
Activation of Chaperone-Mediated Autophagy Inhibits the Aryl Hydrocarbon Receptor Function by Degrading This Receptor in Human Lung Epithelial Carcinoma A549 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Subsection
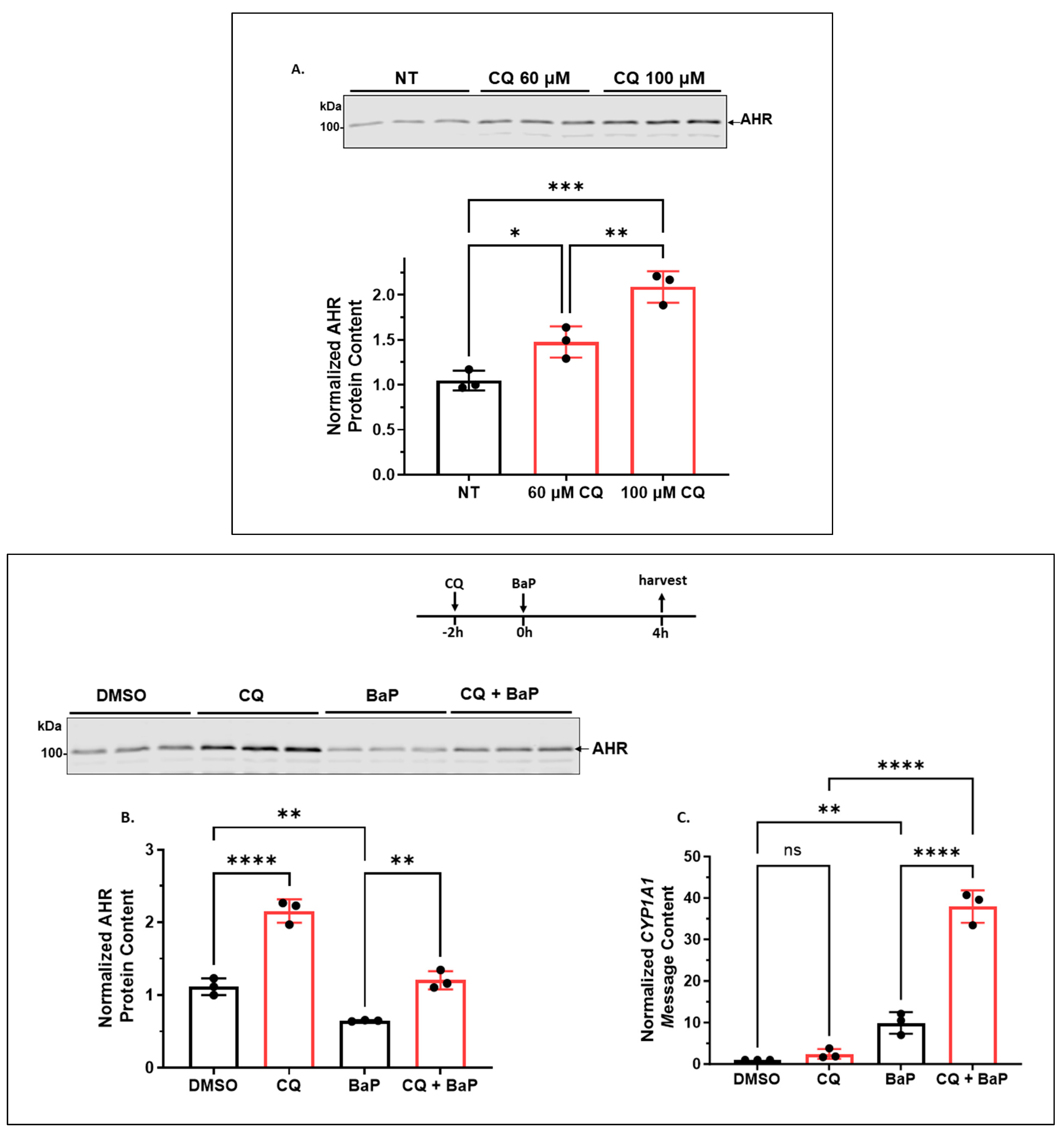
2.1.1. CQ Increases the AHR Protein Levels of A549 Cells in a Functionally Relevant Manner
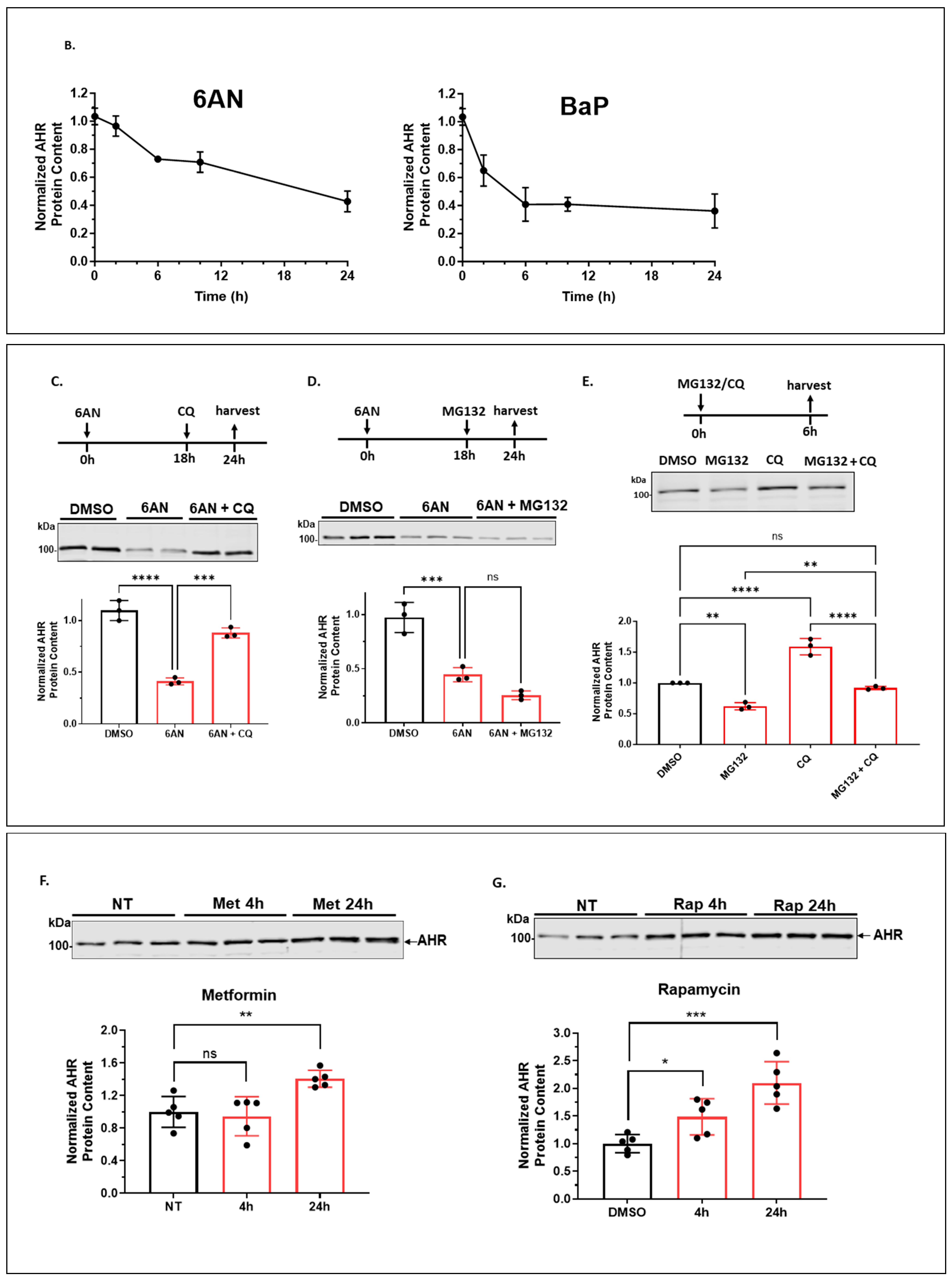
2.1.2. 6-AN Reduces the AHR Protein Content of A549 Cells in a Dose- and Time-Dependent Manner with Functional Relevance
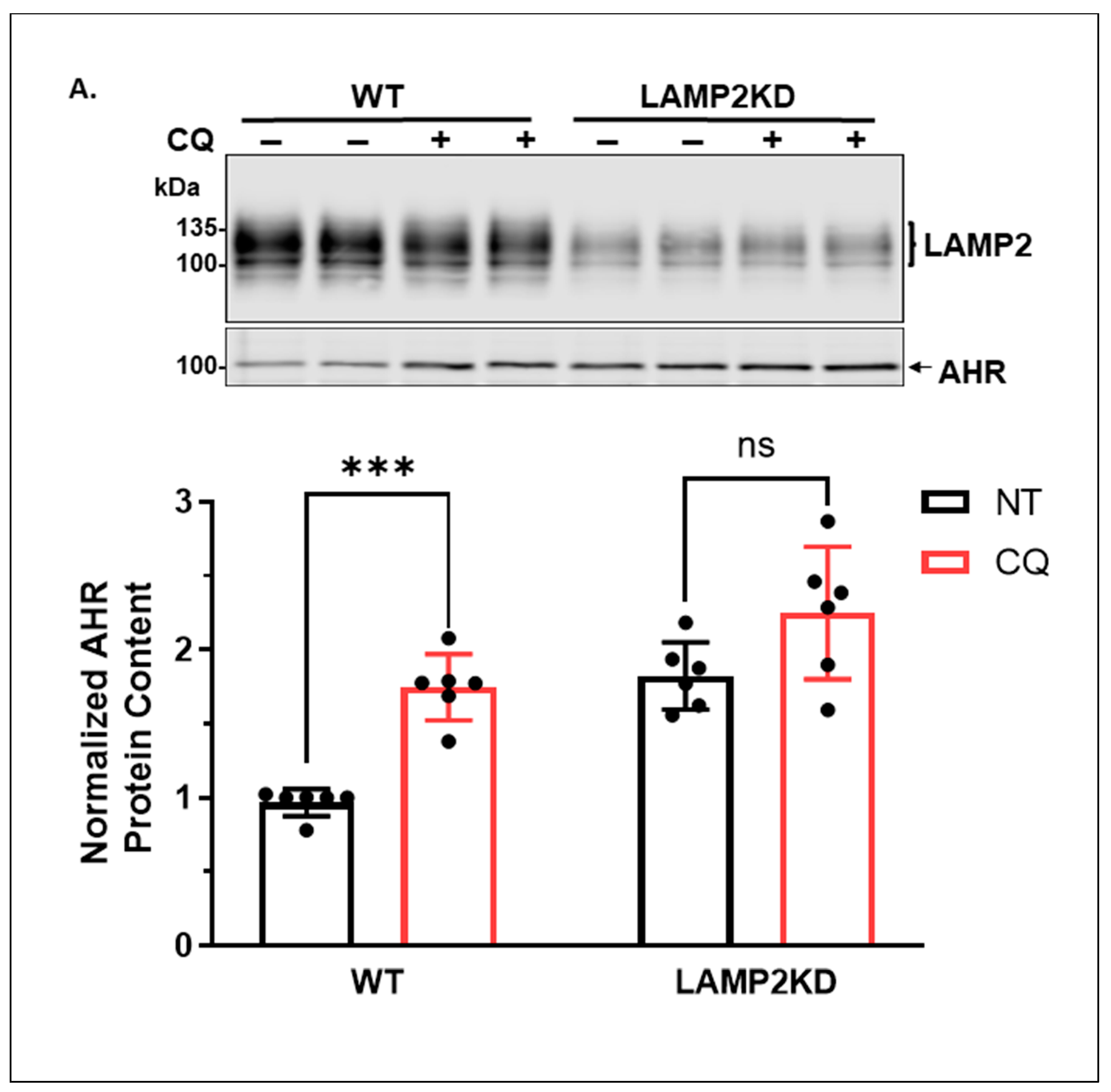
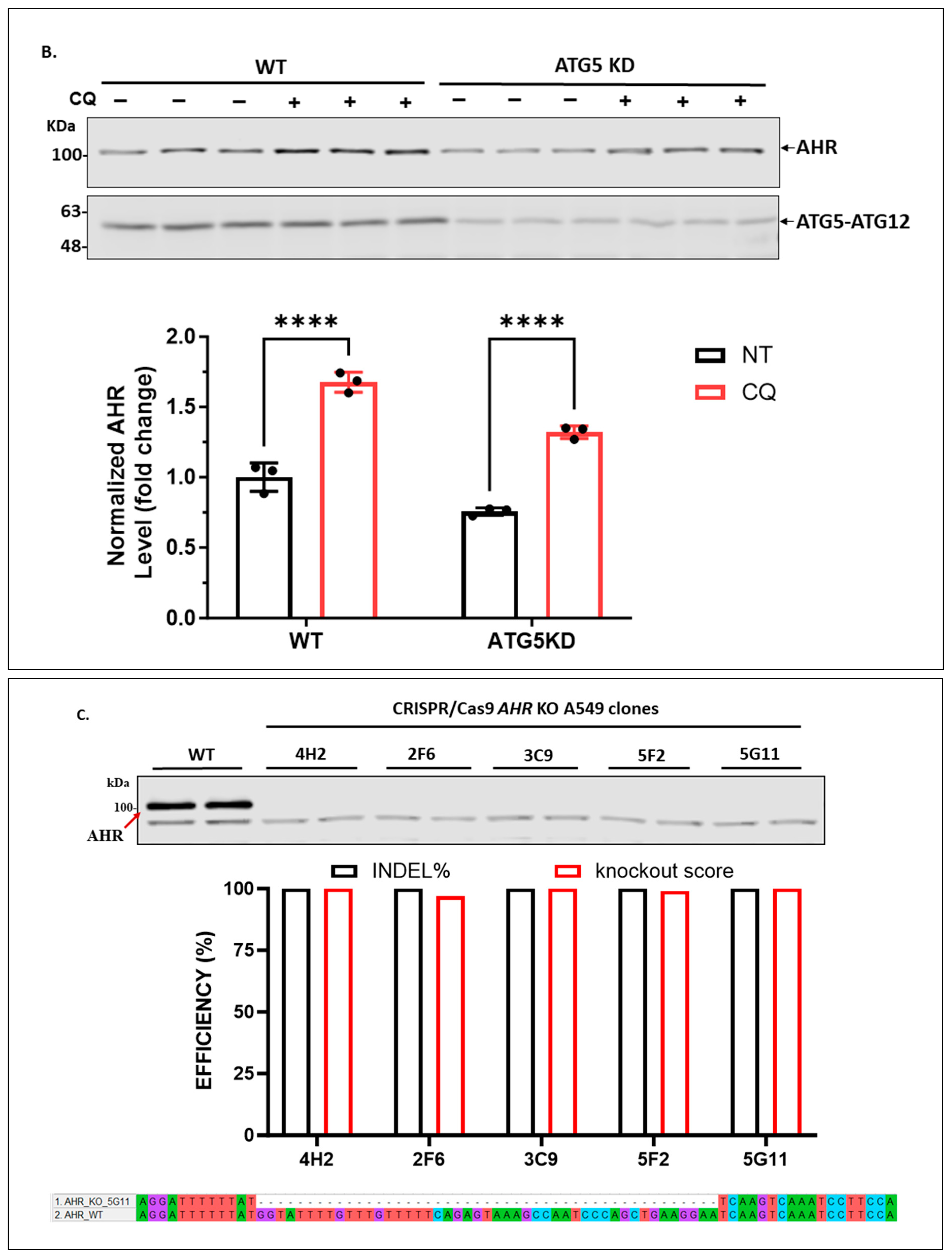
2.1.3. Knockdown of LAMP2 in A549 Cells Abolishes the CQ-Mediated Increase of the AHR Protein Content
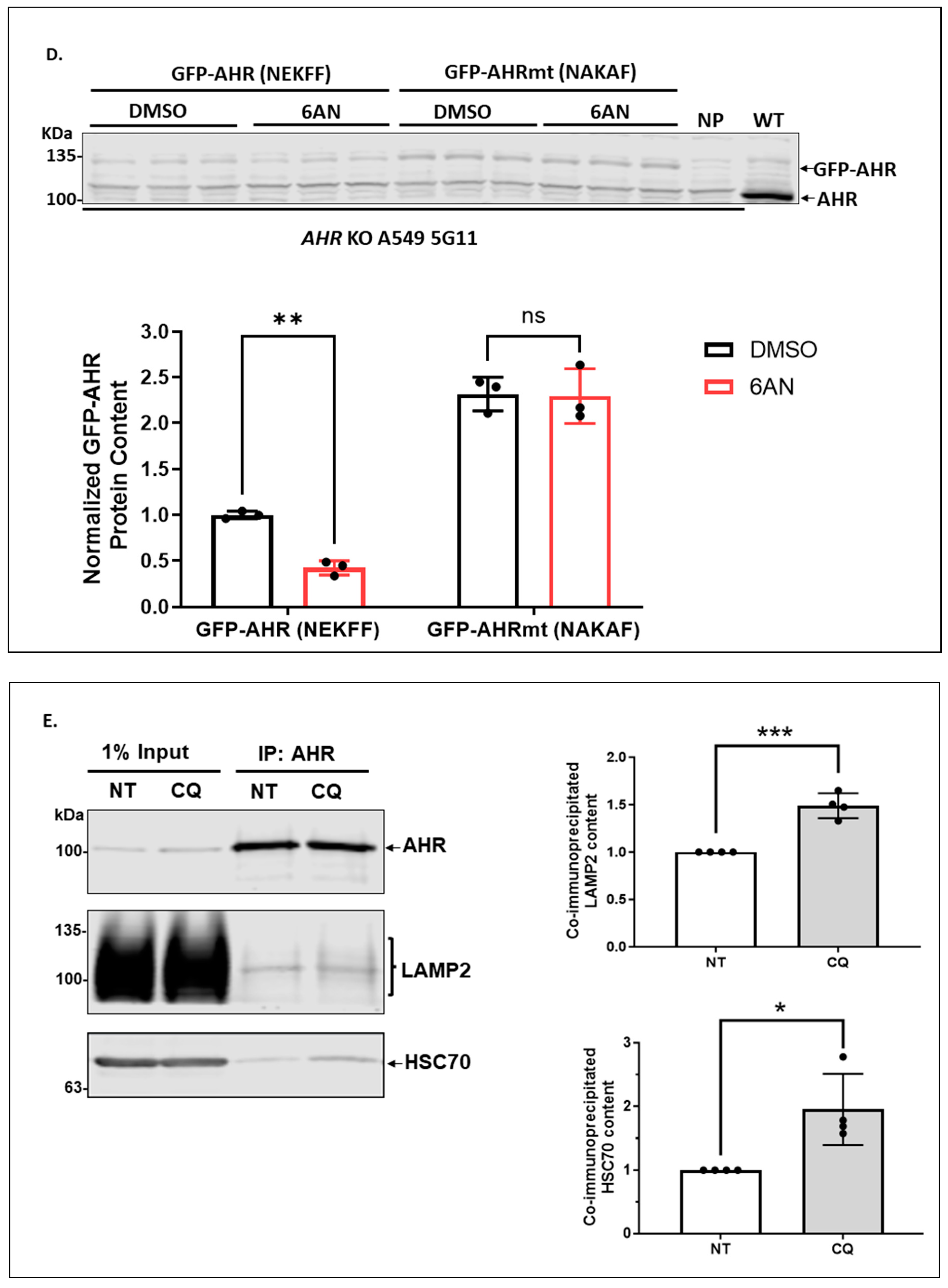
2.1.4. Degradation of AHR via CMA Is Dependent on the NEKFF Motif of AHR in A549 Cells
2.1.5. Autophagy of AHR Is Ongoing in the Background While AHR Is Undergoing Rapid Degradation via the Ubiquitin–Proteasome System after Treatment with an AHR Ligand in A549 Cells
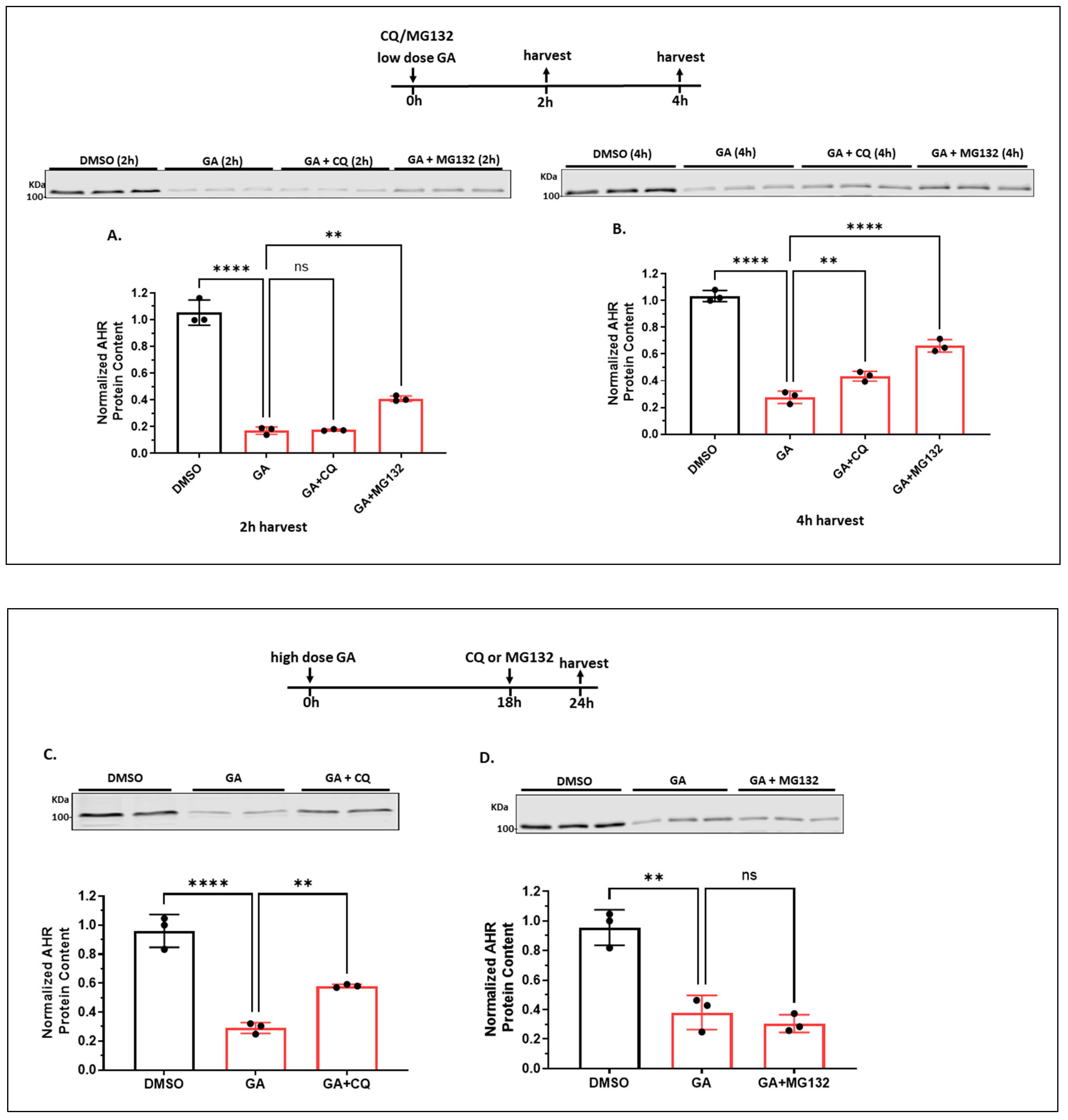
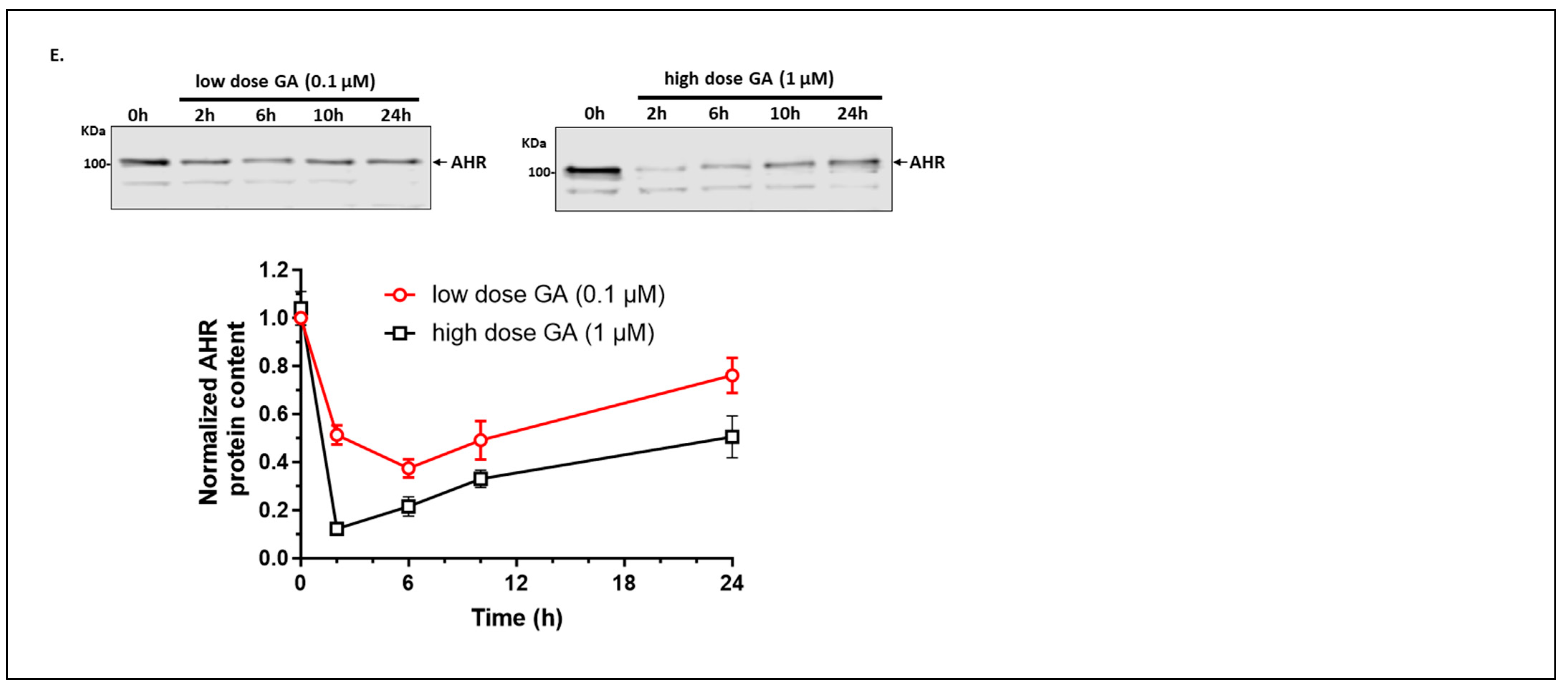
2.1.6. Autophagy Is Not Involved in the Quick-Onset Degradation of AHR Triggered by a Low Dose of Geldanamycin (GA) But Is Involved in Controlling the AHR Levels after Both Low and High Doses of GA Treatment in A549 Cells
2.1.7. 6-AN Is Not an AHR Ligand
2.1.8. AHR Promotes Migration of A549 Cells in a Wound Healing Assay
2.1.9. AHR Promotes EMT in A549 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Preparation of Whole Cell Extract and Western Blot Analysis
4.4. RNA Extraction and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.5. Generation of ATG5, LAMP2 Stable Knockdown A549 Cells Using Lentivirus
4.6. CRISPR/Cas9-Mediated AHR Knockout in A549 Cells
4.7. Transient Transfection
4.8. Co-Immunoprecipitation
4.9. Ligand Dependent, DRE-Driven Expression of GFP in H4G1.1c3 Cells
4.10. Wound Healing Assay
4.11. Transwell Migration and Invasion Assay
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, L.; Hatzakis, E.; Nichols, R.G.; Hao, R.; Correll, J.; Smith, P.B.; Chiaro, C.R.; Perdew, G.H.; Patterson, A.D. Metabolomics reveals that aryl hydrocarbon receptor activation by environmental chemicals induces systemic metabolic dysfunction in mice. Environ. Sci. Technol. 2015, 49, 8067–8077. [Google Scholar] [CrossRef] [PubMed]
- Goya-Jorge, E.; Jorge Rodriguez, M.E.; Veitia, M.S.; Giner, R.M. Plant occurring flavonoids as modulators of the aryl hydrocarbon receptor. Molecules 2021, 26, 2315. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and tryptophan metabolism: Endogenous and dietary routes to Ah receptor activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and Th17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Perdew, G.H.; Esser, C.; Snyder, M.; Sherr, D.H.; van den Bogaard, E.H.; McGovern, K.; Fernandez-Salguero, P.M.; Coumoul, X.; Patterson, A.D. The Ah receptor from toxicity to therapeutics: Report from the 5th AHR Meeting at Penn State University, USA, June 2022. Int. J. Mol. Sci. 2023, 24, 5550. [Google Scholar] [CrossRef]
- Haque, N.; Ojo, E.S.; Krager, S.L.; Tischkau, S.A. Deficiency of adipose aryl hydrocarbon receptor protects against diet-induced metabolic dysfunction through sexually dimorphic mechanisms. Cells 2023, 12, 1748. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Li, Z.; Remes-Lenicov, F.; Davola, M.E.; Elizalde, M.; Paletta, A.; Ashkar, A.A.; Mossman, K.L.; Dugour, A.V.; Figueroa, J.M.; et al. AHR signaling is induced by infection with coronaviruses. Nat. Commun. 2021, 12, 5148. [Google Scholar] [CrossRef]
- Shi, J.; Du, T.; Wang, J.; Tang, C.; Lei, M.; Yu, W.; Yang, Y.; Ma, Y.; Huang, P.; Chen, H.; et al. Aryl hydrocarbon receptor is a proviral host factor and a candidate pan-SARS-CoV-2 therapeutic target. Sci. Adv. 2023, 9, eadf0211. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Li, Y.; Li, L. Promoting epithelial-to-mesenchymal transition by D-kynurenine via activating aryl hydrocarbon receptor. Mol. Cell. Biochem. 2018, 448, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Nucci, N.; Menicali, E.; Morelli, S.; Bini, V.; Colella, R.; Mandarano, M.; Sidoni, A.; Puxeddu, E. The aryl hydrocarbon receptor is expressed in thyroid carcinoma and appears to mediate epithelial-mesenchymal transition. Cancers 2020, 12, 145. [Google Scholar] [CrossRef]
- Gao, H.; Ye, G.; Lin, Y.; Chi, Y.; Dong, S. Benzo[a]pyrene at human blood equivalent level induces human lung epithelial cell invasion and migration via aryl hydrocarbon receptor signaling. J. Appl. Toxicol. 2020, 40, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Selvam, P.; Cheng, C.M.; Dahms, H.U.; Ponnusamy, V.K.; Sun, Y.Y. AhR mediated activation of pro-inflammatory response of RAW 264.7 cells modulate the epithelial-mesenchymal transition. Toxics 2022, 10, 642. [Google Scholar] [CrossRef]
- Tsai, C.H.; Li, C.H.; Cheng, Y.W.; Lee, C.C.; Liao, P.L.; Lin, C.H.; Huang, S.H.; Kang, J.J. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 2017, 7, 41927. [Google Scholar] [CrossRef] [PubMed]
- Nothdurft, S.; Thumser-Henner, C.; Breitenbucher, F.; Okimoto, R.A.; Dorsch, M.; Opitz, C.A.; Sadik, A.; Esser, C.; Holzel, M.; Asthana, S.; et al. Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis. Oncogenesis 2020, 9, 102. [Google Scholar] [CrossRef]
- Enan, E.; Matsumura, F. Identification of c-Src as the integral component of the cytosolic Ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem. Pharmacol. 1996, 52, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.K.; Perdew, G.H. Characterization of the AhR-hsp90-XAP2 core complex and the role of the immunophilin-related protein XAP2 in AhR stabilization. Biochemistry 1999, 38, 8907–8917. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chan, W.K. Selective autophagy maintains the aryl hydrocarbon receptor levels in HeLa cells: A mechanism that is dependent on the p23 co-chaperone. Int. J. Mol. Sci. 2020, 21, 3449. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Li, P.; Ji, M.; Lu, F.; Zhang, J.; Li, H.; Cui, T.; Li Wang, X.; Tang, D.; Ji, C. Degradation of AF1Q by chaperone-mediated autophagy. Exp. Cell Res. 2014, 327, 48–56. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.; Russu, W.A.; Chan, W.K. The aryl hydrocarbon receptor undergoes chaperone-mediated autophagy in triple-negative breast cancer cells. Int. J. Mol. Sci. 2021, 22, 1654. [Google Scholar] [CrossRef] [PubMed]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1999, 274, 28708–28715. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.J.; Whitelaw, M.L. Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. J. Biol. Chem. 1999, 274, 36351–36356. [Google Scholar] [CrossRef] [PubMed]
- Giannone, J.V.; Li, W.; Probst, M.; Okey, A.B. Prolonged depletion of AH receptor without alteration of receptor mRNA levels after treatment of cells in culture with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol. 1998, 55, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Gu, Y.; Ta, L.; Wang, K.; Xu, Z. Induction of autophagy by the MG132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF7 cells. Mol. Med. Rep. 2016, 13, 796–804. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Levy, N. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Shen, Y.F.; Tang, Y.; Zhang, X.J.; Huang, K.X.; Le, W.D. Adaptive changes in autophagy after UPS impairment in Parkinson’s disease. Acta Pharmacol. Sin. 2013, 34, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Pollenz, R.S. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation, and gene regulation. Mol. Pharmacol. 2002, 62, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Uesugi, M.; Takase, K.; Miyamoto, N.; Sawada, K. Hsp90 inhibitor geldanamycin attenuates the cytotoxicity of sunitinib in cardiomyocytes via inhibition of the autophagy pathway. Toxicol. Appl. Pharmacol. 2017, 329, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Q.; Ji, D.; Wei, Y.; Chen, H.; Li, T.; Wan, B.; Yuan, L.; Huang, R.; Chen, G. Inhibition of pentose phosphate pathway suppresses acute myelogenous leukemia. Tumour Biol. 2016, 37, 6027–6034. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, H.; Remmers, N.; Hollingsworth, M.A. Loss of E-cadherin and epithelial to mesenchymal transition is not required for cell motility in tissues or for metastasis. Tissue Barriers 2014, 2, e969112. [Google Scholar] [CrossRef]
- Liu, H.; Yong, Y.; Li, X.; Ye, P.; Tao, K.; Peng, G.; Mo, M.; Guo, W.; Chen, X.; Luo, Y.; et al. Chaperone-mediated autophagy regulates cell growth by targeting SMAD3 in glioma. Neurosci. Bull. 2022, 38, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Pan, Y.; Zheng, S.; Liu, Y.; Xu, J.; Peng, Y.; Zhang, Z.; Wang, Y.; Xiong, Y.; Xu, L.; et al. Inactivation of EGLN3 hydroxylase facilitates Erk3 degradation via autophagy and impedes lung cancer growth. Oncogene 2022, 41, 1752–1766. [Google Scholar] [CrossRef]
- Su, C.M.; Hsu, T.W.; Chen, H.A.; Wang, W.Y.; Huang, C.Y.; Hung, C.C.; Yeh, M.H.; Su, Y.H.; Huang, M.T.; Liao, P.H. Chaperone-mediated autophagy degrade Dicer to promote breast cancer metastasis. J. Cell. Physiol. 2023, 238, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yue, T.; Du, X.; Tang, Z.; Cui, J.; Wang, W.; Xia, W.; Ren, B.; Kan, S.; Li, C.; et al. HSC70 mediated autophagic degradation of oxidized PRL2 is responsible for osteoclastogenesis and inflammatory bone destruction. Cell Death Differ. 2023, 30, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, e12692. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef]
- Sahoo, S.; Padhy, A.A.; Kumari, V.; Mishra, P. Role of ubiquitin-proteasome and autophagy-lysosome pathways in alpha-synuclein aggregate clearance. Mol. Neurobiol. 2022, 59, 5379–5407. [Google Scholar] [CrossRef]
- Dvorak, Z. Pelargonidin and its glycosides as dietary chemopreventives attenuating inflammatory bowel disease symptoms through the aryl hydrocarbon receptor. Eur. J. Nutr. 2020, 59, 3863–3864. [Google Scholar] [CrossRef]
- Retnakumar, S.V.; Geesala, R.; Bretin, A.; Tourneur-Marsille, J.; Ogier-Denis, E.; Maretzky, T.; Nguyen, H.T.T.; Muller, S. Targeting the endo-lysosomal autophagy pathway to treat inflammatory bowel diseases. J. Autoimmun. 2022, 128, 102814. [Google Scholar] [CrossRef]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef]
- Kim, J.; de Haro, M.; Al-Ramahi, I.; Garaicoechea, L.L.; Jeong, H.H.; Sonn, J.Y.; Tadros, B.; Liu, Z.; Botas, J.; Zoghbi, H.Y. Evolutionarily conserved regulators of tau identify targets for new therapies. Neuron 2023, 111, 824–838. [Google Scholar] [CrossRef]
- Bento, C.F.; Fernandes, R.; Ramalho, J.; Marques, C.; Shang, F.; Taylor, A.; Pereira, P. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1alpha for degradation in the presence of methylglyoxal. PLoS ONE 2010, 5, e15062. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1α degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.L.; Perdew, G.H. Carboxyl terminus of hsc70-interacting protein (CHIP) can remodel mature aryl hydrocarbon receptor complexes and mediate ubiquitination of both the AhR and the 90 kDa heat-shock protein (hsp90) in vitro. Biochemistry 2007, 46, 610–621. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Ouyang, L.; Zhou, Y.; Lai, W.; Chen, Y.; Wang, Z.; Yan, B.; Zhang, Z.; Zhou, Y.; Peng, X.; et al. AhR promotes the development of non-small cell lung cancer by inducing SLC7A11-dependent antioxidant function. J. Cancer 2023, 14, 821–834. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Yeh, G.C. Inhibition of aryl hydrocarbon-induced cytochrome P-450 1A1 enzyme activity and CYP1A1 expression by resveratrol. Mol. Pharmacol. 1999, 56, 760–767. [Google Scholar]
- Wang, H.; Zhang, H.; Tang, L.; Chen, H.; Wu, C.; Zhao, M.; Yang, Y.; Chen, X.; Liu, G. Resveratrol inhibits TGF-beta1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology 2013, 303, 139–146. [Google Scholar] [CrossRef]
- Ouyang, L.; Yan, B.; Liu, Y.; Mao, C.; Wang, M.; Liu, N.; Wang, Z.; Liu, S.; Shi, Y.; Chen, L.; et al. The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor. Signal Transduct. Target Ther. 2020, 5, 78. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, X.; Zhang, Y.; Wu, B.; Fang, L.; Wang, N.; Yi, H.; Chang, N.; Chen, L.; Zhang, J. Aryl hydrocarbon receptor mediates Jak2/STAT3 signaling for non-small cell lung cancer stem cell maintenance. Exp. Cell Res. 2020, 396, 112288. [Google Scholar] [CrossRef]
- Gerashchenko, T.S.; Novikov, N.M.; Krakhmal, N.V.; Zolotaryova, S.Y.; Zavyalova, M.V.; Cherdyntseva, N.V.; Denisov, E.V.; Perelmuter, V.M. Markers of Cancer Cell Invasion: Are They Good Enough? J. Clin. Med. 2019, 8, 1092. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Karki, K.; Cheng, Y.; Michelhaugh, S.K.; Mittal, S.; Safe, S. The aryl hydrocarbon receptor is a tumor suppressor-like gene in glioblastoma. J. Biol. Chem. 2019, 294, 11342–11353. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, R.; Shao, D.; Do, S.; Chan, W.K. Activation of Chaperone-Mediated Autophagy Inhibits the Aryl Hydrocarbon Receptor Function by Degrading This Receptor in Human Lung Epithelial Carcinoma A549 Cells. Int. J. Mol. Sci. 2023, 24, 15116. https://doi.org/10.3390/ijms242015116
Xiong R, Shao D, Do S, Chan WK. Activation of Chaperone-Mediated Autophagy Inhibits the Aryl Hydrocarbon Receptor Function by Degrading This Receptor in Human Lung Epithelial Carcinoma A549 Cells. International Journal of Molecular Sciences. 2023; 24(20):15116. https://doi.org/10.3390/ijms242015116
Chicago/Turabian StyleXiong, Rui, Dan Shao, Sandra Do, and William K. Chan. 2023. "Activation of Chaperone-Mediated Autophagy Inhibits the Aryl Hydrocarbon Receptor Function by Degrading This Receptor in Human Lung Epithelial Carcinoma A549 Cells" International Journal of Molecular Sciences 24, no. 20: 15116. https://doi.org/10.3390/ijms242015116
APA StyleXiong, R., Shao, D., Do, S., & Chan, W. K. (2023). Activation of Chaperone-Mediated Autophagy Inhibits the Aryl Hydrocarbon Receptor Function by Degrading This Receptor in Human Lung Epithelial Carcinoma A549 Cells. International Journal of Molecular Sciences, 24(20), 15116. https://doi.org/10.3390/ijms242015116