Sridhar, S.; Habermann, S.; Snell, L.B.; Mangtani, P.; Adetifa, I.; Lalvani, A.; et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: Systematic review and meta-analysis. BMJ 2014, 349, g4643. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.; Denkinger, C.M.; Kik, S.V.; Rangaka, M.X.; Zwerling, A.; Oxlade, O.; Metcalfe, J.Z.; Cattamanchi, A.; Dowdy, D.W.; Dheda, K.; et al. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin. Microbiol. Rev. 2014, 27, 3–20. [Google Scholar] [CrossRef]
- Katelaris, A.L.; Jackson, C.; Southern, J.; Gupta, R.; Drobniewski, F.; Lalvani, A.; Lipman, M.; Mangtani, P.; Abubakar, I. Effectiveness of BCG Vaccination Against Mycobacterium tuberculosis Infection in Adults: A Cross-sectional Analysis of a UK-Based Cohort. J. Infect. Dis. 2020, 221, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Sulis, G.; Roggi, A.; Matteelli, A.; Raviglione, M.C. Tuberculosis: Epidemiology and control. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014070. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. End TB Strategy; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis Module 4: Treatment Drug-Susceptible Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 1, Prevention: Tuberculosis Preventive Treatment; World Health Organization: Geneva, Switzerland, February 2020. [Google Scholar]
- Balu, S.; Reljic, R.; Lewis, M.J.; Pleass, R.J.; McIntosh, R.; van Kooten, C.; van Egmond, M.; Challacombe, S.; Woof, J.M.; Ivanyi, J. A Novel Human IgA Monoclonal Antibody Protects against Tuberculosis. J. Immunol. 2011, 186, 3113–3119. [Google Scholar] [CrossRef]
- López, Y.; Yero, D.; Falero-Diaz, G.; Olivares, N.; Sarmiento, M.E.; Sifontes, S.; Solis, R.L.; Barrios, J.A.; Aguilar, D.; Hernández-Pando, R.; et al. Induction of a protective response with an IgA monoclonal antibody against Mycobacterium tuberculosis 16 kDa protein in a model of progressive pulmonary infection. Int. J. Med. Microbiol. 2009, 299, 447–452. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Shin, E.-C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; e Sousa, C.R. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Shanmuganathan, G.; Orujyan, D.; Narinyan, W.; Poladian, N.; Dhama, S.; Parthasarathy, A.; Ha, A.; Tran, D.; Velpuri, P.; Nguyen, K.H.; et al. Role of Interferons in Mycobacterium tuberculosis Infection. Clin. Pract. 2022, 12, 788–796. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Mach, B.; Steimle, V.; Martinez-Soria, E.; Reith, W. REGULATION OF MHC CLASS II GENES: Lessons from a Disease. Annu. Rev. Immunol. 1996, 14, 301–331. [Google Scholar] [CrossRef]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. THE IFNγ RECEPTOR: A Paradigm for Cytokine Receptor Signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef]
- Ramana, C.V.; Gil, M.P.; Schreiber, R.D.; Stark, G.R. Stat1-dependent and -independent pathways in IFN-γ-dependent signaling. Trends Immunol. 2002, 23, 96–101. [Google Scholar] [CrossRef]
- Casanova, J.-L.; Abel, L. Genetic Dissection of Immunity to Mycobacteria: The Human Model. Annu. Rev. Immunol. 2002, 20, 581–620. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; DiFazio, R.; Flynn, J.L. IFN-γ from CD4 T Cells Is Essential for Host Survival and Enhances CD8 T Cell Function during Mycobacterium tuberculosis Infection. J. Immunol. 2013, 190, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, Y.V.N.; Brelaz, M.C.A.; de Neves, J.K.A.L.; Ferraz, J.C.; Pereira, V.R.A. Role of TNF-Alpha, IFN-Gamma, and IL-10 in the Development of Pulmonary Tuberculosis. Pulm. Med. 2012, 2012, 745483. [Google Scholar] [CrossRef] [PubMed]
- Ladel, C.H.; Szalay, G.; Riedel, D.; Kaufmann, S.H. Interleukin-12 secretion by Mycobacterium tuberculosis-infected macrophages. Infect. Immun. 1997, 65, 1936–1938. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, G.; Megjugorac, N.J.; Yu, R.Y.; Eskdale, J.; Siegel, R.; Tollar, E. The Lambda Interferons: Guardians of the Immune–Epithelial Interface and the T-helper 2 Response. J. Interferon Cytokine Res. 2010, 30, 603–615. [Google Scholar] [CrossRef]
- Khan, T.A.; Mazhar, H.; Saleha, S.; Tipu, H.N.; Muhammad, N.; Abbas, M.N. Interferon-Gamma Improves Macrophages Function against M. tuberculosis in Multidrug-Resistant Tuberculosis Patients. Chemother. Res. Pract. 2016, 2016, 7295390. [Google Scholar] [CrossRef]
- Bae, H.; Barlow, A.T.; Young, H.; Valencia, J.C. Interferon γ: An Overview of Its Functions in Health and Disease. In Encyclopedia of Immunobiology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 494–500. [Google Scholar] [CrossRef]
- Cooper, A.M.; Adams, L.B.; Dalton, D.K.; Appelberg, R.; Ehlers, S. IFN-γ and NO in mycobacterial disease: New jobs for old hands. Trends Microbiol. 2002, 10, 221–226. [Google Scholar] [CrossRef]
- Saito, S.; Nakano, M. Nitric oxide production by peritoneal macrophages of Mycobacterium bovis BCG-infected or non-infected mice: Regulatory roles of T lymphocytes and cytokines. J. Leukoc. Biol. 1996, 59, 908–915. [Google Scholar] [CrossRef]
- Zuñiga, J.; Torres-García, D.; Santos-Mendoza, T.; Rodriguez-Reyna, T.S.; Granados, J.; Yunis, E.J. Cellular and Humoral Mechanisms Involved in the Control of Tuberculosis. Clin. Dev. Immunol. 2012, 2012, 193923. [Google Scholar] [CrossRef]
- Fabri, M.; Stenger, S.; Shin, D.-M.; Yuk, J.-M.; Liu, P.T.; Realegeno, S.; Lee, H.-M.; Krutzik, S.R.; Schenk, M.; Sieling, P.A.; et al. Vitamin D Is Required for IFN-γ–Mediated Antimicrobial Activity of Human Macrophages. Sci. Transl. Med. 2011, 3, 104ra102. [Google Scholar] [CrossRef]
- Yuk, J.-M.; Yoshimori, T.; Jo, E.-K. Autophagy and bacterial infectious diseases. Exp. Mol. Med. 2012, 44, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Yuk, J.-M.; Lee, H.-M.; Lee, S.-H.; Son, J.W.; Harding, C.V.; Kim, J.-M.; Modlin, R.L.; Jo, E.-K. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol. 2010, 12, 1648–1665. [Google Scholar] [CrossRef] [PubMed]
- Korbel, D.S.; Schneider, B.E.; Schaible, U.E. Innate immunity in tuberculosis: Myths and truth. Microbes Infect. 2008, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J. Immune evasion by Mycobacterium tuberculosis: Living with the enemy. Curr. Opin. Immunol. 2003, 15, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Kleinnijenhuis, J.; Oosting, M.; Joosten, L.A.B.; Netea, M.G.; van Crevel, R. Innate Immune Recognition of Mycobacterium tuberculosis. Clin. Dev. Immunol. 2011, 2011, 405310. [Google Scholar] [CrossRef]
- Saiga, H.; Shimada, Y.; Takeda, K. Innate Immune Effectors in Mycobacterial Infection. Clin. Dev. Immunol. 2011, 2011, 347594. [Google Scholar] [CrossRef]
- Schwander, S.; Dheda, K. Human Lung Immunity against Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 2011, 183, 696–707. [Google Scholar] [CrossRef]
- Means, T.K.; Wang, S.; Lien, E.; Yoshimura, A.; Golenbock, D.T.; Fenton, M.J. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 1999, 163, 3920–3927. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 Is an Adaptor Protein in the hToll/IL-1 Receptor Family Signaling Pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 2001, 1, 20–30. [Google Scholar] [CrossRef]
- Cooper, A.M.; Mayer-Barber, K.D.; Sher, A. Role of innate cytokines in mycobacterial infection. Mucosal. Immunol. 2011, 4, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Martin, C.J.; Nunes-Alves, C.; Divangahi, M.; Remold, H.G. Lipids, apoptosis, and cross-presentation: Links in the chain of host defense against Mycobacterium tuberculosis. Microbes Infect. 2011, 13, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, A.; Alt, J.; Perera, V.; Behar, S.M. Activation of NKT Cells Protects Mice from Tuberculosis. Infect. Immun. 2002, 70, 6302–6309. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-T.; Cambier, C.J.; Davis, J.M.; Hall, C.J.; Crosier, P.S.; Ramakrishnan, L. Neutrophils Exert Protection in the Early Tuberculous Granuloma by Oxidative Killing of Mycobacteria Phagocytosed from Infected Macrophages. Cell Host Microbe 2012, 12, 301–312. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Chang, Q.; Ma, P.; Hu, Y.; Cao, X. Type III Interferons in Viral Infection and Antiviral Immunity. Cell. Physiol. Biochem. 2018, 51, 173–185. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda Interferon (IFN-λ), a Type III IFN, Is Induced by Viruses and IFNs and Displays Potent Antiviral Activity against Select Virus Infections In Vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef]
- Iversen, M.B.; Paludan, S.R. Mechanisms of Type III Interferon Expression. J. Interferon Cytokine Res. 2010, 30, 573–578. [Google Scholar] [CrossRef]
- Tezuka, Y.; Endo, S.; Matsui, A.; Sato, A.; Saito, K.; Semba, K.; Takahashi, M.; Murakami, T. Potential anti-tumor effect of IFN-λ2 (IL-28A) against human lung cancer cells. Lung Cancer 2012, 78, 185–192. [Google Scholar] [CrossRef]
- Marcello, T.; Grakoui, A.; Spaeth, G.B.; Machlin, E.S.; Kotenko, S.V.; Macdonald, M.R.; Rice, C.M. Interferons α and λ Inhibit Hepatitis C Virus Replication with Distinct Signal Transduction and Gene Regulation Kinetics. Gastroenterology 2006, 131, 1887–1898. [Google Scholar] [CrossRef]
- Travar, M.; Vucic, M.; Petkovic, M. Interferon Lambda-2 Levels in Sputum of Patients with Pulmonary Mycobacterium tuberculosis Infection. Scand. J. Immunol. 2014, 80, 43–49. [Google Scholar] [CrossRef]
- Bierne, H.; Travier, L.; Mahlakoiv, T.; Tailleux, L.; Subtil, A.; Lebreton, A.; Paliwal, A.; Gicquel, B.; Staeheli, P.; Lecuit, M.; et al. Activation of Type III Interferon Genes by Pathogenic Bacteria in Infected Epithelial Cells and Mouse Placenta. PLoS ONE 2012, 7, e39080. [Google Scholar] [CrossRef]
- Kotenko, S.V. IFN-λs. Curr. Opin. Immunol. 2011, 23, 583–590. [Google Scholar] [CrossRef]
- Barnes, P.F.; Abrams, J.S.; Lu, S.; Sieling, P.A.; Rea, T.H.; Modlin, R.L. Patterns of cytokine production by mycobacterium-reactive human T-cell clones. Infect. Immun. 1993, 61, 197–203. [Google Scholar] [CrossRef]
- Liew, F.Y.; Li, Y.; Millott, S. Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J. Immunol. 1990, 145, 4306–4310. [Google Scholar] [CrossRef]
- Gong, J.H.; Zhang, M.; Modlin, R.L.; Linsley, P.S.; Iyer, D.; Lin, Y.; Barnes, P.F. Interleukin-10 downregulates Mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infect. Immun. 1996, 64, 913–918. [Google Scholar] [CrossRef]
- Orme, I.M.; Cooper, A.M. Cytokine/chemokine cascades in immunity to tuberculosis. Immunol. Today 1999, 20, 307–312. [Google Scholar] [CrossRef]
- Fraziano, M.; Cappelli, G.; Santucci, M.; Mariani, F.; Amicosante, M.; Casarini, M.; Giosuè, S.; Bisetti, A.; Colizzi, V. Expression of CCR5 Is Increased in Human Monocyte-Derived Macrophages and Alveolar Macrophages in the Course of in Vivo and in Vitro Mycobacterium tuberculosis Infection. AIDS Res. Hum. Retrovir. 1999, 15, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Venketaraman, V.; Dayaram, Y.K.; Amin, A.G.; Ngo, R.; Green, R.M.; Talaue, M.T.; Mann, J.; Connell, N.D. Role of Glutathione in Macrophage Control of Mycobacteria. Infect. Immun. 2003, 71, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Venketaraman, V.; Dayaram, Y.K.; Talaue, M.T.; Connell, N.D. Glutathione and Nitrosoglutathione in Macrophage Defense against Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Millman, A.C.; Salman, M.; Dayaram, Y.K.; Connell, N.D.; Venketaraman, V. Natural Killer Cells, Glutathione, Cytokines, and Innate Immunity against Mycobacterium tuberculosis. J. Interferon Cytokine Res. 2008, 28, 153–165. [Google Scholar] [CrossRef]
- Guerra, C.; Morris, D.; Sipin, A.; Kung, S.; Franklin, M.; Gray, D.; Tanzil, M.; Guilford, F.; Khasawneh, F.T.; Venketaraman, V. Glutathione and Adaptive Immune Responses against Mycobacterium tuberculosis Infection in Healthy and HIV Infected Individuals. PLoS ONE 2011, 6, e28378. [Google Scholar] [CrossRef]
- Ashley, D.; Hernandez, J.; Cao, R.; To, K.; Yegiazaryan, A.; Abrahem, R.; Nguyen, T.; Owens, J.; Lambros, M.; Subbian, S.; et al. Antimycobacterial Effects of Everolimus in a Human Granuloma Model. J. Clin. Med. 2020, 9, 2043. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Jagannath, C.; Lindsey, D.R.; Dhandayuthapani, S.; Xu, Y.; Hunter, R.L.; Eissa, N.T. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med. 2009, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Vitamin D Inhibits Human Immunodeficiency Virus Type 1 and Mycobacterium tuberculosis Infection in Macrophages through the Induction of Autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Regulation of Phagosome Maturation by Signals from Toll-Like Receptors. Science 2004, 304, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Bafica, A.; Scanga, C.A.; Serhan, C.; Machado, F.; White, S.; Sher, A.; Aliberti, J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase–dependent lipoxin production. J. Clin. Investig. 2005, 115, 1601–1606. [Google Scholar] [CrossRef]
- Shivakoti, R.; Dalli, J.; Kadam, D.; Gaikwad, S.; Barthwal, M.; Colas, R.A.; Mazzacuva, F.; Lokhande, R.; Dharmshale, S.; Bharadwaj, R.; et al. Lipid mediators of inflammation and Resolution in individuals with tuberculosis and tuberculosis-Diabetes. Prostaglandins Other Lipid Mediat. 2020, 147, 106398. [Google Scholar] [CrossRef]
- Berry, M.P.R.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Ottenhoff, T.H.M.; Dass, R.H.; Yang, N.; Zhang, M.; Wong, H.E.E.; Sahiratmadja, E.; Khor, C.C.; Alisjahbana, B.; Van Crevel, R.; Marzuki, S.; et al. Genome-Wide Expression Profiling Identifies Type 1 Interferon Response Pathways in Active Tuberculosis. PLoS ONE 2012, 7, e45839. [Google Scholar] [CrossRef]
- Bloom, C.I.; Graham, C.M.; Berry, M.P.R.; Rozakeas, F.; Redford, P.S.; Wang, Y.; Xu, Z.; Wilkinson, K.A.; Wilkinson, R.J.; Kendrick, Y.; et al. Transcriptional Blood Signatures Distinguish Pulmonary Tuberculosis, Pulmonary Sarcoidosis, Pneumonias and Lung Cancers. PLoS ONE 2013, 8, e70630. [Google Scholar] [CrossRef]
- Wang, J.; Hussain, T.; Zhang, K.; Liao, Y.; Yao, J.; Song, Y.; Sabir, N.; Cheng, G.; Dong, H.; Li, M.; et al. Inhibition of type I interferon signaling abrogates early Mycobacterium bovis infection. BMC Infect. Dis. 2019, 19, 1031. [Google Scholar] [CrossRef] [PubMed]
- Zak, D.E.; Penn-Nicholson, A.; Scriba, T.J.; Thompson, E.; Suliman, S.; Amon, L.M.; Mahomed, H.; Erasmus, M.; Whatney, W.; Hussey, G.D.; et al. A blood RNA signature for tuberculosis disease risk: A prospective cohort study. Lancet 2016, 387, 2312–2322. [Google Scholar] [CrossRef]
- Scriba, T.J.; Penn-Nicholson, A.; Shankar, S.; Hraha, T.; Thompson, E.G.; Sterling, D.; Nemes, E.; Darboe, F.; Suliman, S.; Amon, L.M.; et al. Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLoS Pathog. 2017, 13, e1006687. [Google Scholar] [CrossRef]
- Singhania, A.; Verma, R.; Graham, C.M.; Lee, J.; Tran, T.; Richardson, M.; Lecine, P.; Leissner, P.; Berry, M.P.R.; Wilkinson, R.J.; et al. A modular transcriptional signature identifies phenotypic heterogeneity of human tuberculosis infection. Nat. Commun. 2018, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Teixeira, L.; Mayer-Barber, K.; Sher, A.; O’Garra, A. Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med. 2018, 215, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.L.; Schultz, T.E.; Duke, T.J.; Blumenthal, A. Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front. Immunol. 2017, 8, 1633. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V.; Kalra, P.; Goel, M.; Khilnani, G.C.; Saini, V.; Prasad, G.B.K.S.; Gupta, U.D.; Prasad, H.K. Impact and prognosis of the expression of IFN-α among tuberculosis patients. PLoS ONE 2020, 15, e0235488. [Google Scholar] [CrossRef]
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J.; Jörg, S.; Heinemann, E.; Oberbeck-Müller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Deweerd, N.A.; Stifter, S.A.; Liu, L.; Zhou, B.; Wang, W.; Zhou, Y.; Ying, B.; Hu, X.; Matthews, A.Y.; et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat. Commun. 2018, 9, 85. [Google Scholar] [CrossRef]
- Bogunovic, D.; Byun, M.; Durfee, L.A.; Abhyankar, A.; Sanal, O.; Mansouri, D.; Salem, S.; Radovanovic, I.; Grant, A.V.; Adimi, P.; et al. Mycobacterial Disease and Impaired IFN-γ Immunity in Humans with Inherited ISG15 Deficiency. Science 2012, 337, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Sabbatani, S.; Manfredi, R.; Marinacci, G.; Pavoni, M.; Cristoni, L.; Chiodo, F. Reactivation of severe, acute pulmonary tuberculosis during treatment with pegylated interferon-alpha and ribavirin for chronic HCV hepatitis. Scand. J. Infect. Dis. 2006, 38, 205–208. [Google Scholar] [CrossRef]
- Farah, R.; Awad, J. The association of interferon with the development of pulmonary tuberculosis. Int. J. Clin. Pharmacol. Ther. 2007, 45, 598–600. [Google Scholar] [CrossRef]
- Telesca, C.; Angelico, M.; Piccolo, P.; Nosotti, L.; Morrone, A.; Longhi, C.; Carbone, M.; Baiocchi, L. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J. Infect. 2007, 54, e223–e226. [Google Scholar] [CrossRef]
- Belkahla, N.; Kchir, H.; Maamouri, N.; Ouerghi, H.; Hariz, F.; Chouaib, S.; Chaabouni, H.; Mami, N. Réactivation d’une tuberculose sous bithérapie interféron-pégylé et ribavirine pour une hépatite chronique C. Rev. Med. Interne 2010, 31, e1–e3. [Google Scholar] [CrossRef]
- De Uehara, S.N.O.; Emori, C.T.; Perez, R.M.; Mendes-Correa, M.C.J.; Ferreira, A.D.S.P.; Feldner, A.C.D.C.A.; Silva, A.E.B.; Filho, R.J.C.; Silva, I.S.D.S.E.; Ferraz, M.L.C.G. High incidence of tuberculosis in patients treated for hepatitis C chronic infection. Braz. J. Infect. Dis. 2016, 20, 205–209. [Google Scholar] [CrossRef]
- Matsuoka, S.; Fujikawa, H.; Hasegawa, H.; Ochiai, T.; Watanabe, Y.; Moriyama, M. Onset of Tuberculosis from a Pulmonary Latent Tuberculosis Infection during Antiviral Triple Therapy for Chronic Hepatitis C. Intern. Med. 2016, 55, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.; Cardone, M.; Thompson, R.; Shenderov, K.; Kirschman, K.D.; Mayer-Barber, K.D.; Myers, T.G.; Rabin, R.L.; Trinchieri, G.; Sher, A.; et al. Mycobacterium tuberculosis Triggers Host Type I IFN Signaling to Regulate IL-1β Production in Human Macrophages. J. Immunol. 2011, 187, 2540–2547. [Google Scholar] [CrossRef]
- Pandey, A.K.; Yang, Y.; Jiang, Z.; Fortune, S.M.; Coulombe, F.; Behr, M.A.; Fitzgerald, K.; Sassetti, C.M.; Kelliher, M.A. NOD2, RIP2 and IRF5 Play a Critical Role in the Type I Interferon Response to Mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000500. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Johndrow, J.E.; Manzanillo, P.; Cox, J.S. The Type I IFN Response to Infection with Mycobacterium tuberculosis Requires ESX-1-Mediated Secretion and Contributes to Pathogenesis. J. Immunol. 2007, 178, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Manzanillo, P.S.; Shiloh, M.U.; Portnoy, D.A.; Cox, J.S. Mycobacterium tuberculosis Activates the DNA-Dependent Cytosolic Surveillance Pathway within Macrophages. Cell Host Microbe 2012, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015, 17, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Wiens, K.E.; Ernst, J.D. The Mechanism for Type I Interferon Induction by Mycobacterium tuberculosis is Bacterial Strain-Dependent. PLoS Pathog. 2016, 12, e1005809. [Google Scholar] [CrossRef]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef]
- Dey, B.; Dey, R.J.; Cheung, L.S.; Pokkali, S.; Guo, H.; Lee, J.-H.; Bishai, W.R. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat. Med. 2015, 21, 401–406. [Google Scholar] [CrossRef]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef]
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.-D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015, 17, 820–828. [Google Scholar] [CrossRef]
- Stamm, C.E.; Collins, A.C.; Shiloh, M.U. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunol. Rev. 2015, 264, 204–219. [Google Scholar] [CrossRef]
- Moreira-Teixeira, L.; Sousa, J.; McNab, F.W.; Torrado, E.; Cardoso, F.; Machado, H.; Castro, F.; Cardoso, V.; Gaifem, J.; Wu, X.; et al. Type I IFN Inhibits Alternative Macrophage Activation during Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-γ Signaling. J. Immunol. 2016, 197, 4714–4726. [Google Scholar] [CrossRef]
- Carmona, J.; Cruz, A.; Moreira-Teixeira, L.; Sousa, C.; Sousa, J.; Osorio, N.S.; Saraiva, A.L.; Svenson, S.; Kallenius, G.; Pedrosa, J.; et al. Mycobacterium tuberculosis Strains Are Differentially Recognized by TLRs with an Impact on the Immune Response. PLoS ONE 2013, 8, e67277. [Google Scholar] [CrossRef]
- Leber, J.H.; Crimmins, G.T.; Raghavan, S.; Meyer-Morse, N.P.; Cox, J.S.; Portnoy, D.A. Distinct TLR- and NLR-Mediated Transcriptional Responses to an Intracellular Pathogen. PLoS Pathog. 2008, 4, e6. [Google Scholar] [CrossRef]
- de Paus, R.A.; van Wengen, A.; Schmidt, I.; Visser, M.; Verdegaal, E.M.; van Dissel, J.T.; van de Vosse, E. Inhibition of the type I immune responses of human monocytes by IFN-α and IFN-β. Cytokine 2013, 61, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Teles, R.M.B.; Graeber, T.G.; Krutzik, S.R.; Montoya, D.; Schenk, M.; Lee, D.J.; Komisopoulou, E.; Kelly-Scumpia, K.; Chun, R.; Iyer, S.S.; et al. Type I Interferon Suppresses Type II Interferon-Triggered Human Anti-Mycobacterial Responses. Science 2013, 339, 1448–1453. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Barber, D.L.; Hieny, S.; Feng, C.; Caspar, P.; Oland, S.; Gordon, S.; Sher, A. Innate and Adaptive Interferons Suppress IL-1α and IL-1β Production by Distinct Pulmonary Myeloid Subsets during Mycobacterium tuberculosis Infection. Immunity 2011, 35, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E.; Freedman, V.H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757. [Google Scholar] [CrossRef]
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing Strains Upregulate Type I IFNs and Increase Expression of Negative Regulators of the Jak-Stat Pathway. J. Interf. Cytokine Res. 2005, 25, 694–701. [Google Scholar] [CrossRef]
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The Hypervirulent Mycobacterium tuberculosis Strain HN878 Induces a Potent TH1 Response followed by Rapid Down-Regulation. J. Immunol. 2007, 179, 522–531. [Google Scholar] [CrossRef]
- McNab, F.W.; Ewbank, J.; Howes, A.; Moreira-Teixeira, L.; Martirosyan, A.; Ghilardi, N.; Saraiva, M.; O’Garra, A. Type I IFN Induces IL-10 Production in an IL-27–Independent Manner and Blocks Responsiveness to IFN-γ for Production of IL-12 and Bacterial Killing in Mycobacterium tuberculosis–Infected Macrophages. J. Immunol. 2014, 193, 3600–3612. [Google Scholar] [CrossRef]
- Antonelli, L.R.; Rothfuchs, A.; Gonçalves, R.; Roffê, E.; Cheever, A.W.; Bafica, A.; Salazar, A.M.; Feng, C.; Sher, A. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Investig. 2010, 120, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat. Commun. 2020, 11, 5566. [Google Scholar] [CrossRef]
- Fremond, C.M.; Togbe, D.; Doz, E.; Rose, S.; Vasseur, V.; Maillet, I.; Jacobs, M.; Ryffel, B.; Quesniaux, V.F.J. IL-1 Receptor-Mediated Signal Is an Essential Component of MyD88-Dependent Innate Response to Mycobacterium tuberculosis Infection. J. Immunol. 2007, 179, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Barber, K.D.; Barber, D.L.; Shenderov, K.; White, S.D.; Wilson, M.S.; Cheever, A.; Kugler, D.; Hieny, S.; Caspar, P.; Núñez, G.; et al. Cutting Edge: Caspase-1 Independent IL-1β Production Is Critical for Host Resistance to Mycobacterium tuberculosis and Does Not Require TLR Signaling In Vivo. J. Immunol. 2010, 184, 3326–3330. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, P.; Sada-Ovalle, I.; Nishimura, T.; Anderson, A.C.; Kuchroo, V.K.; Remold, H.G.; Behar, S.M. IL-1β Promotes Antimicrobial Immunity in Macrophages by Regulating TNFR Signaling and Caspase-3 Activation. J. Immunol. 2013, 190, 4196–4204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhou, B.; Li, S.; Yue, J.; Yang, H.; Wen, Y.; Zhan, S.; Wang, W.; Liao, M.; Zhang, M.; et al. Allele-Specific Induction of IL-1β Expression by C/EBPβ and PU.1 Contributes to Increased Tuberculosis Susceptibility. PLoS Pathog. 2014, 10, e1004426. [Google Scholar] [CrossRef]
- Mishra, B.B.; Rathinam, V.A.K.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; A Fitzgerald, K.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome–dependent processing of IL-1β. Nat. Immunol. 2012, 14, 52–60. [Google Scholar] [CrossRef]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Sher, A. Cytokine and lipid mediator networks in tuberculosis. Immunol. Rev. 2015, 264, 264–275. [Google Scholar] [CrossRef]
- Ward, C.M.; Jyonouchi, H.; Kotenko, S.V.; Smirnov, S.V.; Patel, R.; Aguila, H.; McSherry, G.; Dashefsky, B.; Holland, S.M. Adjunctive treatment of disseminated Mycobacterium avium complex infection with interferon alpha-2b in a patient with complete interferon-gamma receptor R1 deficiency. Eur. J. Pediatr. 2006, 166, 981–985. [Google Scholar] [CrossRef]
- Bax, H.I.; Freeman, A.F.; Ding, L.; Hsu, A.P.; Marciano, B.; Kristosturyan, E.; Jancel, T.; Spalding, C.; Pechacek, J.; Olivier, K.N.; et al. Interferon Alpha Treatment of Patients with Impaired Interferon Gamma Signaling. J. Clin. Immunol. 2013, 33, 991–1001. [Google Scholar] [CrossRef]
- Desvignes, L.; Wolf, A.J.; Ernst, J.D. Dynamic Roles of Type I and Type II IFNs in Early Infection with Mycobacterium tuberculosis. J. Immunol. 2012, 188, 6205–6215. [Google Scholar] [CrossRef]
- Rivas-Santiago, C.E.; Guerrero, G.G. IFN-α Boosting of Mycobacterium bovis Bacillus Calmette Güerin-Vaccine Promoted Th1 Type Cellular Response and Protection against M. tuberculosis Infection. Biomed. Res. Int. 2017, 2017, 8796760. [Google Scholar] [CrossRef]
- O’Riordan, M.; Yi, C.H.; Gonzales, R.; Lee, K.-D.; Portnoy, D.A. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 13861–13866. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Age/Sex Group | Number of Cases | Percent Total |
| Adult men | 6,000,000 | 56.5% |
| Adult women | 3,400,000 | 32.5% |
| Children | 1,200,000 | 11% |
| Region | Number of Incidences | Percent Total |
| Southeast Asia | 630,000 | 45% |
| Africa | 322,000 | 23% |
| Western Pacific | 252,000 | 18% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mundra, A.; Yegiazaryan, A.; Karsian, H.; Alsaigh, D.; Bonavida, V.; Frame, M.; May, N.; Gargaloyan, A.; Abnousian, A.; Venketaraman, V. Pathogenicity of Type I Interferons in Mycobacterium tuberculosis. Int. J. Mol. Sci. 2023, 24, 3919. https://doi.org/10.3390/ijms24043919
Mundra A, Yegiazaryan A, Karsian H, Alsaigh D, Bonavida V, Frame M, May N, Gargaloyan A, Abnousian A, Venketaraman V. Pathogenicity of Type I Interferons in Mycobacterium tuberculosis. International Journal of Molecular Sciences. 2023; 24(4):3919. https://doi.org/10.3390/ijms24043919
Chicago/Turabian StyleMundra, Akaash, Aram Yegiazaryan, Haig Karsian, Dijla Alsaigh, Victor Bonavida, Mitchell Frame, Nicole May, Areg Gargaloyan, Arbi Abnousian, and Vishwanath Venketaraman. 2023. "Pathogenicity of Type I Interferons in Mycobacterium tuberculosis" International Journal of Molecular Sciences 24, no. 4: 3919. https://doi.org/10.3390/ijms24043919
APA StyleMundra, A., Yegiazaryan, A., Karsian, H., Alsaigh, D., Bonavida, V., Frame, M., May, N., Gargaloyan, A., Abnousian, A., & Venketaraman, V. (2023). Pathogenicity of Type I Interferons in Mycobacterium tuberculosis. International Journal of Molecular Sciences, 24(4), 3919. https://doi.org/10.3390/ijms24043919