

Impact of Microbiome–Brain Communication on Neuroinflammation and Neurodegeneration
 ,
, {kind=link}
Abstract
:1. Introduction
2. The Gut and the Brain—Neurological Disorders and Intestinal Inflammation
2.1. Multiple Sclerosis
2.2. Alzheimer’s Disease
2.3. Parkinson’s Disease
2.4. Inflammatory Bowel Disease
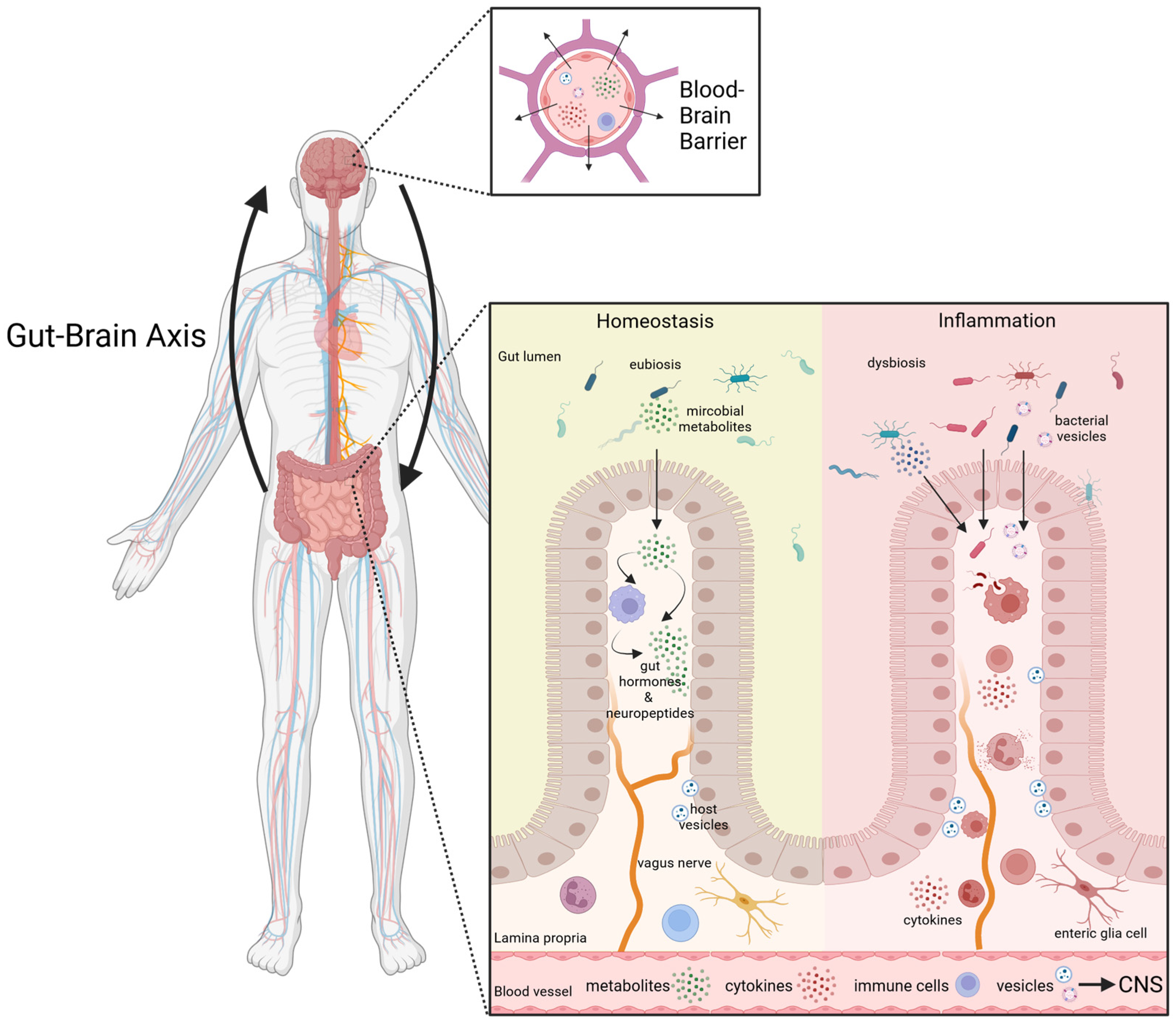
3. Mechanisms of Microbe-Host Communication
4. Microbial Dysbiosis: Implications for Pathogenesis and Therapeutic Strategies
5. Bacterial Cell Wall Components and Bacterial Extracellular Vesical as Novel Aspect within the Microbe–Host Interaction
5.1. LPS as Inducer of Neuroinflammation and Neurodegeneration
5.2. Bacterial Extracellular Vesicles as Promoter of Neurodegenerative Diseases
Author Contributions
Funding
Conflicts of Interest
References
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Scarano, F.; Nucera, S.; Scicchitano, M.; Oppedisano, F.; Bosco, F.; Ruga, S.; et al. The Contribution of Gut Microbiota–Brain Axis in the Development of Brain Disorders. Front. Neurosci. 2021, 15, 616883. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Schreiber, H.L., IV; Mazmanian, S.K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Günther, C.; Rothhammer, V.; Karow, M.; Neurath, M.F.; Winner, B. The Gut-Brain Axis in Inflammatory Bowel Disease—Current and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 8870. [Google Scholar] [CrossRef]
- Masanetz, R.K.; Winkler, J.; Winner, B.; Günther, C.; Süß, P. The Gut–Immune–Brain Axis: An Important Route for Neuropsychiatric Morbidity in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 11111. [Google Scholar] [CrossRef]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Doboszewska, U.; Szopa, A.; Serefko, A.; Włodarczyk, M.; Zielińska, A.; Poleszak, E.; Fichna, J.; Wlaź, P. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharmacol. Res. 2021, 172, 105840. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Haghikia, A.; Wilck, N.; Müller, D.N.; Linker, R.A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Fettig, N.M.; Osborne, L.C. Direct and indirect effects of microbiota-derived metabolites on neuroinflammation in multiple sclerosis. Microbes Infect. 2021, 23, 104814. [Google Scholar] [CrossRef] [PubMed]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Cosorich, I.; Dalla-Costa, G.; Sorini, C.; Ferrarese, R.; Messina, M.J.; Dolpady, J.; Radice, E.; Mariani, A.; Testoni, P.A.; Canducci, F.; et al. High frequency of intestinal T H 17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci. Adv. 2017, 3, e1700492. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.-W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Krishnamoorthy, G. Microbial view of central nervous system autoimmunity. FEBS Lett. 2014, 588, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Riccio, P.; Rossano, R. Diet, Gut Microbiota, and Vitamins D + A in Multiple Sclerosis. Neurotherapeutics 2018, 15, 75–91. [Google Scholar] [CrossRef]
- Bjurstöm, H.; Wang, J.; Ericsson, I.; Bengtsson, M.; Liu, Y.; Kumar-Mendu, S.; Issazadeh-Navikas, S.; Birnir, B. GABA, a natural immunomodulator of T lymphocytes. J. Neuroimmunol. 2008, 205, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef]
- Krantis, A.; Fried, D.E.; Watson, R.E.; Robson, S.C.; Gulbransen, B.D.; Li, Y.; Xiang, Y.-Y.; Lu, W.-Y.; Liu, C.; Li, J. GABA in the Mammalian Enteric Nervous System. Physiology 2000, 15, 284–290. [Google Scholar] [CrossRef]
- Bhat, R.; Axtell, R.; Mitra, A.; Miranda, M.; Lock, C.; Tsien, R.W.; Steinman, L. Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2580–2585. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Pokusaeva, K.; Johnson, C.; Luk, B.; Uribe, G.; Fu, Y.; Oezguen, N.; Matsunami, R.K.; Lugo, M.; Major, A.; Mori-Akiyama, Y.; et al. GABA-producing Bifidobacterium dentium modulates visceral sensitivity in the intestine. Neurogastroenterol. Motil. 2017, 29, e12904. [Google Scholar] [CrossRef]
- Szabo, C. Gaseotransmitters: New frontiers for translational science. Sci. Transl. Med. 2010, 2, 59ps54. [Google Scholar] [CrossRef] [PubMed]
- Oleskin, A.V.; Shenderov, B.A. Probiotics and Psychobiotics: The Role of Microbial Neurochemicals. Probiotics Antimicrob. Proteins 2019, 11, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Oleskin, A.V.; Shenderov, B.A. Neuromodulatory effects and targets of the SCFAs and gasotransmitters produced by the human symbiotic microbiota. Microb. Ecol. Health Dis. 2016, 27, 30971. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Sebastian Monasor, L.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, Y.; Hase, K. Partners in Leaky Gut Syndrome: Intestinal Dysbiosis and Autoimmunity. Front. Immunol. 2021, 12, 673708. [Google Scholar] [CrossRef] [PubMed]
- Fleck, A.-K.; Schuppan, D.; Wiendl, H.; Klotz, L. Gut–CNS-Axis as Possibility to Modulate Inflammatory Disease Activity—Implications for Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 1526. [Google Scholar] [CrossRef]
- Kadowaki, A.; Quintana, F.J. The Gut–CNS Axis in Multiple Sclerosis. Trends Neurosci. 2020, 43, 622–634. [Google Scholar] [CrossRef]
- Legroux, L.; Arbour, N. Multiple Sclerosis and T Lymphocytes: An Entangled Story. J. Neuroimmune Pharmacol. 2015, 10, 528–546. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef] [PubMed]
- Perneczky, R.; Jessen, F.; Grimmer, T.; Levin, J.; Flöel, A.; Peters, O.; Froelich, L. Anti-amyloid antibody therapies in Alzheimer’s disease. Brain 2023, 146, 842–849. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Day, G.S. Anti-amyloid therapies for Alzheimer disease: Finally, good news for patients. Mol. Neurodegener. 2023, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.H.; Yahaya, M.F.; Mohamed, W.; Teoh, S.L.; Hui, C.K.; Kumar, J. Pharmacotherapy of Alzheimer’s Disease: Seeking Clarity in a Time of Uncertainty. Front. Pharmacol. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Zhuang, Z.-Q.; Shen, L.-L.; Li, W.-W.; Fu, X.; Zeng, F.; Gui, L.; Lü, Y.; Cai, M.; Zhu, C.; Tan, Y.-L.; et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1337–1346. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, S.; Shin, S.J.; Park, Y.H.; Nam, Y.; Kim, C.W.; Lee, K.W.; Kim, S.-M.; Jung, I.D.; Yang, H.D.; et al. Gram-negative bacteria and their lipopolysaccharides in Alzheimer’s disease: Pathologic roles and therapeutic implications. Transl. Neurodegener. 2021, 10, 49. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-S.; Li, X.-H.; Liu, P.; Li, J.; Liu, L. The relationship between Alzheimer’s disease and intestinal microflora structure and inflammatory factors. Front. Aging Neurosci. 2022, 14, 972982. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Sarnataro, D. Probiotics, prebiotics and their role in Alzheimer’s disease. Neural Regen. Res. 2021, 16, 1768–1769. [Google Scholar] [CrossRef] [PubMed]
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Wang, L.-K.; Wu, H.-Y.; Jiao, L. Effects of prebiotic galacto-oligosaccharide on postoperative cognitive dysfunction and neuroinflammation through targeting of the gut-brain axis. BMC Anesthesiol. 2018, 18, 177. [Google Scholar] [CrossRef]
- Chen, D.; Yang, X.; Yang, J.; Lai, G.; Yong, T.; Tang, X.; Shuai, O.; Zhou, G.; Xie, Y.; Wu, Q. Prebiotic Effect of Fructooligosaccharides from Morinda officinalis on Alzheimer’s Disease in Rodent Models by Targeting the Microbiota-Gut-Brain Axis. Front. Aging Neurosci. 2017, 9, 403. [Google Scholar] [CrossRef]
- Liu, Q.; Xi, Y.; Wang, Q.; Liu, J.; Li, P.; Meng, X.; Liu, K.; Chen, W.; Liu, X.; Liu, Z. Mannan oligosaccharide attenuates cognitive and behavioral disorders in the 5xFAD Alzheimer’s disease mouse model via regulating the gut microbiota-brain axis. Brain Behav. Immun. 2021, 95, 330–343. [Google Scholar] [CrossRef]
- Chandra, S.; Sisodia, S.S.; Vassar, R.J. The gut microbiome in Alzheimer’s disease: What we know and what remains to be explored. Mol. Neurodegener. 2023, 18, 9. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Valencia, J.; Ferreira, M.; Merino-Torres, J.F.; Marcilla, A.; Soriano, J.M. The Potential Roles of Extracellular Vesicles as Biomarkers for Parkinson’s Disease: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 11508. [Google Scholar] [CrossRef] [PubMed]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2016, 139 Pt 2, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Rani, K.; Mukherjee, R.; Singh, E.; Kumar, S.; Sharma, V.; Vishwakarma, P.; Bharti, P.S.; Nikolajeff, F.; Dinda, A.K.; Goyal, V.; et al. Neuronal exosomes in saliva of Parkinson’s disease patients: A pilot study. Park. Relat. Disord. 2019, 67, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Han, C.L.; Wang, K.L.; Sui, Y.P.; Li, Z.B.; Chen, N.; Fan, S.Y.; Shimabukuro, M.; Wang, F.; Meng, F.G. Integrated analysis of exosomal lncRNA and mRNA expression profiles reveals the involvement of lnc-MKRN2-42:1 in the pathogenesis of Parkinson’s disease. CNS Neurosci. Ther. 2020, 26, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-Y.; Lu, J.-M.; Zhao, Z.-Q.; Li, M.-C.; Lu, T.; An, X.-S.; Xue, L.-J. MicroRNA biomarkers of Parkinson’s disease in serum exosome-like microvesicles. Neurosci. Lett. 2017, 644, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-H.; Chen, Z.-T.; Zhou, R.-L.; Zhang, X.; Ye, Q.-Y.; Wang, Y.-Z. Increased DJ-1 and α-Synuclein in Plasma Neural-Derived Exosomes as Potential Markers for Parkinson’s Disease. Front. Aging Neurosci. 2019, 10, 438. [Google Scholar] [CrossRef]
- Stuendl, A.; Kraus, T.; Chatterjee, M.; Zapke, B.; Sadowski, B.; Moebius, W.; Hobert, M.A.; Deuschle, C.; Brockmann, K.; Maetzler, W.; et al. α-Synuclein in Plasma-Derived Extracellular Vesicles Is a Potential Biomarker of Parkinson’s Disease. Mov. Disord. 2021, 36, 2508–2518. [Google Scholar] [CrossRef]
- Kluge, A.; Bunk, J.; Schaeffer, E.; Drobny, A.; Xiang, W.; Knacke, H.; Bub, S.; Lückstädt, W.; Arnold, P.; Lucius, R.; et al. Detection of neuron-derived pathological α-synuclein in blood. Brain 2022, 145, 3058–3071. [Google Scholar] [CrossRef]
- Beach, T.G.; Corbillé, A.-G.; Letournel, F.; Kordower, J.H.; Kremer, T.; Munoz, D.G.; Intorcia, A.; Hentz, J.; Adler, C.H.; Sue, L.I.; et al. Multicenter Assessment of Immunohistochemical Methods for Pathological Alpha-Synuclein in Sigmoid Colon of Autopsied Parkinson’s Disease and Control Subjects. J. Park. Dis. 2016, 6, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.M.; Keshavarzian, A.; Mutlu, E.; Dodiya, H.B.; Daian, D.; Jaglin, J.A.; Kordower, J.H. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. 2012, 27, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.J.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Resnikoff, H.; Metzger, J.M.; Lopez, M.; Bondarenko, V.; Mejia, A.; Simmons, H.A.; Emborg, M.E. Colonic inflammation affects myenteric alpha-synuclein in nonhuman primates. J. Inflamm. Res. 2019, 12, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lim, K.-L.; Tan, E.-K. Intestine-derived α-synuclein initiates and aggravates pathogenesis of Parkinson’s disease in Drosophila. Transl. Neurodegener. 2022, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Q.; Haikal, C.; Li, W.; Li, M.T.; Wang, Z.Y.; Li, J.Y. Age-dependent alpha-synuclein accumulation and aggregation in the colon of a transgenic mouse model of Parkinson’s disease. Transl. Neurodegener. 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Zhong, C.B.; Chen, Q.Q.; Haikal, C.; Li, W.; Svanbergsson, A.; Diepenbroek, M.; Li, J.Y. Age-Dependent Alpha-Synuclein Accumulation and Phosphorylation in the Enteric Nervous System in a Transgenic Mouse Model of Parkinson’s Disease. Neurosci. Bull. 2017, 33, 483–492. [Google Scholar] [CrossRef]
- Liu, D.; Guo, J.J.; Su, J.H.; Svanbergsson, A.; Yuan, L.; Haikal, C.; Li, W.; Gouras, G.; Li, J.Y. Differential seeding and propagating efficiency of alpha-synuclein strains generated in different conditions. Transl. Neurodegener. 2021, 10, 20. [Google Scholar] [CrossRef]
- Kurnik, M.; Gil, K.; Gajda, M.; Thor, P.; Bugajski, A. Neuropathic alterations of the myenteric plexus neurons following subacute intraperitoneal administration of salsolinol. Folia Histochem. Cytobiol. 2015, 53, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamgüney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.F.; De Oliveira, H.L.; Yamada, E.S.; Neves, B.C.; Pereira, A., Jr. The Gut and Parkinson’s Disease-A Bidirectional Pathway. Front. Neurol. 2019, 10, 574. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.A. Differential diagnosis of inflammatory bowel disease: What is the role of colonoscopy? Clin. Endosc. 2012, 45, 254–262. [Google Scholar] [CrossRef] [PubMed]
- de Lange, K.M.; Barrett, J.C. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 2015, 64, 91–100. [Google Scholar] [CrossRef]
- Barberio, B.; Zamani, M.; Black, C.J.; Savarino, E.V.; Ford, A.C. Prevalence of symptoms of anxiety and depression in patients with inflammatory bowel disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Gracie, D.J.; Guthrie, E.A.; Hamlin, P.J.; Ford, A.C. Bi-directionality of Brain-Gut Interactions in Patients with Inflammatory Bowel Disease. Gastroenterology 2018, 154, 1635–1646.e3. [Google Scholar] [CrossRef]
- Wang, X.; Wan, J.; Wang, M.; Zhang, Y.; Wu, K.; Yang, F. Multiple sclerosis and inflammatory bowel disease: A systematic review and meta-analysis. Ann. Clin. Transl. Neurol. 2022, 9, 132–140. [Google Scholar] [CrossRef]
- Kosmidou, M.; Katsanos, A.H.; Katsanos, K.H.; Kyritsis, A.P.; Tsivgoulis, G.; Christodoulou, D.; Giannopoulos, S. Multiple sclerosis and inflammatory bowel diseases: A systematic review and meta-analysis. J. Neurol. 2017, 264, 254–259. [Google Scholar] [CrossRef]
- Parkes, M.; Cortes, A.; van Heel, D.A.; Brown, M.A. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 2013, 14, 661–673. [Google Scholar] [CrossRef]
- Yang, Y.; Musco, H.; Simpson-Yap, S.; Zhu, Z.; Wang, Y.; Lin, X.; Zhang, J.; Taylor, B.; Gratten, J.; Zhou, Y. Investigating the shared genetic architecture between multiple sclerosis and inflammatory bowel diseases. Nat. Commun. 2021, 12, 5641. [Google Scholar] [CrossRef]
- Camara-Lemarroy, C.R.; Metz, L.; Meddings, J.B.; Sharkey, K.A.; Yong, V.W. The intestinal barrier in multiple sclerosis: Implications for pathophysiology and therapeutics. Brain 2018, 141, 1900–1916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, H.E.; Bai, Y.-M.; Tsai, S.-J.; Su, T.-P.; Chen, T.-J.; Wang, Y.-P.; Chen, M.-H. Inflammatory bowel disease is associated with higher dementia risk: A nationwide longitudinal study. Gut 2021, 70, 85–91. [Google Scholar] [CrossRef]
- Zingel, R.; Bohlken, J.; Kostev, K. Association Between Inflammatory Bowel Disease and Dementia: A Retrospective Cohort Study. J. Alzheimer’s Dis. 2021, 80, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Alkhayyat, M.; Abou Saleh, M.; Sarmini, M.T.; Singh, A.; Garg, R.; Garg, P.; Mansoor, E.; Padival, R.; Cohen, B.L. Alzheimer Disease Occurs More Frequently In Patients with Inflammatory Bowel Disease: Insight from a Nationwide Study. J. Clin. Gastroenterol. 2023, 57, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Zhang, C.; Zhang, K.; Liu, Y.Z.; Peng, X.X.; Zong, Q. Inflammatory bowel disease and risk of Parkinson’s disease: Evidence from a meta-analysis of 14 studies involving more than 13.4 million individuals. Front. Med. 2023, 10, 1137366. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yuan, M.; Liu, Y.; Yang, F.; Chen, W.Z.; Xu, Z.Z.; Xiang, Z.B.; Xu, R.S. Association between inflammatory bowel diseases and Parkinson’s disease: Systematic review and meta-analysis. Neural Regen. Res. 2022, 17, 344–353. [Google Scholar] [PubMed]
- Wang, D.; Zhang, X.; Du, H. Inflammatory bowel disease: A potential pathogenic factor of Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 119, 110610. [Google Scholar] [CrossRef]
- Zeng, L.; White, C.C.; Bennett, D.A.; Klein, H.U.; De Jager, P.L. Genetic insights into the association between inflammatory bowel disease and Alzheimer’s disease. medRxiv 2023. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lobbestael, E.; Vermeire, S.; Sabino, J.; Cleynen, I. Inflammatory bowel disease and Parkinson’s disease: Common pathophysiological links. Gut 2021, 70, 408–417. [Google Scholar] [CrossRef]
- Kang, X.; Ploner, A.; Wang, Y.; Ludvigsson, J.F.; Williams, D.M.; Pedersen, N.L.; Wirdefeldt, K. Genetic overlap between Parkinson’s disease and inflammatory bowel disease. Brain Commun. 2023, 5, fcad002. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Mues, M.; Koutrolos, M.; Al Rasbi, Z.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Ther. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Hernandez, S.; Gerlach, N.; Fletcher, J.M.; Biros, E.; Mack, M.; Körner, H.; Baxter, A.G. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Hernandez, S.; Baxter, A.G. Role of toll-like receptors in multiple sclerosis. Am. J. Clin. Exp. Immunol. 2013, 2, 75–93. [Google Scholar]
- Cossu, D.; Yokoyama, K.; Hattori, N. Bacteria–Host Interactions in Multiple Sclerosis. Front. Microbiol. 2018, 9, 2966. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, L.; Peng, L.; Qiu, W. TLR9 and its signaling pathway in multiple sclerosis. J. Neurol. Sci. 2017, 373, 95–99. [Google Scholar] [CrossRef]
- Seno, A.; Maruhashi, T.; Kaifu, T.; Yabe, R.; Fujikado, N.; Ma, G.; Ikarashi, T.; Kakuta, S.; Iwakura, Y. Exacerbation of experimental autoimmune encephalomyelitis in mice deficient for DCIR, an inhibitory C-type lectin receptor. Exp. Anim. 2015, 64, 109–119. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Gris, K.V.; Mahvelati, T.; Amrani, A.; Lukens, J.R.; Gris, D. NLR-Dependent Regulation of Inflammation in Multiple Sclerosis. Front. Immunol. 2018, 8, 2012. [Google Scholar] [CrossRef] [PubMed]
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I.; et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef] [PubMed]
- Wijeyekoon, R.S.; Kronenberg-Versteeg, D.; Scott, K.M.; Hayat, S.; Kuan, W.-L.; Evans, J.R.; Breen, D.P.; Cummins, G.; Jones, J.L.; Clatworthy, M.R.; et al. Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav. Immun. 2020, 87, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.C.; Rasley, A.; Tranguch, S.L.; Marriott, I. Cultured astrocytes express toll-like receptors for bacterial products. Glia 2003, 43, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.B.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 2020, 9, e53111. [Google Scholar] [CrossRef]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, A.M.; Anyaegbu, C.C.; Anderton, R.S. TLR2 and TLR4 in Parkinson’s disease pathogenesis: The environment takes a toll on the gut. Transl. Neurodegener. 2021, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef]
- Pike, A.F.; Szabò, I.; Veerhuis, R.; Bubacco, L. The potential convergence of NLRP3 inflammasome, potassium, and dopamine mechanisms in Parkinson’s disease. npj Park. Dis. 2022, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Kim, H.D.; Maxwell, J.A.; Li, L.; Fukuchi, K.I. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2008, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K.I. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhu, T.; Ni, W.; Zhou, C.; Zhou, H.; Lin, L.; Hu, Y.; Sun, X.; Han, J.; Zhou, Y.; et al. Early activation of Toll-like receptor-3 reduces the pathological progression of Alzheimer’s disease in APP/PS1 mouse. Alzheimer’s Res. Ther. 2023, 15, 33. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Perry, G.; Rezaei, N. Toll-like receptors in Alzheimer’s disease. J. Neuroimmunol. 2020, 348, 577362. [Google Scholar] [CrossRef]
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain 2006, 129 Pt 11, 3006–3019. [Google Scholar] [CrossRef]
- Billiau, A.; Matthys, P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar] [CrossRef]
- Brocke, S.; Gaur, A.; Piercy, C.; Gautam, A.; Gijbels, K.; Fathman, C.G.; Steinman, L. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature 1993, 365, 642–644. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; de Vos, E.A. Role of the intestinal microbiome in health and disease: From correlation to causation. Nutr. Rev. 2012, 70 (Suppl. S1), S45–S56. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xie, L.; Li, Y.; Wei, C. More than 9,000,000 unique genes in human gut bacterial community: Estimating gene numbers inside a human body. PLoS ONE 2009, 4, e6074. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef]
- Obata, Y.; Pachnis, V. The Effect of Microbiota and the Immune System on the Development and Organization of the Enteric Nervous System. Gastroenterology 2016, 151, 836–844. [Google Scholar] [CrossRef]
- Kamada, N.; Kim, Y.-G.; Sham, H.P.; Vallance, B.A.; Puente, J.L.; Martens, E.C.; Núñez, G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 2012, 336, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Boertien, J.M.; Pereira, P.A.; Aho, V.T.; Scheperjans, F. Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review. J. Park. Dis. 2019, 9 (Suppl. S2), S297–S312. [Google Scholar] [CrossRef] [PubMed]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2019, 34, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Lubomski, M.; Tan, A.H.; Lim, S.-Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2020, 267, 2507–2523. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Zhu, F.; Hart, J.; Roalstad, S.; Graves, J.; Lynch, S.; Waubant, E.; US Network of Pediatric MS Centers. Gut microbiota in early pediatric multiple sclerosis: A case-control study. Eur. J. Neurol. 2016, 23, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Hart, J.; Roalstad, S.; Graves, J.; Spencer, C.M.; Lynch, S.V.; Zamvil, S.S.; Waubant, E.; et al. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol. 2016, 16, 182. [Google Scholar] [CrossRef]
- Montgomery, T.L.; Künstner, A.; Kennedy, J.J.; Fang, Q.; Asarian, L.; Culp-Hill, R.; D’alessandro, A.; Teuscher, C.; Busch, H.; Krementsov, D.N. Interactions between host genetics and gut microbiota determine susceptibility to CNS autoimmunity. Proc. Natl. Acad. Sci. USA 2020, 117, 27516–27527. [Google Scholar] [CrossRef] [PubMed]
- Pröbstel, A.-K.; Baranzini, S.E. The Role of the Gut Microbiome in Multiple Sclerosis Risk and Progression: Towards Characterization of the “MS Microbiome”. Neurotherapeutics 2018, 15, 126–134. [Google Scholar] [CrossRef]
- Cox, L.M.; Maghzi, A.H.; Liu, S.; Tankou, S.K.; Dhang, F.H.; Willocq, V.; Song, A.; Wasén, C.; Tauhid, S.; Chu, R.; et al. Gut Microbiome in Progressive Multiple Sclerosis. Ann. Neurol. 2021, 89, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- iMSMS Consortium. Gut microbiome of multiple sclerosis patients and paired household healthy controls reveal associations with disease risk and course. Cell 2022, 185, 3467–3486.e16. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, N.; Tan, H.-Y.; Li, S.; Zhang, C.; Feng, Y. Function of Akkermansia muciniphila in Obesity: Interactions with Lipid Metabolism, Immune Response and Gut Systems. Front. Microbiol. 2020, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, S.Y.; Kostopoulos, I.; De Vos, W.M.; Belzer, C. Akkermansia muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef]
- Liu, S.; Rezende, R.M.; Moreira, T.G.; Tankou, S.K.; Cox, L.M.; Wu, M.; Song, A.; Dhang, F.H.; Wei, Z.; Costamagna, G.; et al. Oral Administration of miR-30d from Feces of MS Patients Suppresses MS-like Symptoms in Mice by Expanding Akkermansia muciniphila. Cell Host Microbe 2019, 26, 779–794.e8. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Zheng, Y.; Huang, Y.; Feng, Y.; Xu, K.; Zhang, W.; Wang, Y.; Nie, K.; Qin, M. Excessive consumption of mucin by over-colonized Akkermansia muciniphila promotes intestinal barrier damage during malignant intestinal environment. Front. Microbiol. 2023, 14, 1111911. [Google Scholar] [CrossRef]
- Bian, X.; Wu, W.; Yang, L.; Lv, L.; Wang, Q.; Li, Y.; Ye, J.; Fang, D.; Wu, J.; Jiang, X.; et al. Administration of Akkermansia muciniphila Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice. Front. Microbiol. 2019, 10, 2259. [Google Scholar] [CrossRef]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef]
- Ghezzi, L.; Cantoni, C.; Pinget, G.V.; Zhou, Y.; Piccio, L. Targeting the gut to treat multiple sclerosis. J. Clin. Investig. 2021, 131, e143774. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.N.; Shahi, S.K.; Mangalam, A.K. The “Gut Feeling”: Breaking Down the Role of Gut Microbiome in Multiple Sclerosis. Neurotherapeutics 2018, 15, 109–125. [Google Scholar] [CrossRef]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Telesford, K.M.; Yan, W.; Ochoa-Reparaz, J.; Pant, A.; Kircher, C.; Christy, M.A.; Begum-Haque, S.; Kasper, D.L.; Kasper, L.H. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39+Foxp3+ T cells and Treg function. Gut Microbes 2015, 6, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Wang, Y.; Begum-Haque, S.; Dasgupta, S.; Kasper, D.L.; Kasper, L.H. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 2010, 3, 487–495. [Google Scholar] [CrossRef]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Ditrio, L.E.; Burroughs, A.R.; Foureau, D.M.; Haque-Begum, S.; Kasper, L.H. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2009, 183, 6041–6050. [Google Scholar] [CrossRef]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.-C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Jiang, J.; Chu, C.; Wu, C.; Wang, C.; Zhang, C.; Li, T.; Zhai, Q.; Yu, L.; Tian, F.; Chen, W. Efficacy of probiotics in multiple sclerosis: A systematic review of preclinical trials and meta-analysis of randomized controlled trials. Food Funct. 2021, 12, 2354–2377. [Google Scholar] [CrossRef]
- Wing, A.C.; Kremenchutzky, M. Multiple sclerosis and faecal microbiome transplantation: Are you going to eat that? Benef. Microbes 2019, 10, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Lavasani, S.; Dzhambazov, B.; Nouri, M.; Fåk, F.; Buske, S.; Molin, G.; Thorlacius, H.; Alenfall, J.; Jeppsson, B.; Weström, B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE 2010, 5, e9009. [Google Scholar] [CrossRef] [PubMed]
- Secher, T.; Kassem, S.; Benamar, M.; Bernard, I.; Boury, M.; Barreau, F.; Oswald, E.; Saoudi, A. Oral Administration of the Probiotic Strain Escherichia coli Nissle 1917 Reduces Susceptibility to Neuroinflammation and Repairs Experimental Autoimmune Encephalomyelitis-Induced Intestinal Barrier Dysfunction. Front. Immunol. 2017, 8, 1096. [Google Scholar] [CrossRef] [PubMed]
- Mangalam, A.; Shahi, S.K.; Luckey, D.; Karau, M.; Marietta, E.; Luo, N.; Choung, R.S.; Ju, J.; Sompallae, R.; Gibson-Corley, K.; et al. Human Gut-Derived Commensal Bacteria Suppress CNS Inflammatory and Demyelinating Disease. Cell Rep. 2017, 20, 1269–1277. [Google Scholar] [CrossRef]
- Shahi, S.K.; Freedman, S.N.; Mangalam, A.K. Gut microbiome in multiple sclerosis: The players involved and the roles they play. Gut Microbes 2017, 8, 607–615. [Google Scholar] [CrossRef]
- Iljazovic, A.; Amend, L.; Galvez, E.J.; de Oliveira, R.; Strowig, T. Modulation of inflammatory responses by gastrointestinal Prevotella spp.—From associations to functional studies. Int. J. Med. Microbiol. 2021, 311, 151472. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Qiu, Y.; Wang, J.; Fang, Y.; Zhang, Y.; Chen, H.; Du, Q.; Zhao, Z.; Yan, C.; Yang, M.; et al. Dysbiosis of gut microbiota in patients with neuromyelitis optica spectrum disorders: A cross sectional study. J. Neuroimmunol. 2020, 339, 577126. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. npj Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Li, Z.; Liang, H.; Hu, Y.; Lu, L.; Zheng, C.; Fan, Y.; Wu, B.; Zou, T.; Luo, X.; Zhang, X.; et al. Gut bacterial profiles in Parkinson’s disease: A systematic review. CNS Neurosci. Ther. 2023, 29, 140–157. [Google Scholar] [CrossRef]
- Heeney, D.D.; Gareau, M.G.; Marco, M.L. Intestinal Lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotechnol. 2018, 49, 140–147. [Google Scholar] [CrossRef]
- Tan, A.H.; Hor, J.W.; Chong, C.W.; Lim, S.Y. Probiotics for Parkinson’s disease: Current evidence and future directions. JGH Open 2021, 5, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-T.; Chen, J.-H.; Huang, T.-W. Probiotics treatment for Parkinson disease: A systematic review and meta-analysis of clinical trials. Aging 2022, 14, 7014–7025. [Google Scholar] [CrossRef] [PubMed]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. 2017, 32, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Schwiertz, A.; Unger, M.M.; Becker, A.; Faßbender, K.; Ratering, S.; Kohl, M.; Schnell, S.; Schäfer, K.-H.; Egert, M. Effect of Parkinson’s disease and related medications on the composition of the fecal bacterial microbiota. NPJ Park. Dis. 2019, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, G.; Li, Z.; Wu, B.; Luo, E.; Qiu, X.; Guo, J.; Xia, Z.; Zheng, C.; Su, Q.; et al. Altered Actinobacteria and Firmicutes Phylum Associated Epitopes in Patients with Parkinson’s Disease. Front. Immunol. 2021, 12, 632482. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Garretti, F.; Monahan, C.; Sloan, N.; Bergen, J.; Shahriar, S.; Kim, S.W.; Sette, A.; Cutforth, T.; Kanter, E.; Agalliu, D.; et al. Interaction of an α-synuclein epitope with HLA-DRB1*15:01 triggers enteric features in mice reminiscent of prodromal Parkinson’s disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Derkinderen, P.; Shannon, K.M.; Brundin, P. Gut feelings about smoking and coffee in Parkinson’s disease. Mov. Disord. 2014, 29, 976–979. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.F.; Zhu, Y.L.; Zhou, Z.L.; Jia, X.B.; Xu, Y.D.; Yang, Q.; Cui, C.; Shen, Y.Q. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F.; Liu, S.; Du, J.; Hu, X.; Xiong, J.; Fang, R.; Chen, W.; Sun, J. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J. Neurol. Sci. 2017, 381, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Paiva, I.; Pinho, R.; Pavlou, M.A.; Hennion, M.; Wales, P.; Schütz, A.-L.; Rajput, A.; Szegő, É.M.; Kerimoglu, C.; Gerhardt, E.; et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 2017, 26, 2231–2246. [Google Scholar] [CrossRef] [PubMed]
- St. Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R.; Singh, S. Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: Modulation of histone deacetylase activity. Behav. Brain Res. 2015, 291, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Salvá, M.; Van Rompuy, A.S.; Heeman, B.; Van den Haute, C.; Baekelandt, V. Loss-of-function rodent models for parkin and PINK1. J. Park. Dis. 2011, 1, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Hinterleitner, R.; Meisel, M.; Zhang, C.; Leone, V.; Zhang, X.; Oyler-Castrillo, P.; Zhang, X.; Musch, M.W.; Shen, X.; et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APP(SWE)/PS1(DeltaE9) murine model of Alzheimer’s disease. Sci. Rep. 2017, 7, 10411. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.B.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Aβ amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Kuntz, T.; Shaik, S.M.; Baufeld, C.; Leibowitz, J.; Zhang, X.; Gottel, N.; Zhang, X.; Butovsky, O.; Gilbert, J.A.; et al. Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J. Exp.Med. 2019, 216, 1542–1560. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Lutz, H.L.; Weigle, I.Q.; Patel, P.; Michalkiewicz, J.; Roman-Santiago, C.J.; Zhang, C.M.; Liang, Y.; Srinath, A.; Zhang, X.; et al. Gut microbiota-driven brain Aβ amyloidosis in mice requires microglia. J. Exp. Med. 2022, 219, e20200895. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Frith, M.; Sidebottom, A.; Cao, Y.; Koval, J.; Chang, E.; Sisodia, S.S. Synergistic depletion of gut microbial consortia, but not individual antibiotics, reduces amyloidosis in APPPS1-21 Alzheimer’s transgenic mice. Sci. Rep. 2020, 10, 8183. [Google Scholar] [CrossRef]
- Mezö, C.; Dokalis, N.; Mossad, O.; Staszewski, O.; Neuber, J.; Yilmaz, B.; Schnepf, D.; de Agüero, M.G.; Ganal-Vonarburg, S.C.; Macpherson, A.J.; et al. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.-F.; Huang, L.; et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef]
- Escribano, B.M.; Medina-Fernández, F.J.; Aguilar-Luque, M.; Agüera, E.; Feijoo, M.; Garcia-Maceira, F.I.; Lillo, R.; Vieyra-Reyes, P.; Giraldo, A.I.; Luque, E.; et al. Lipopolysaccharide Binding Protein and Oxidative Stress in a Multiple Sclerosis Model. Neurotherapeutics 2017, 14, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Erickson, M.A. The blood–brain barrier and immune function and dysfunction. Neurobiol. Dis. 2010, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Wispelwey, B.; Lesse, A.J.; Hansen, E.J.; Scheld, W.M. Haemophilus influenzae lipopolysaccharide-induced blood brain barrier permeability during experimental meningitis in the rat. J. Clin. Investig. 1988, 82, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfiled, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-Derived Lipopolysaccharide Enriched in the Perinuclear Region of Alzheimer’s Disease Brain. Front. Immunol. 2017, 8, 1064. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer’s Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front. Cell. Infect. Microbiol. 2017, 7, 318. [Google Scholar] [CrossRef]
- Castaño, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef]
- Zhang, J.; Stanton, D.; Nguyen, X.; Liu, M.; Zhang, Z.; Gash, D.; Bing, G. Intrapallidal lipopolysaccharide injection increases iron and ferritin levels in glia of the rat substantia nigra and induces locomotor deficits. Neuroscience 2005, 135, 829–838. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.Y.; Liu, M.; Hunter, R.L.; Cass, W.A.; Pandya, J.D.; Sullivan, P.G.; Shin, E.J.; Kim, H.C.; Gash, D.M.; Bing, G. Striatal neuroinflammation promotes Parkinsonism in rats. PLoS ONE 2009, 4, e5482. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bing, G. Lipopolysaccharide animal models for Parkinson’s disease. Park. Dis. 2011, 2011, 327089. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, S.; Feinstein, D.; Rivest, S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol. 1998, 8, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef]
- Acioglu, C.; Heary, R.F.; Elkabes, S. Roles of neuronal toll-like receptors in neuropathic pain and central nervous system injuries and diseases. Brain Behav. Immun. 2022, 102, 163–178. [Google Scholar] [CrossRef]
- Mitchell, L.; Smith, S.H.; Braun, J.S.; Herzog, K.; Weber, J.R.; Tuomanen, E.I. Dual phases of apoptosis in pneumococcal meningitis. J. Infect. Dis. 2004, 190, 2039–2046. [Google Scholar] [CrossRef]
- Arentsen, T.; Qian, Y.; Gkotzis, S.; Femenia, T.; Wang, T.; Udekwu, K.; Forssberg, H.; Heijtz, R.D. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol. Psychiatry 2017, 22, 257–266. [Google Scholar] [CrossRef]
- Acarin, L.; González, B.; Castellano, B. Neuronal, astroglial and microglial cytokine expression after an excitotoxic lesion in the immature rat brain. Eur. J. Neurosci. 2000, 12, 3505–3520. [Google Scholar] [CrossRef]
- Bittel, M.; Reichert, P.; Sarfati, I.; Dressel, A.; Leikam, S.; Uderhardt, S.; Stolzer, I.; Phu, T.A.; Ng, M.; Vu, N.K.; et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J. Extracell. Vesicles 2021, 10, e12159. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, J.; Park, J.; Gho, Y.S. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin. Cell Dev. Biol. 2015, 40, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, M.; Schild, S.; Kaparakis-Liaskos, M.; Eberl, L. Composition and functions of bacterial membrane vesicles. Nat. Rev. Microbiol. 2023, 21, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Park, A.-M.; Tsunoda, I. Helicobacter pylori infection in the stomach induces neuroinflammation: The potential roles of bacterial outer membrane vesicles in an animal model of Alzheimer’s disease. Inflamm. Regen. 2022, 42, 39. [Google Scholar] [CrossRef] [PubMed]
- D’anca, M.; Fenoglio, C.; Buccellato, F.R.; Visconte, C.; Galimberti, D.; Scarpini, E. Extracellular Vesicles in Multiple Sclerosis: Role in the Pathogenesis and Potential Usefulness as Biomarkers and Therapeutic Tools. Cells 2021, 10, 1733. [Google Scholar] [CrossRef]
- Lee, K.-E.; Kim, J.-K.; Han, S.-K.; Lee, D.Y.; Lee, H.-J.; Yim, S.-V.; Kim, D.-H. The extracellular vesicle of gut microbial Paenalcaligenes hominis is a risk factor for vagus nerve-mediated cognitive impairment. Microbiome 2020, 8, 107. [Google Scholar] [CrossRef]
- Wei, S.; Peng, W.; Mai, Y.; Li, K.; Wei, W.; Hu, L.; Zhu, S.; Zhou, H.; Jie, W.; Wei, Z.; et al. Outer membrane vesicles enhance tau phosphorylation and contribute to cognitive impairment. J. Cell. Physiol. 2020, 235, 4843–4855. [Google Scholar] [CrossRef]
- Banks, W.A.; Robinson, S.M. Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain Behav. Immun. 2010, 24, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Pirolli, N.H.; Bentley, W.E.; Jay, S.M. Bacterial Extracellular Vesicles and the Gut-Microbiota Brain Axis: Emerging Roles in Communication and Potential as Therapeutics. Adv. Biol. 2021, 5, e2000540. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-S.; Choi, K.-H.; Kim, Y.-S.; Hong, B.S.; Kim, O.Y.; Kim, J.H.; Yoon, C.M.; Koh, G.-Y.; Kim, Y.-K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS ONE 2010, 5, e11334. [Google Scholar] [CrossRef]
- Kim, J.H.; Yoon, Y.J.; Lee, J.; Choi, E.-J.; Yi, N.; Park, K.-S.; Park, J.; Lötvall, J.; Kim, Y.-K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli up-regulate expression of endothelial cell adhesion molecules in vitro and in vivo. PLoS ONE 2013, 8, e59276. [Google Scholar] [CrossRef] [PubMed]
- Veith, P.D.; Chen, Y.-Y.; Gorasia, D.G.; Chen, D.; Glew, M.D.; O’brien-Simpson, N.M.; Cecil, J.D.; Holden, J.A.; Reynolds, E.C. Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J. Proteome Res. 2014, 13, 2420–2432. [Google Scholar] [CrossRef]
- Nakao, R.; Takashiba, S.; Kosono, S.; Yoshida, M.; Watanabe, H.; Ohnishi, M.; Senpuku, H. Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain-mediated detachment of cultured oral epithelial cells and immune responses. Microbes Infect. 2014, 16, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Kadowaki, T.; Nakanishi, H. Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins. Neurochem. Int. 2022, 154, 105282. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Nara, P.L.; Sindelar, D.; Penn, M.S.; Potempa, J.; Griffin, W.S.T. Porphyromonas gingivalis Outer Membrane Vesicles as the Major Driver of and Explanation for Neuropathogenesis, the Cholinergic Hypothesis, Iron Dyshomeostasis, and Salivary Lactoferrin in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, 1417–1450. [Google Scholar] [CrossRef]
- Muraca, M.; Putignani, L.; Fierabracci, A.; Teti, A.; Perilongo, G. Gut microbiota-derived outer membrane vesicles: Under-recognized major players in health and disease? Discov. Med. 2015, 19, 343–348. [Google Scholar]
- Aldick, T.; Bielaszewska, M.; Uhlin, B.E.; Humpf, H.U.; Wai, S.N.; Karch, H. Vesicular stabilization and activity augmentation of enterohaemorrhagic Escherichia coli haemolysin. Mol. Microbiol. 2009, 71, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Rüter, C.; Kunsmann, L.; Greune, L.; Bauwens, A.; Zhang, W.; Kuczius, T.; Kim, K.S.; Mellmann, A.; Schmidt, M.A.; et al. Enterohemorrhagic Escherichia coli hemolysin employs outer membrane vesicles to target mitochondria and cause endothelial and epithelial apoptosis. PLOS Pathog. 2013, 9, e1003797. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Austin, C.; Lewinski, M.; Stahl, R.A.K. Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat. Rev. Nephrol. 2012, 8, 658–669. [Google Scholar] [CrossRef]
- Xie, J.; Cools, L.; Van Imschoot, G.; Van Wonterghem, E.; Pauwels, M.J.; Vlaeminck, I.; De Witte, C.; El Andaloussi, S.; Wierda, K.; De Groef, L.; et al. Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. J. Extracell. Vesicles 2023, 12, e12306. [Google Scholar] [CrossRef]
- Liu, P.; Wang, X.; Yang, Q.; Yan, X.; Fan, Y.; Zhang, S.; Wei, Y.; Huang, M.; Jiang, L.; Feng, L. Collaborative Action of Microglia and Astrocytes Mediates Neutrophil Recruitment to the CNS to Defend against Escherichia coli K1 Infection. Int. J. Mol. Sci. 2022, 23, 6540. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.Y.; Choi, S.-Y.; Lee, J.H.; Hong, S.-H.; Lee, H.-J. Delivery of Periodontopathogenic Extracellular Vesicles to Brain Monocytes and Microglial IL-6 Promotion by RNA Cargo. Front. Mol. Biosci. 2020, 7, 596366. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Han, E.-C.; Choi, S.-Y.; Park, J.-W.; Hong, S.-H.; Lee, H.-J. Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J. 2019, 33, 13412–13422. [Google Scholar]
- Wei, S.-C.; Wei, W.; Peng, W.-J.; Liu, Z.; Cai, Z.-Y.; Zhao, B. Metabolic Alterations in the Outer Membrane Vesicles of Patients with Alzheimer’s Disease: An LC-MS/MS-based Metabolomics Analysis. Curr. Alzheimer Res. 2019, 16, 1183–1195. [Google Scholar] [CrossRef]
- Park, J.-Y.; Choi, J.; Lee, Y.; Lee, J.-E.; Lee, E.-H.; Kwon, H.-J.; Yang, J.; Jeong, B.-R.; Kim, Y.-K.; Han, P.-L. Metagenome Analysis of Bodily Microbiota in a Mouse Model of Alzheimer Disease Using Bacteria-derived Membrane Vesicles in Blood. Exp. Neurobiol. 2017, 26, 369–379. [Google Scholar] [CrossRef]
- Man, A.L.; Bertelli, E.; Rentini, S.; Regoli, M.; Briars, G.; Marini, M.; Watson, A.J.M.; Nicoletti, C. Age-associated modifications of intestinal permeability and innate immunity in human small intestine. Clin. Sci. 2015, 129, 515–527. [Google Scholar] [CrossRef]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.-A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’driscoll, C.M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Manni, G.; Buratta, S.; Pallotta, M.T.; Chiasserini, D.; Di Michele, A.; Emiliani, C.; Giovagnoli, S.; Pascucci, L.; Romani, R.; Bellezza, I.; et al. Extracellular Vesicles in Aging: An Emerging Hallmark? Cells 2023, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, B.; Gao, H.; He, C.; Hua, R.; Liang, C.; Zhang, S.; Wang, Y.; Xin, S.; Xu, J. Vagus Nerve and Underlying Impact on the Gut Microbiota-Brain Axis in Behavior and Neurodegenerative Diseases. J. Inflamm. Res. 2022, 15, 6213–6230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Dai, N.; Sheng, K.; Lu, H.; Wang, J.; Chen, L.; Wang, Y. Gut bacterial extracellular vesicles: Important players in regulating intestinal microenvironment. Gut Microbes 2022, 14, 2134689. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zeng, J.; Yang, Y.; Xiong, Y.; Zhang, Z.H.; Qiu, M.; Yan, X.; Sun, X.Y.; Tuo, Q.Z.; Liu, R.; et al. Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3β. J. Alzheimer’s Dis. 2015, 43, 153–165. [Google Scholar] [CrossRef]
- Iyaswamy, A.; Lu, K.; Guan, X.-J.; Kan, Y.; Su, C.; Liu, J.; Jaganathan, R.; Vasudevan, K.; Paul, J.; Thakur, A.; et al. Impact and Advances in the Role of Bacterial Extracellular Vesicles in Neurodegenerative Disease and Its Therapeutics. Biomedicines 2023, 11, 2056. [Google Scholar] [CrossRef]
- Bitto, N.J.; Kaparakis-Liaskos, M. The Therapeutic Benefit of Bacterial Membrane Vesicles. Int. J. Mol. Sci. 2017, 18, 1287. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, Z.; Yang, Z.; Zhang, Y.; Chen, H.; Yang, X.; Fang, X.; Zhu, Y.; Zhang, J.; Ouyang, F.; et al. Lactobacillus plantarum-derived extracellular vesicles protect against ischemic brain injury via the microRNA-101a-3p/c-Fos/TGF-β axis. Pharmacol. Res. 2022, 182, 106332. [Google Scholar] [CrossRef] [PubMed]
- Alpdundar Bulut, E.; Bayyurt Kocabas, B.; Aykut, G.; Guler, U.; Salih, B.; Surucu Yilmaz, N.; Ayanoglu, I.C.; Polat, M.M.; Akcali, K.C.; Gursel, I.; et al. Human Gut Commensal Membrane Vesicles Modulate Inflammation by Generating M2-like Macrophages and Myeloid-Derived Suppressor Cells. J. Immunol. 2020, 205, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Moon, C.M.; Shin, T.-S.; Kim, E.K.; McDowell, A.; Jo, M.-K.; Joo, Y.H.; Kim, S.-E.; Jung, H.-K.; Shim, K.-N.; et al. Lactobacillus paracasei-derived extracellular vesicles attenuate the intestinal inflammatory response by augmenting the endoplasmic reticulum stress pathway. Exp. Mol. Med. 2020, 52, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Park, E.; Ko, S.; Choi, E.; Kim, S. Therapeutic effects of kefir grain Lactobacillus-derived extracellular vesicles in mice with 2,4,6-trinitrobenzene sulfonic acid-induced inflammatory bowel disease. J. Dairy Sci. 2018, 101, 8662–8671. [Google Scholar] [CrossRef] [PubMed]
- West, C.L.; Stanisz, A.M.; Mao, Y.-K.; Champagne-Jorgensen, K.; Bienenstock, J.; Kunze, W.A. Microvesicles from Lactobacillus reuteri (DSM-17938) completely reproduce modulation of gut motility by bacteria in mice. PLoS ONE 2020, 15, e0225481. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, Y.-K.; Han, P.-L. Extracellular Vesicles Derived from Lactobacillus plantarum Increase BDNF Expression in Cultured Hippocampal Neurons and Produce Antidepressant-like Effects in Mice. Exp. Neurobiol. 2019, 28, 158–171. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolzer, I.; Scherer, E.; Süß, P.; Rothhammer, V.; Winner, B.; Neurath, M.F.; Günther, C. Impact of Microbiome–Brain Communication on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 14925. https://doi.org/10.3390/ijms241914925
Stolzer I, Scherer E, Süß P, Rothhammer V, Winner B, Neurath MF, Günther C. Impact of Microbiome–Brain Communication on Neuroinflammation and Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(19):14925. https://doi.org/10.3390/ijms241914925
Chicago/Turabian StyleStolzer, Iris, Eveline Scherer, Patrick Süß, Veit Rothhammer, Beate Winner, Markus F. Neurath, and Claudia Günther. 2023. "Impact of Microbiome–Brain Communication on Neuroinflammation and Neurodegeneration" International Journal of Molecular Sciences 24, no. 19: 14925. https://doi.org/10.3390/ijms241914925
APA StyleStolzer, I., Scherer, E., Süß, P., Rothhammer, V., Winner, B., Neurath, M. F., & Günther, C. (2023). Impact of Microbiome–Brain Communication on Neuroinflammation and Neurodegeneration. International Journal of Molecular Sciences, 24(19), 14925. https://doi.org/10.3390/ijms241914925