
Early Alterations in Structural and Functional Properties in the Neuromuscular Junctions of Mutant FUS Mice
 , , ,
, , ,
Abstract
1. Introduction
2. Results
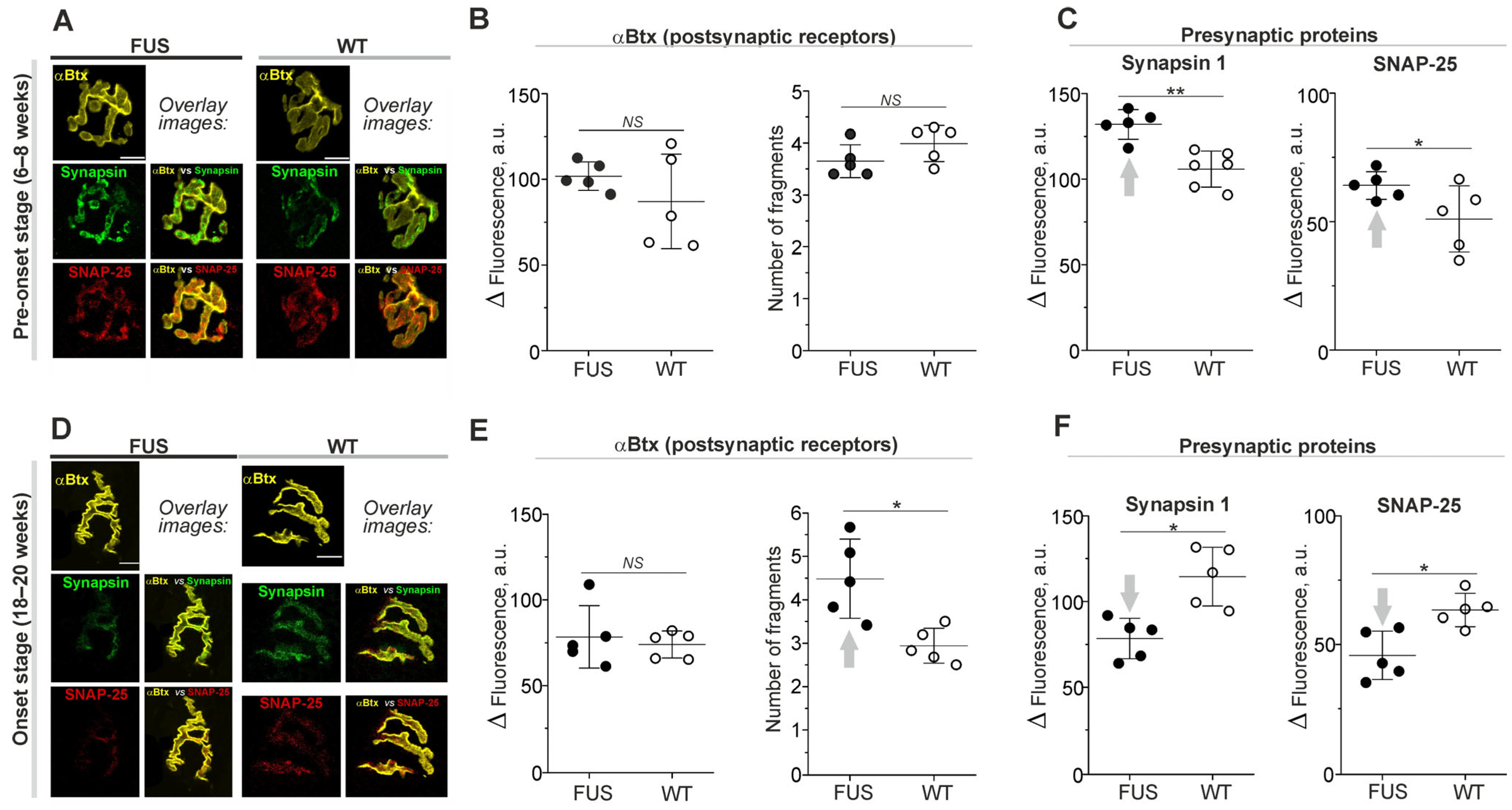
2.1. Structural Characterization of NMJs: Analysis of End-Plate Fragmentation and Immunolabeling of Presynaptic Proteins
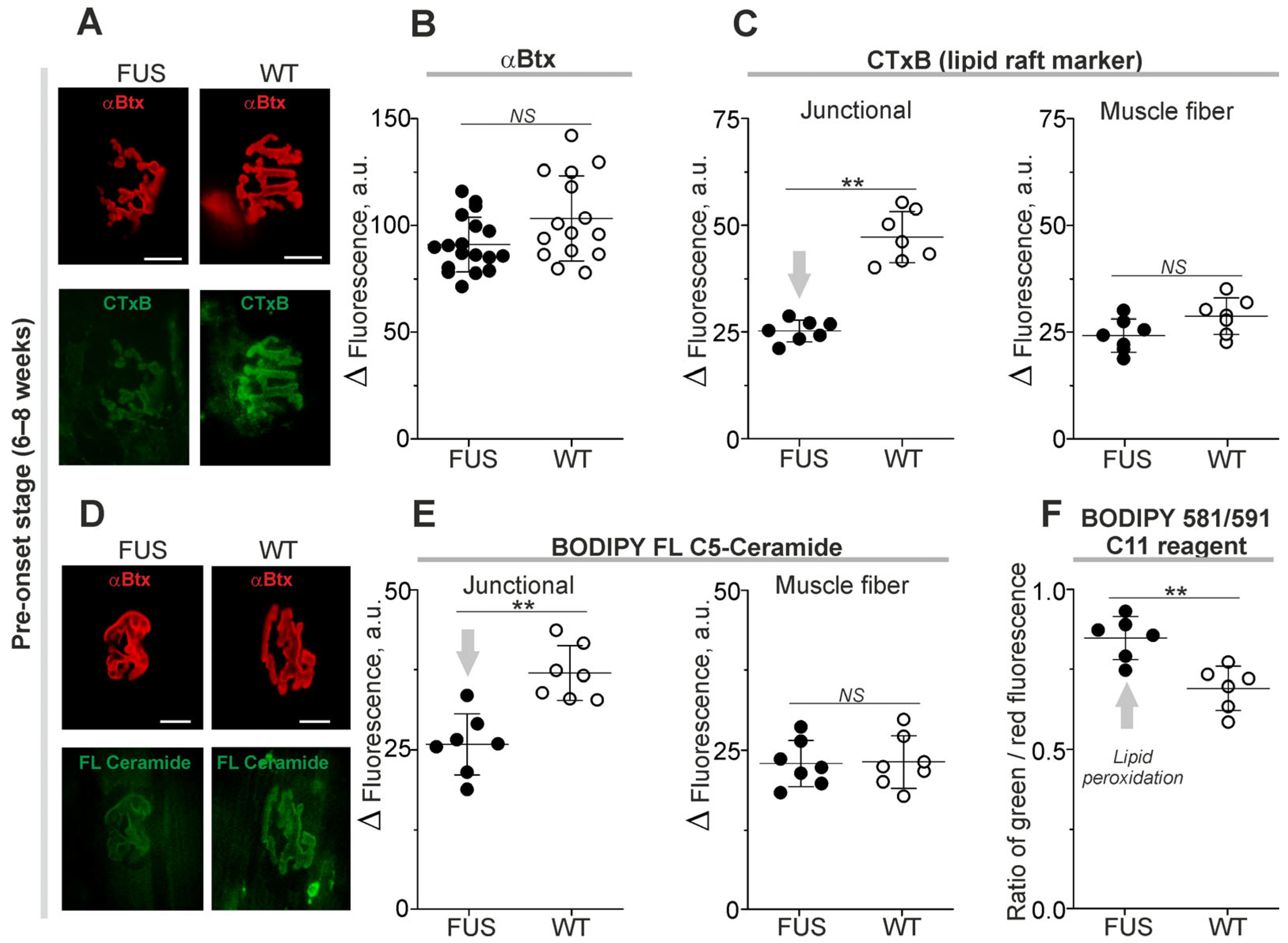
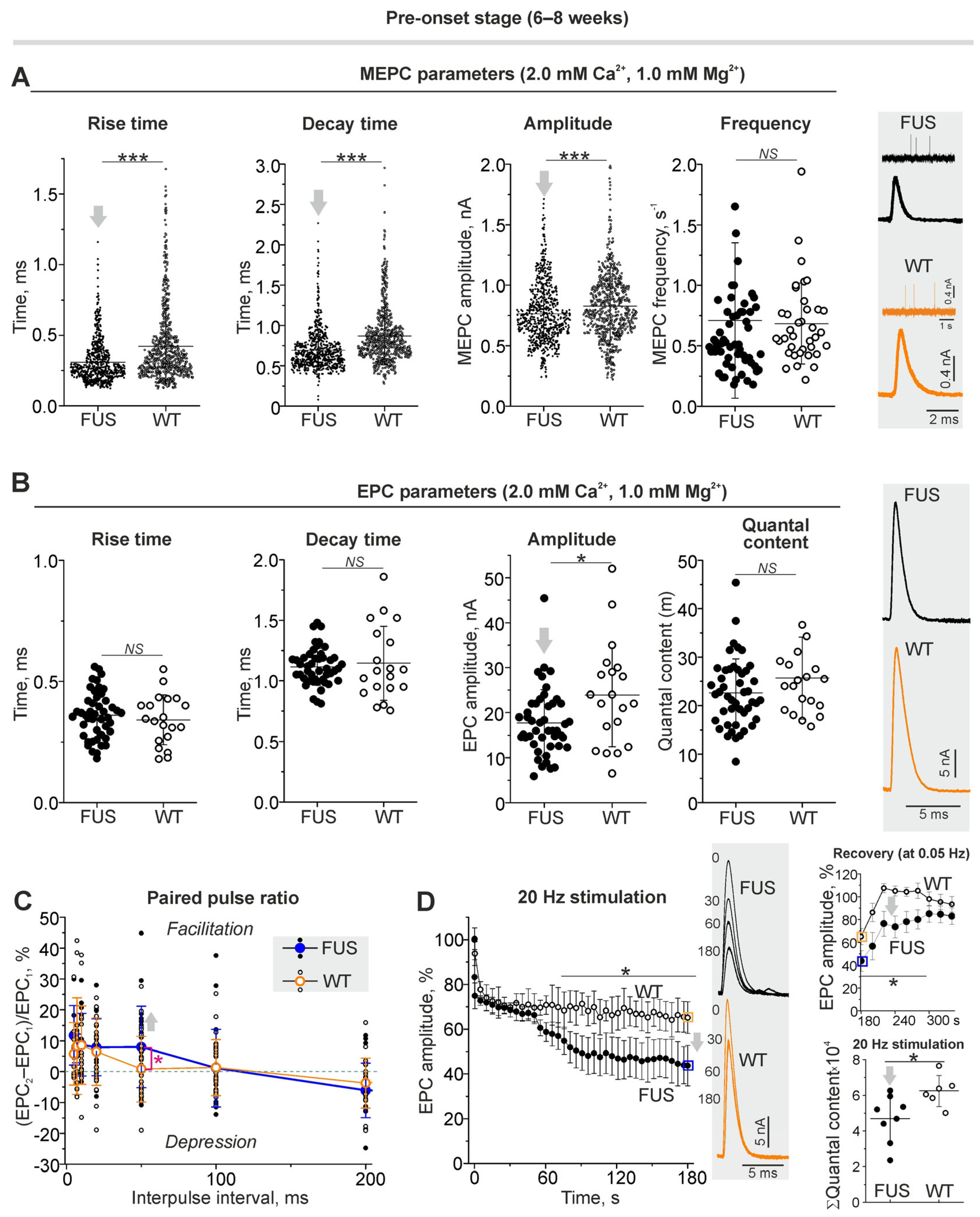
2.2. Structural Characterization of NMJs: The Changes in Synaptic Membrane Properties at Pre-Onset Stage
2.3. Functional Characterization of NMJs in the FUS Mice: Neurotransmitter Reception and Release under Low Calcium in Extracellular Solution
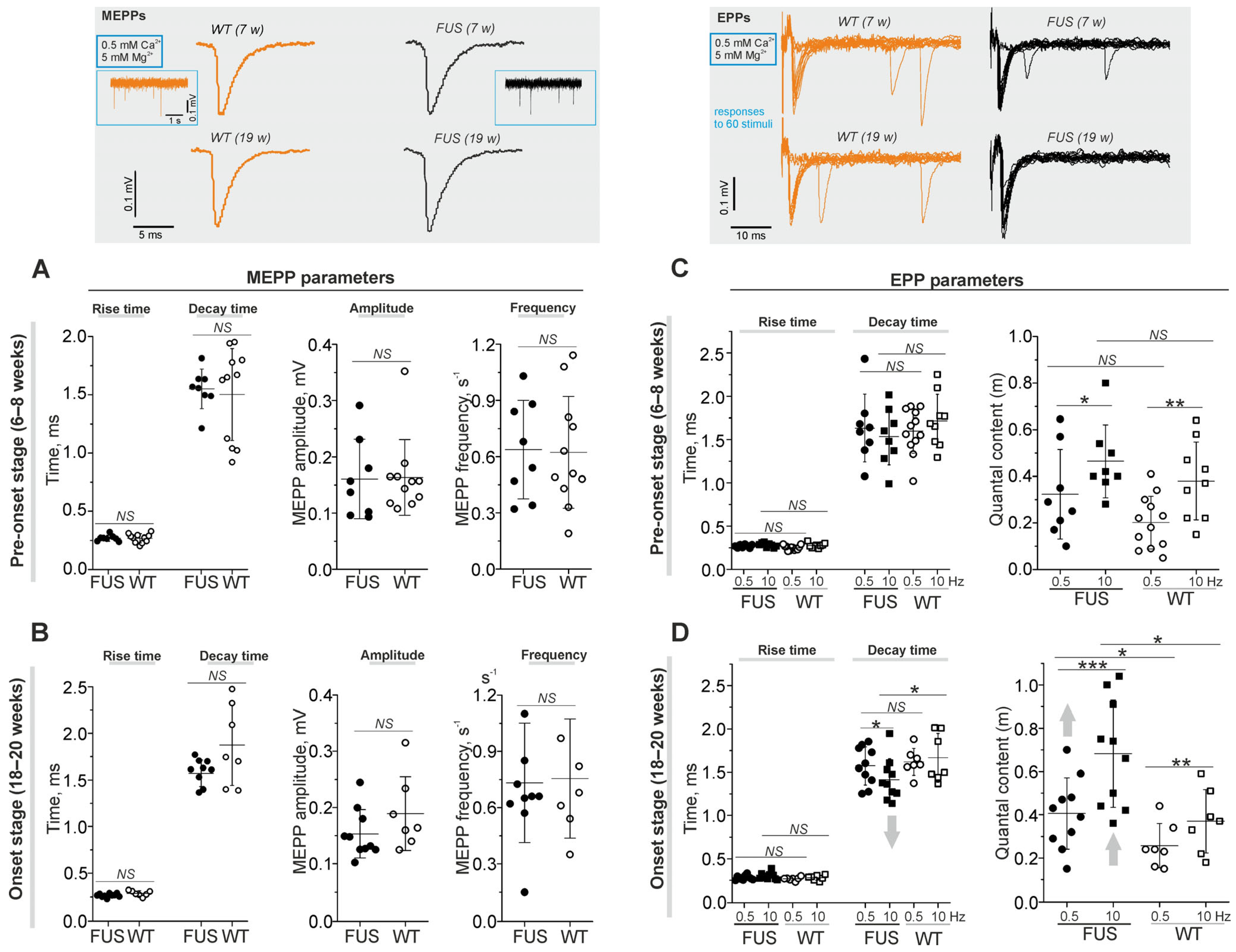
2.3.1. Spontaneous and Evoked Postsynaptic Responses
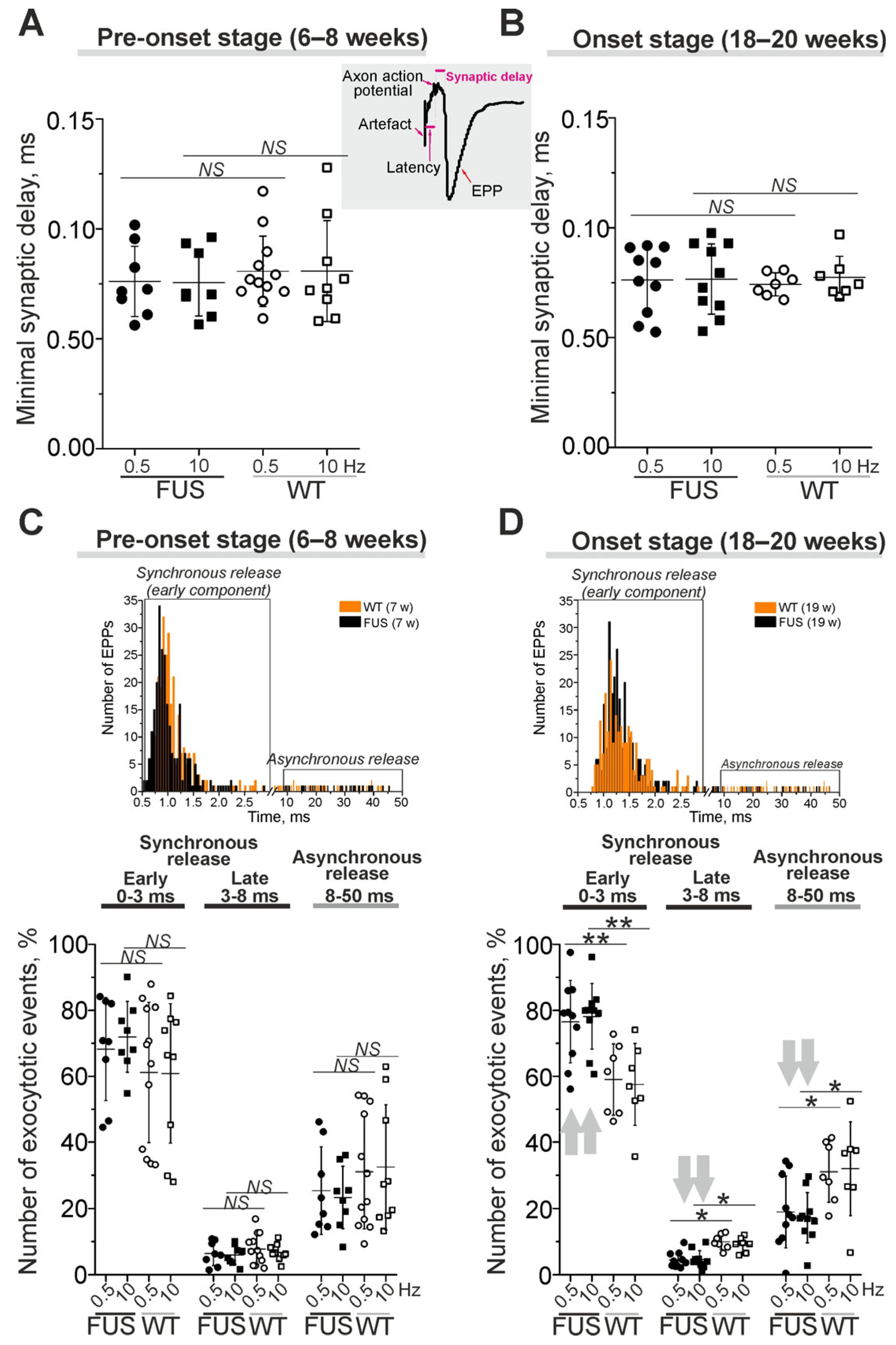
2.3.2. Estimation of the Real Synaptic Delay and the Kinetics of Neurotransmitter Release
2.4. Functional Characterization of NMJs in the FUS Mice: Neurotransmitter Reception and Release under Normal External Calcium in Extracellular Solution
2.4.1. Spontaneous and Evoked Neurotransmitter Release
2.4.2. Short-Term Plasticity and the Kinetics of Neurotransmitter Release upon Intense Nerve Stimulation
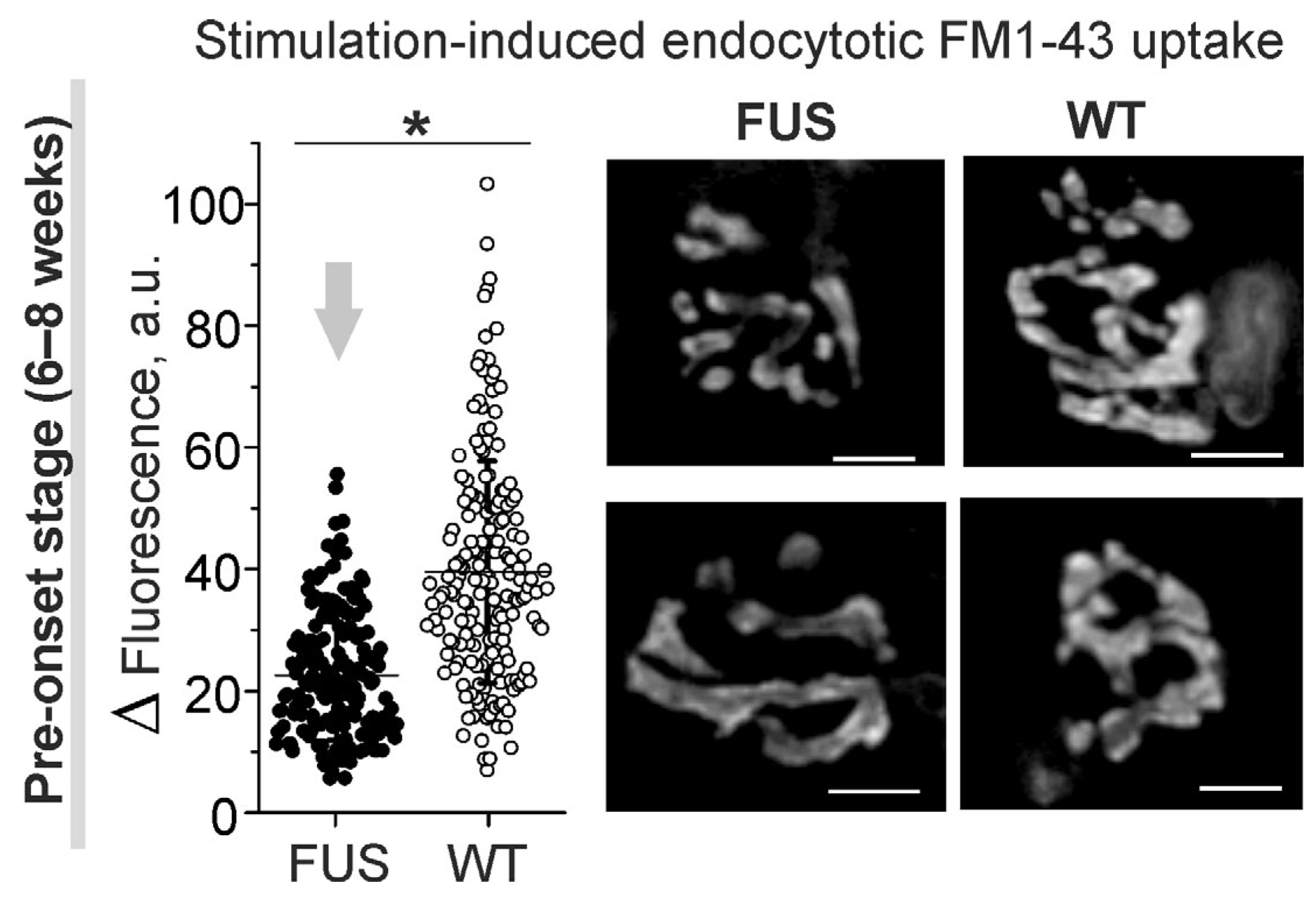
2.5. Functional Characterization of NMJs in the FUS Mice: Estimation of Synaptic Vesicle Endocytosis
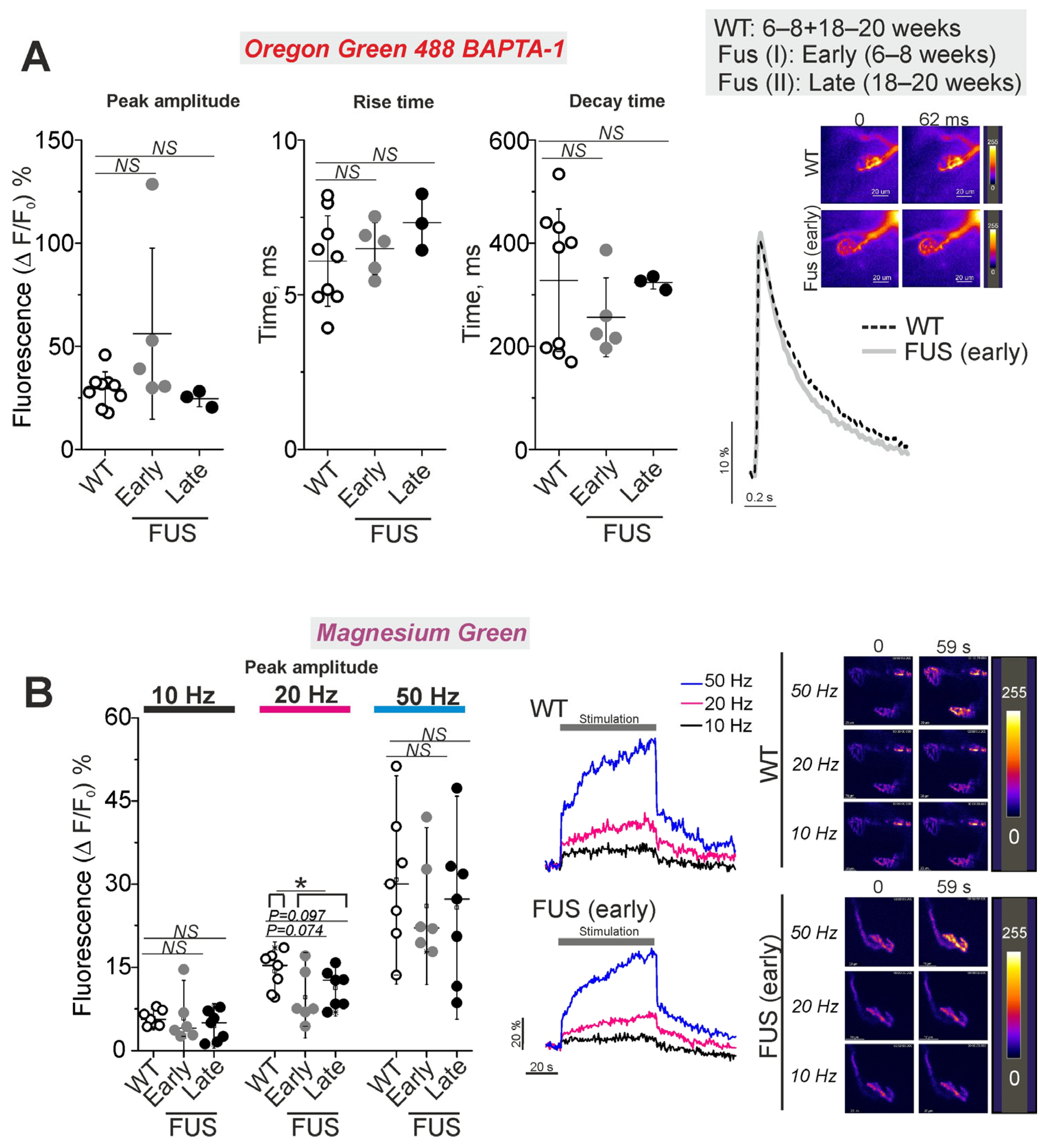
2.6. Functional Characterization of NMJs in the FUS Mice: Intraterminal Ca2+ Dynamics upon Nerve Stimulation at Different Frequencies
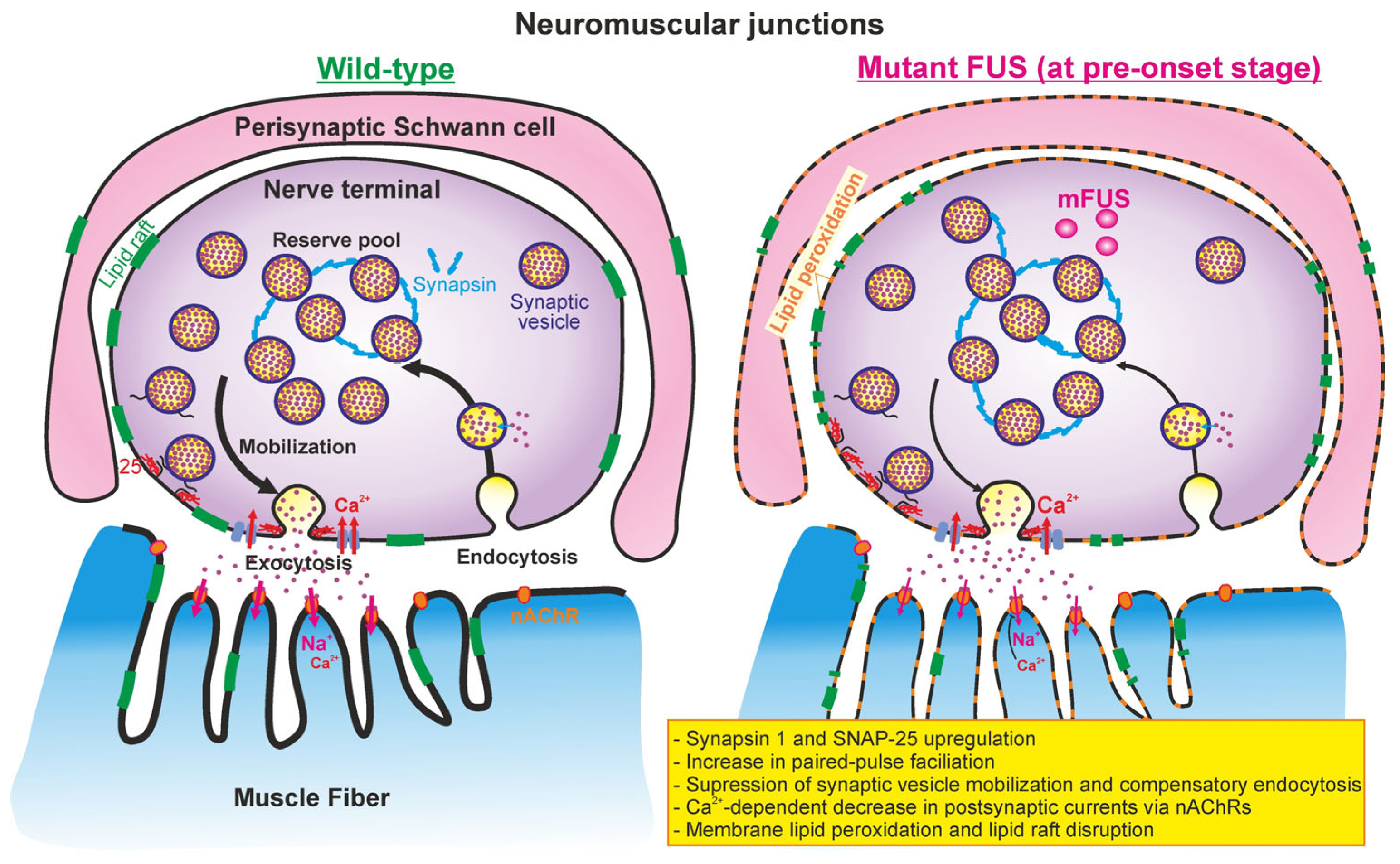
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Microelectrode Recordings
4.3. Immunostaining
4.4. Lipid Assays
4.5. Assessment of Synaptic Vesicle Endocytosis
4.6. Calcium Imaging
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, X.; Smith, V.; Jackson, M.; Tran, M.; Thomas, M.; Patel, A.; Lorusso, E.; Nimbalkar, S.; Cai, Y.; McAleer, C.W.; et al. A Human-Based Functional NMJ System for Personalized ALS Modeling and Drug Testing. Adv. Ther. 2020, 3, 2000133. [Google Scholar] [CrossRef]
- Alhindi, A.; Boehm, I.; Chaytow, H. Small junction, big problems: Neuromuscular junction pathology in mouse models of amyotrophic lateral sclerosis (ALS). J. Anat. 2022, 241, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Gelon, P.A.; Dutchak, P.A.; Sephton, C.F. Synaptic dysfunction in ALS and FTD: Anatomical and molecular changes provide insights into mechanisms of disease. Front. Mol. Neurosci. 2022, 15, 1000183. [Google Scholar] [CrossRef]
- Picchiarelli, G.; Demestre, M.; Zuko, A.; Been, M.; Higelin, J.; Dieterle, S.; Goy, M.A.; Mallik, M.; Sellier, C.; Scekic-Zahirovic, J.; et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 2019, 22, 1793–1805. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef]
- Mukhamedyarov, M.A.; Petrov, A.M.; Grigoryev, P.N.; Giniatullin, A.R.; Petukhova, E.O.; Zefirov, A.L. Amyotrophic Lateral Sclerosis: Modern Views on the Pathogenesis and Experimental Models. Zhurnal Vyss. Nervn. Deyatelnosti Im. IP Pavlov. 2018, 68, 551–566. [Google Scholar] [CrossRef]
- Chen, L. FUS mutation is probably the most common pathogenic gene for JALS, especially sporadic JALS. Rev. Neurol. 2021, 177, 333–340. [Google Scholar] [CrossRef]
- Assoni, A.F.; Foijer, F.; Zatz, M. Amyotrophic Lateral Sclerosis, FUS and Protein Synthesis Defects. Stem Cell Rev. Rep. 2023, 19, 625–638. [Google Scholar] [CrossRef]
- Fujii, R.; Okabe, S.; Urushido, T.; Inoue, K.; Yoshimura, A.; Tachibana, T.; Nishikawa, T.; Hicks, G.G.; Takumi, T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 2005, 15, 587–593. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Salam, S.; Tacconelli, S.; Smith, B.N.; Mitchell, J.C.; Glennon, E.; Nikolaou, N.; Houart, C.; Vance, C. Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci. Rep. 2021, 11, 13613. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Krasnow, E.N.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Kerstens, A.; Giacomazzi, G.; Pavie, B.; Rossaert, E.; Beckers, J.; et al. Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Rep. 2021, 16, 2213–2227. [Google Scholar] [CrossRef]
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Fogarty, M.J.; Sieck, G.C. Evolution and Functional Differentiation of the Diaphragm Muscle of Mammals. Compr. Physiol. 2019, 9, 715–766. [Google Scholar] [CrossRef]
- Sieck, D.C.; Zhan, W.Z.; Fang, Y.H.; Ermilov, L.G.; Sieck, G.C.; Mantilla, C.B. Structure-activity relationships in rodent diaphragm muscle fibers vs. neuromuscular junctions. Respir. Physiol. Neurobiol. 2012, 180, 88–96. [Google Scholar] [CrossRef]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastiao, A.M.; Ribeiro, J.A. Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS ONE 2013, 8, e73846. [Google Scholar] [CrossRef]
- Zakyrjanova, G.F.; Giniatullin, A.R.; Mukhutdinova, K.A.; Kuznetsova, E.A.; Petrov, A.M. Early differences in membrane properties at the neuromuscular junctions of ALS model mice: Effects of 25-hydroxycholesterol. Life Sci. 2021, 273, 119300. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.; Sebastiao, A.M.; Ribeiro, J.A. Presymptomatic and symptomatic ALS SOD1(G93A) mice differ in adenosine A1 and A2A receptor-mediated tonic modulation of neuromuscular transmission. Purinergic Signal. 2015, 11, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Arbour, D.; Tremblay, E.; Martineau, E.; Julien, J.P.; Robitaille, R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J. Neurosci. 2015, 35, 688–706. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Li, A.; Li, X.; Park, K.; Zhou, X.; Yi, F.; Xiao, Y.; Yoon, D.; Tan, T.; Ostrow, L.W.; et al. MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression. Antioxidants 2021, 10, 1522. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Funikov, S.Y.; Rezvykh, A.P.; Mazin, P.V.; Morozov, A.V.; Maltsev, A.V.; Chicheva, M.M.; Vikhareva, E.A.; Evgen’ev, M.B.; Ustyugov, A.A. FUS(1-359) transgenic mice as a model of ALS: Pathophysiological and molecular aspects of the proteinopathy. Neurogenetics 2018, 19, 189–204. [Google Scholar] [CrossRef]
- Probert, F.; Gorlova, A.; Deikin, A.; Bettendorff, L.; Veniaminova, E.; Nedorubov, A.; Chaprov, K.D.; Ivanova, T.A.; Anthony, D.C.; Strekalova, T. In FUS [1-359]-tg mice O,S-dibenzoyl thiamine reduces muscle atrophy, decreases glycogen synthase kinase 3 beta, and normalizes the metabolome. Biomed. Pharmacother. 2022, 156, 113986. [Google Scholar] [CrossRef]
- Sambon, M.; Gorlova, A.; Demelenne, A.; Alhama-Riba, J.; Coumans, B.; Lakaye, B.; Wins, P.; Fillet, M.; Anthony, D.C.; Strekalova, T.; et al. Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration. Biomedicines 2020, 8, 361. [Google Scholar] [CrossRef]
- Crivello, M.; Hogg, M.C.; Jirstrom, E.; Halang, L.; Woods, I.; Rayner, M.; Coughlan, K.S.; Lewandowski, S.A.; Prehn, J.H.M. Vascular regression precedes motor neuron loss in the FUS (1-359) ALS mouse model. Dis. Model. Mech. 2019, 12, dmm040238. [Google Scholar] [CrossRef]
- Lysikova, E.A.; Kukharsky, M.S.; Chaprov, K.D.; Vasilieva, N.A.; Roman, A.Y.; Ovchinnikov, R.K.; Deykin, A.V.; Ninkina, N.; Buchman, V.L. Behavioural impairments in mice of a novel FUS transgenic line recapitulate features of frontotemporal lobar degeneration. Genes Brain Behav. 2019, 18, e12607. [Google Scholar] [CrossRef]
- de Munter, J.; Shafarevich, I.; Liundup, A.; Pavlov, D.; Wolters, E.C.; Gorlova, A.; Veniaminova, E.; Umriukhin, A.; Kalueff, A.; Svistunov, A.; et al. Neuro-Cells therapy improves motor outcomes and suppresses inflammation during experimental syndrome of amyotrophic lateral sclerosis in mice. CNS Neurosci. Ther. 2020, 26, 504–517. [Google Scholar] [CrossRef]
- Hogg, M.C.; Halang, L.; Woods, I.; Coughlan, K.S.; Prehn, J.H.M. Riluzole does not improve lifespan or motor function in three ALS mouse models. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 438–445. [Google Scholar] [CrossRef]
- Zhai, J.; Strom, A.L.; Kilty, R.; Venkatakrishnan, P.; White, J.; Everson, W.V.; Smart, E.J.; Zhu, H. Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. FEBS J. 2009, 276, 3308–3323. [Google Scholar] [CrossRef]
- Fernandez-Beltran, L.C.; Godoy-Corchuelo, J.M.; Losa-Fontangordo, M.; Williams, D.; Matias-Guiu, J.; Corrochano, S. A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. Int. J. Mol. Sci. 2021, 22, 9553. [Google Scholar] [CrossRef]
- Bouscary, A.; Quessada, C.; Mosbach, A.; Callizot, N.; Spedding, M.; Loeffler, J.P.; Henriques, A. Ambroxol Hydrochloride Improves Motor Functions and Extends Survival in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2019, 10, 883. [Google Scholar] [CrossRef]
- Wang, S.; Ichinomiya, T.; Savchenko, P.; Wang, D.; Sawada, A.; Li, X.; Duong, T.; Li, W.; Bonds, J.A.; Kim, E.J.; et al. Subpial delivery of adeno-associated virus 9-synapsin-caveolin-1 (AAV9-SynCav1) preserves motor neuron and neuromuscular junction morphology, motor function, delays disease onset, and extends survival in hSOD1(G93A) mice. Theranostics 2022, 12, 5389–5403. [Google Scholar] [CrossRef]
- Sawada, A.; Wang, S.; Jian, M.; Leem, J.; Wackerbarth, J.; Egawa, J.; Schilling, J.M.; Platoshyn, O.; Zemljic-Harpf, A.; Roth, D.M.; et al. Neuron-targeted caveolin-1 improves neuromuscular function and extends survival in SOD1(G93A) mice. FASEB J. 2019, 33, 7545–7554. [Google Scholar] [CrossRef]
- DiPasquale, M.; Deering, T.G.; Desai, D.; Sharma, A.K.; Amin, S.; Fox, T.E.; Kester, M.; Katsaras, J.; Marquardt, D.; Heberle, F.A. Influence of ceramide on lipid domain stability studied with small-angle neutron scattering: The role of acyl chain length and unsaturation. Chem. Phys. Lipids 2022, 245, 105205. [Google Scholar] [CrossRef]
- Tsentsevitsky, A.N.; Gafurova, C.R.; Mukhutdinova, K.A.; Giniatullin, A.R.; Fedorov, N.S.; Malomouzh, A.I.; Petrov, A.M. Sphingomyelinase modulates synaptic vesicle mobilization at the mice neuromuscular junctions. Life Sci. 2023, 318, 121507. [Google Scholar] [CrossRef]
- Petrov, A.M.; Shalagina, M.N.; Protopopov, V.A.; Sergeev, V.G.; Ovechkin, S.V.; Ovchinina, N.G.; Sekunov, A.V.; Zefirov, A.L.; Zakirjanova, G.F.; Bryndina, I.G. Changes in Membrane Ceramide Pools in Rat Soleus Muscle in Response to Short-Term Disuse. Int. J. Mol. Sci. 2019, 20, 4860. [Google Scholar] [CrossRef]
- Tsentsevitsky, A.N.; Gafurova, C.R.; Petrov, A.M. K(ATP) channels as ROS-dependent modulator of neurotransmitter release at the neuromuscular junctions. Life Sci. 2022, 310, 121120. [Google Scholar] [CrossRef] [PubMed]
- Tsentsevitsky, A.N.; Zakyrjanova, G.F.; Petrov, A.M. Cadmium desynchronizes neurotransmitter release in the neuromuscular junction: Key role of ROS. Free. Radic. Biol. Med. 2020, 155, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Khuzakhmetova, V.; Samigullin, D.; Nurullin, L.; Vyskocil, F.; Nikolsky, E.; Bukharaeva, E. Kinetics of neurotransmitter release in neuromuscular synapses of newborn and adult rats. Int. J. Dev. Neurosci. 2014, 34, 9–18. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.M. The statistics of transmitter release at chemical synapses. Int. Rev. Physiol. 1978, 17, 49–117. [Google Scholar]
- Tsentsevitsky, A.N.; Khaziev, E.F.; Kovyazina, I.V.; Petrov, A.M. GIRK channel as a versatile regulator of neurotransmitter release via L-type Ca2+ channel-dependent mechanism in the neuromuscular junction. Neuropharmacology 2022, 209, 109021. [Google Scholar] [CrossRef]
- Bukharaeva, E.A.; Skorinkin, A.I.; Samigullin, D.V.; Petrov, A.M. Presynaptic Acetylcholine Receptors Modulate the Time Course of Action Potential-Evoked Acetylcholine Quanta Secretion at Neuromuscular Junctions. Biomedicines 2022, 10, 1771. [Google Scholar] [CrossRef]
- Ginebaugh, S.P.; Badawi, Y.; Tarr, T.B.; Meriney, S.D. Neuromuscular Active Zone Structure and Function in Healthy and Lambert-Eaton Myasthenic Syndrome States. Biomolecules 2022, 12, 740. [Google Scholar] [CrossRef]
- Grassi, F.; Fucile, S. Calcium influx through muscle nAChR-channels: One route, multiple roles. Neuroscience 2020, 439, 117–124. [Google Scholar] [CrossRef]
- Jackman, S.L.; Regehr, W.G. The Mechanisms and Functions of Synaptic Facilitation. Neuron 2017, 94, 447–464. [Google Scholar] [CrossRef]
- Medina-Moreno, A.; Henriquez, J.P. Maturation of a postsynaptic domain: Role of small Rho GTPases in organising nicotinic acetylcholine receptor aggregates at the vertebrate neuromuscular junction. J. Anat. 2022, 241, 1148–1156. [Google Scholar] [CrossRef]
- Rudolf, R.; Straka, T. Nicotinic acetylcholine receptor at vertebrate motor endplates: Endocytosis, recycling, and degradation. Neurosci. Lett. 2019, 711, 134434. [Google Scholar] [CrossRef]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Laine, J.; et al. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. J. Cachexia Sarcopenia Muscle 2022, 13, 1385–1402. [Google Scholar] [CrossRef]
- Romagnoli, C.; Sharma, P.; Zonefrati, R.; Palmini, G.; Lucattelli, E.; Ward, D.T.; Ellinger, I.; Innocenti, M.; Brandi, M.L. Study of the Expression and Function of Calcium-Sensing Receptor in Human Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 7282. [Google Scholar] [CrossRef]
- Volpe, P.; Bosutti, A.; Nori, A.; Filadi, R.; Gherardi, G.; Trautmann, G.; Furlan, S.; Massaria, G.; Sciancalepore, M.; Megighian, A.; et al. Nerve-dependent distribution of subsynaptic type 1 inositol 1,4,5-trisphosphate receptor at the neuromuscular junction. J. Gen. Physiol. 2022, 154, e202213128. [Google Scholar] [CrossRef]
- Sahadevan, S.; Hembach, K.M.; Tantardini, E.; Perez-Berlanga, M.; Hruska-Plochan, M.; Megat, S.; Weber, J.; Schwarz, P.; Dupuis, L.; Robinson, M.D.; et al. Synaptic FUS accumulation triggers early misregulation of synaptic RNAs in a mouse model of ALS. Nat. Commun. 2021, 12, 3027. [Google Scholar] [CrossRef]
- Sansevrino, R.; Hoffmann, C.; Milovanovic, D. Condensate biology of synaptic vesicle clusters. Trends Neurosci. 2023, 46, 293–306. [Google Scholar] [CrossRef]
- Markert, S.M.; Skoruppa, M.; Yu, B.; Mulcahy, B.; Zhen, M.; Gao, S.; Sendtner, M.; Stigloher, C. Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol. Open. 2020, 9, bio055129. [Google Scholar] [CrossRef]
- Coyne, A.N.; Lorenzini, I.; Chou, C.C.; Torvund, M.; Rogers, R.S.; Starr, A.; Zaepfel, B.L.; Levy, J.; Johannesmeyer, J.; Schwartz, J.C.; et al. Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell. Rep. 2017, 21, 110–125. [Google Scholar] [CrossRef]
- Irfan, M.; Gopaul, K.R.; Miry, O.; Hokfelt, T.; Stanton, P.K.; Bark, C. SNAP-25 isoforms differentially regulate synaptic transmission and long-term synaptic plasticity at central synapses. Sci. Rep. 2019, 9, 6403. [Google Scholar] [CrossRef]
- Scullin, C.S.; Tafoya, L.C.; Wilson, M.C.; Partridge, L.D. Presynaptic residual calcium and synaptic facilitation at hippocampal synapses of mice with altered expression of SNAP-25. Brain Res. 2012, 1431, 1–12. [Google Scholar] [CrossRef]
- Feliciano, P.; Matos, H.; Andrade, R.; Bykhovskaia, M. Synapsin II Regulation of GABAergic Synaptic Transmission Is Dependent on Interneuron Subtype. J. Neurosci. 2017, 37, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Augustine, G.J. Synapsins and the Synaptic Vesicle Reserve Pool: Floats or Anchors? Cells 2021, 10, 658. [Google Scholar] [CrossRef] [PubMed]
- Vaden, J.H.; Banumurthy, G.; Gusarevich, E.S.; Overstreet-Wadiche, L.; Wadiche, J.I. The readily-releasable pool dynamically regulates multivesicular release. Elife 2019, 8, e47434. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Xiao, X.; Bi, M.; Tang, T.; Kong, D.; Diao, M.; Jiao, Q.; Chen, X.; Yan, C.; Du, X.; et al. ATP-sensitive potassium channels: A double-edged sword in neurodegenerative diseases. Ageing Res. Rev. 2022, 80, 101676. [Google Scholar] [CrossRef] [PubMed]
- Stoklund Dittlau, K.; Terrie, L.; Baatsen, P.; Kerstens, A.; De Swert, L.; Janky, R.; Corthout, N.; Masrori, P.; Van Damme, P.; Hyttel, P.; et al. FUS-ALS hiPSC-derived astrocytes impair human motor units through both gain-of-toxicity and loss-of-support mechanisms. Mol. Neurodegener. 2023, 18, 5. [Google Scholar] [CrossRef]
- Shahidullah, M.; Le Marchand, S.J.; Fei, H.; Zhang, J.; Pandey, U.B.; Dalva, M.B.; Pasinelli, P.; Levitan, I.B. Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J. Neurosci. 2013, 33, 19590–19598. [Google Scholar] [CrossRef]
- Janse van Mantgem, M.R.; van Rheenen, W.; Hackeng, A.V.; van Es, M.A.; Veldink, J.H.; van den Berg, L.H.; van Eijk, R.P.A. Association between Serum Lipids and Survival in Patients with Amyotrophic Lateral Sclerosis: A Meta-analysis and Population-Based Study. Neurology 2023, 100, e1062–e1071. [Google Scholar] [CrossRef]
- Agrawal, I.; Lim, Y.S.; Ng, S.Y.; Ling, S.C. Deciphering lipid dysregulation in ALS: From mechanisms to translational medicine. Transl. Neurodegener. 2022, 11, 48. [Google Scholar] [CrossRef]
- Odnoshivkina, U.G.; Kuznetsova, E.A.; Petrov, A.M. 25-Hydroxycholesterol as a Signaling Molecule of the Nervous System. Biochemistry 2022, 87, 524–537. [Google Scholar] [CrossRef]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef]
- Vejux, A.; Namsi, A.; Nury, T.; Moreau, T.; Lizard, G. Biomarkers of Amyotrophic Lateral Sclerosis: Current Status and Interest of Oxysterols and Phytosterols. Front. Mol. Neurosci. 2018, 11, 12. [Google Scholar] [CrossRef]
- Dodge, J.C.; Yu, J.; Sardi, S.P.; Shihabuddin, L.S. Sterol auto-oxidation adversely affects human motor neuron viability and is a neuropathological feature of amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 803. [Google Scholar] [CrossRef]
- Gutner, U.A.; Shupik, M.A.; Maloshitskaya, O.A.; Sokolov, S.A.; Rezvykh, A.P.; Funikov, S.Y.; Lebedev, A.T.; Ustyugov, A.A.; Alessenko, A.V. Changes in the Metabolism of Sphingoid Bases in the Brain and Spinal Cord of Transgenic FUS(1-359) Mice, a Model of Amyotrophic Lateral Sclerosis. Biochemistry 2019, 84, 1166–1176. [Google Scholar] [CrossRef]
- Wang, D.; Liang, W.; Huo, D.; Wang, H.; Wang, Y.; Cong, C.; Zhang, C.; Yan, S.; Gao, M.; Su, X.; et al. SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1. Cell Death Differ. 2023, 30, 369–382. [Google Scholar] [CrossRef]
- Mukhutdinova, K.A.; Kasimov, M.R.; Giniatullin, A.R.; Zakyrjanova, G.F.; Petrov, A.M. 24S-hydroxycholesterol suppresses neuromuscular transmission in SOD1(G93A) mice: A possible role of NO and lipid rafts. Mol. Cell. Neurosci. 2018, 88, 308–318. [Google Scholar] [CrossRef]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.; Chandrasekar, A.; Prause, J.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 2014, 5, e1290. [Google Scholar] [CrossRef]
- Petrov, A.M.; Kravtsova, V.V.; Matchkov, V.V.; Vasiliev, A.N.; Zefirov, A.L.; Chibalin, A.V.; Heiny, J.A.; Krivoi, I.I. Membrane lipid rafts are disturbed in the response of rat skeletal muscle to short-term disuse. Am. J. Physiol. Cell. Physiol. 2017, 312, C627–C637. [Google Scholar] [CrossRef]
- Bryndina, I.G.; Shalagina, M.N.; Sekunov, A.V.; Zefirov, A.L.; Petrov, A.M. Clomipramine counteracts lipid raft disturbance due to short-term muscle disuse. Neurosci. Lett. 2018, 664, 1–6. [Google Scholar] [CrossRef]
- Choi, B.J.; Park, K.H.; Park, M.H.; Huang, E.J.; Kim, S.H.; Bae, J.S.; Jin, H.K. Acid sphingomyelinase inhibition improves motor behavioral deficits and neuronal loss in an amyotrophic lateral sclerosis mouse model. BMB Rep. 2022, 55, 621–626. [Google Scholar] [CrossRef]
- Bryndina, I.G.; Shalagina, M.N.; Protopopov, V.A.; Sekunov, A.V.; Zefirov, A.L.; Zakirjanova, G.F.; Petrov, A.M. Early Lipid Raft-Related Changes: Interplay between Unilateral Denervation and Hindlimb Suspension. Int. J. Mol. Sci. 2021, 22, 2239. [Google Scholar] [CrossRef]
- Kasimov, M.R.; Zakyrjanova, G.F.; Giniatullin, A.R.; Zefirov, A.L.; Petrov, A.M. Similar oxysterols may lead to opposite effects on synaptic transmission: Olesoxime versus 5alpha-cholestan-3-one at the frog neuromuscular junction. Biochim. Biophys. Acta 2016, 1861, 606–616. [Google Scholar] [CrossRef] [PubMed]
- West, R.J.H.; Briggs, L.; Perona Fjeldstad, M.; Ribchester, R.R.; Sweeney, S.T. Sphingolipids regulate neuromuscular synapse structure and function in Drosophila. J. Comp. Neurol. 2018, 526, 1995–2009. [Google Scholar] [CrossRef]
- Pato, C.; Stetzkowski-Marden, F.; Gaus, K.; Recouvreur, M.; Cartaud, A.; Cartaud, J. Role of lipid rafts in agrin-elicited acetylcholine receptor clustering. Chem. Biol. Interact. 2008, 175, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Krivoi, I.I.; Petrov, A.M. Cholesterol and the Safety Factor for Neuromuscular Transmission. Int. J. Mol. Sci. 2019, 20, 1046. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Soler-Jover, A.; Blasi, J.; Aguilera, J. Synaptic proteins and SNARE complexes are localized in lipid rafts from rat brain synaptosomes. Biochem. Biophys. Res. Commun. 2005, 329, 117–124. [Google Scholar] [CrossRef]
- Garcia-Martinez, V.; Montes, M.A.; Villanueva, J.; Gimenez-Molina, Y.; de Toledo, G.A.; Gutierrez, L.M. Sphingomyelin derivatives increase the frequency of microvesicle and granule fusion in chromaffin cells. Neuroscience 2015, 295, 117–125. [Google Scholar] [CrossRef]
- Jia, J.Y.; Lamer, S.; Schumann, M.; Schmidt, M.R.; Krause, E.; Haucke, V. Quantitative proteomics analysis of detergent-resistant membranes from chemical synapses: Evidence for cholesterol as spatial organizer of synaptic vesicle cycling. Mol. Cell. Proteom. 2006, 5, 2060–2071. [Google Scholar] [CrossRef]
- Kao, H.T.; Ryoo, K.; Lin, A.; Janoschka, S.R.; Augustine, G.J.; Porton, B. Synapsins regulate brain-derived neurotrophic factor-mediated synaptic potentiation and axon elongation by acting on membrane rafts. Eur. J. Neurosci. 2017, 45, 1085–1101. [Google Scholar] [CrossRef]
- Venkova, K.; Christov, A.; Kamaluddin, Z.; Kobalka, P.; Siddiqui, S.; Hensley, K. Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2014, 73, 702–713. [Google Scholar] [CrossRef]
- Harrison, J.M.; Rafuse, V.F. Muscle fiber-type specific terminal Schwann cell pathology leads to sprouting deficits following partial denervation in SOD1(G93A) mice. Neurobiol. Dis. 2020, 145, 105052. [Google Scholar] [CrossRef]
- Martineau, E.; Arbour, D.; Vallee, J.; Robitaille, R. Properties of Glial Cell at the Neuromuscular Junction Are Incompatible with Synaptic Repair in the SOD1(G37R) ALS Mouse Model. J. Neurosci. 2020, 40, 7759–7777. [Google Scholar] [CrossRef]
- Adey, B.N.; Cooper-Knock, J.; Al Khleifat, A.; Fogh, I.; van Damme, P.; Corcia, P.; Couratier, P.; Hardiman, O.; McLaughlin, R.; Gotkine, M.; et al. Large-scale analyses of CAV1 and CAV2 suggest their expression is higher in post-mortem ALS brain tissue and affects survival. Front. Cell. Neurosci. 2023, 17, 1112405. [Google Scholar] [CrossRef]
- Wang, Y.; Bai, L.; Li, S.; Wen, Y.; Liu, Q.; Li, R.; Liu, Y. Simvastatin Enhances Muscle Regeneration Through Autophagic Defect-Mediated Inflammation and mTOR Activation in G93ASOD1 Mice. Mol. Neurobiol. 2021, 58, 1593–1606. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Zhang, S.; Kenna, K.P.; Moll, T.; Franklin, J.P.; Allen, S.; Nezhad, H.G.; Iacoangeli, A.; Yacovzada, N.Y.; Eitan, C.; et al. Rare variant burden analysis within enhancers identifies CAV1 as an ALS risk gene. Cell. Rep. 2021, 34, 108730. [Google Scholar] [CrossRef]
- Bukcharaeva, E.A.; Kim, K.C.; Moravec, J.; Nikolsky, E.E.; Vyskocil, F. Noradrenaline synchronizes evoked quantal release at frog neuromuscular junctions. J. Physiol. 1999, 517 Pt 3, 879–888. [Google Scholar] [CrossRef]
- Giniatullin, A.; Petrov, A.; Giniatullin, R. Action of Hydrogen Peroxide on Synaptic Transmission at the Mouse Neuromuscular Junction. Neuroscience 2019, 399, 135–145. [Google Scholar] [CrossRef]
- Glavinovic, M.I. Voltage clamping of unparalysed cut rat diaphragm for study of transmitter release. J. Physiol. 1979, 290, 467–480. [Google Scholar] [CrossRef]
- Zakharov, A.V. Elph: An Open-Source Program for Acquisition Control and Analysis of Electrophysiological Signals. Uchenye Zap. Kazan. Univ. Seriya Estestv. Nauk. 2019, 161, 245–254. [Google Scholar] [CrossRef]
- Margheri, G.; D’Agostino, R.; Trigari, S.; Sottini, S.; Del Rosso, M. The beta-subunit of cholera toxin has a high affinity for ganglioside GM1 embedded into solid supported lipid membranes with a lipid raft-like composition. Lipids 2014, 49, 203–206. [Google Scholar] [CrossRef]
- Marks, D.L.; Bittman, R.; Pagano, R.E. Use of Bodipy-labeled sphingolipid and cholesterol analogs to examine membrane microdomains in cells. Histochem. Cell. Biol. 2008, 130, 819–832. [Google Scholar] [CrossRef]
- Grigoryev, P.N.; Khisamieva, G.A.; Zefirov, A.L. Septin Polymerization Slows Synaptic Vesicle Recycling in Motor Nerve Endings. Acta Nat. 2019, 11, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhilyakov, N.; Arkhipov, A.; Malomouzh, A.; Samigullin, D. Activation of Neuronal Nicotinic Receptors Inhibits Acetylcholine Release in the Neuromuscular Junction by Increasing Ca2+ Flux through Cav1 Channels. Int. J. Mol. Sci. 2021, 22, 9031. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | FUS (6–8 w) | WT (6–8 w) | p-Value | FUS (18–20 w) | WT (18–20 w) | p-Value |
|---|---|---|---|---|---|---|
| nAChR area | 197 ± 20 µm | 205 ± 46 µm | 0.835 | 145 ± 29 µm | 189 ± 25 µm | 0.038 * |
| Synapsin 1 area | 97 ± 24 µm | 86 ± 30 µm | 0.403 | 68 ± 17 µm | 83 ± 20 µm | 0.403 |
| SNAP-25 area | 80 ± 31 µm | 84 ± 23 µm | 0.531 | 65 ± 28 µm | 72 ± 10 µm | 1 |
| Synaptophysin area | 113 ± 12 µm | 118 ± 17 µm | 0.676 | 78 ± 9 µm | 121 ± 38 µm | 0.012 * |
| M1 coefficient * | 0.51 ± 0.13 | 0.55 ± 0.13 | 0.362 | 0.35 ± 0.12 | 0.49 ± 0.13 | 0.067 |
| M2 coefficient ** | 0.36 ± 0.09 | 0.32 ± 0.70 | 0.208 | 0.22 ± 0.08 | 0.32 ± 0.06 | 0.024 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukhamedyarov, M.A.; Khabibrakhmanov, A.N.; Khuzakhmetova, V.F.; Giniatullin, A.R.; Zakirjanova, G.F.; Zhilyakov, N.V.; Mukhutdinova, K.A.; Samigullin, D.V.; Grigoryev, P.N.; Zakharov, A.V.; et al. Early Alterations in Structural and Functional Properties in the Neuromuscular Junctions of Mutant FUS Mice. Int. J. Mol. Sci. 2023, 24, 9022. https://doi.org/10.3390/ijms24109022
Mukhamedyarov MA, Khabibrakhmanov AN, Khuzakhmetova VF, Giniatullin AR, Zakirjanova GF, Zhilyakov NV, Mukhutdinova KA, Samigullin DV, Grigoryev PN, Zakharov AV, et al. Early Alterations in Structural and Functional Properties in the Neuromuscular Junctions of Mutant FUS Mice. International Journal of Molecular Sciences. 2023; 24(10):9022. https://doi.org/10.3390/ijms24109022
Chicago/Turabian StyleMukhamedyarov, Marat A., Aydar N. Khabibrakhmanov, Venera F. Khuzakhmetova, Arthur R. Giniatullin, Guzalia F. Zakirjanova, Nikita V. Zhilyakov, Kamilla A. Mukhutdinova, Dmitry V. Samigullin, Pavel N. Grigoryev, Andrey V. Zakharov, and et al. 2023. "Early Alterations in Structural and Functional Properties in the Neuromuscular Junctions of Mutant FUS Mice" International Journal of Molecular Sciences 24, no. 10: 9022. https://doi.org/10.3390/ijms24109022
APA StyleMukhamedyarov, M. A., Khabibrakhmanov, A. N., Khuzakhmetova, V. F., Giniatullin, A. R., Zakirjanova, G. F., Zhilyakov, N. V., Mukhutdinova, K. A., Samigullin, D. V., Grigoryev, P. N., Zakharov, A. V., Zefirov, A. L., & Petrov, A. M. (2023). Early Alterations in Structural and Functional Properties in the Neuromuscular Junctions of Mutant FUS Mice. International Journal of Molecular Sciences, 24(10), 9022. https://doi.org/10.3390/ijms24109022