
A Novel Time-Resolved Fluorescence Resonance Energy Transfer Assay for the Discovery of Small-Molecule Inhibitors of HIV-1 Tat-Regulated Transcription

 ,
,
Abstract
1. Introduction
2. Results
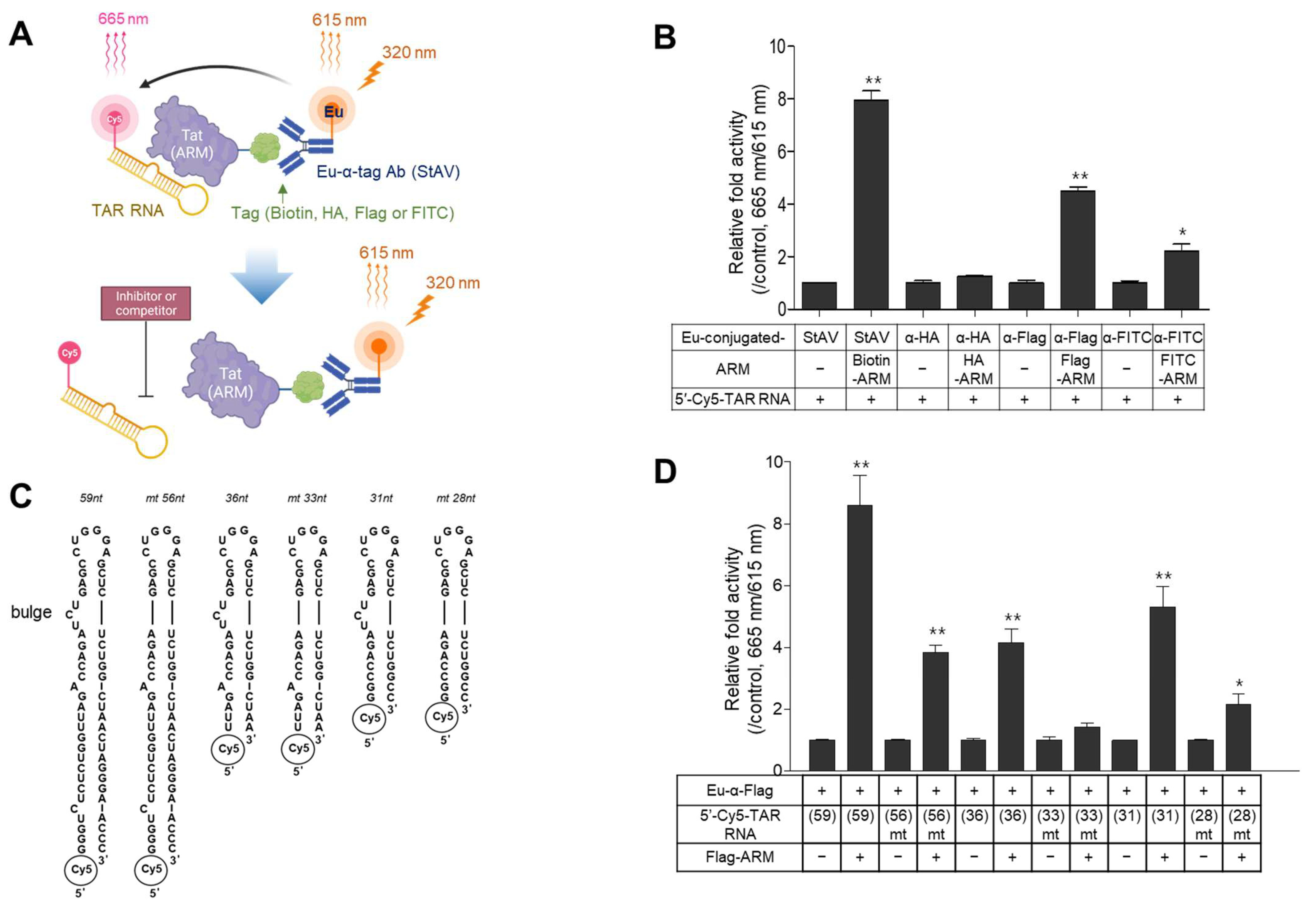
2.1. Design of the TR-FRET Assays and Selection of a Suitable Pair of Components
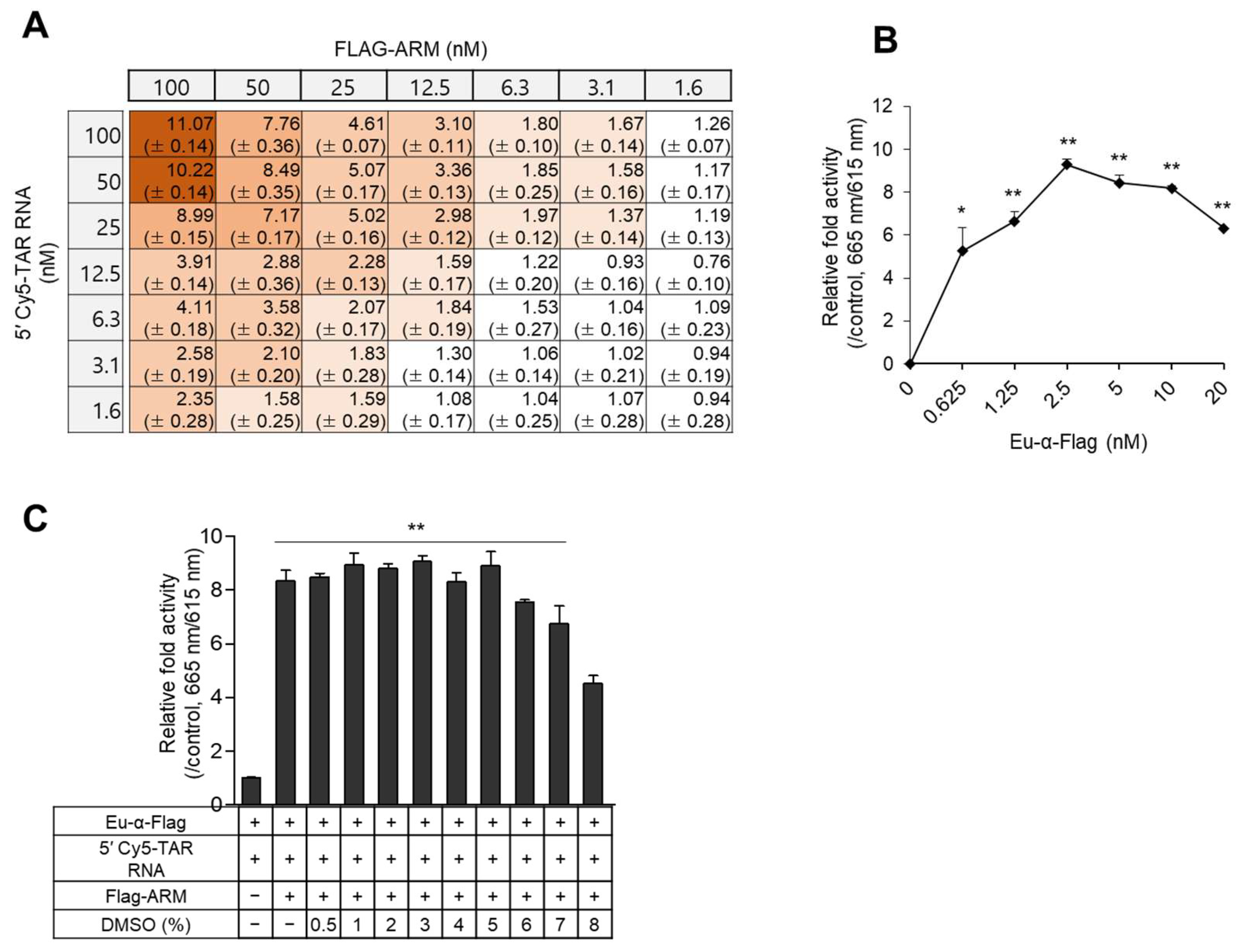
2.2. Optimization of the TR-FRET Assay
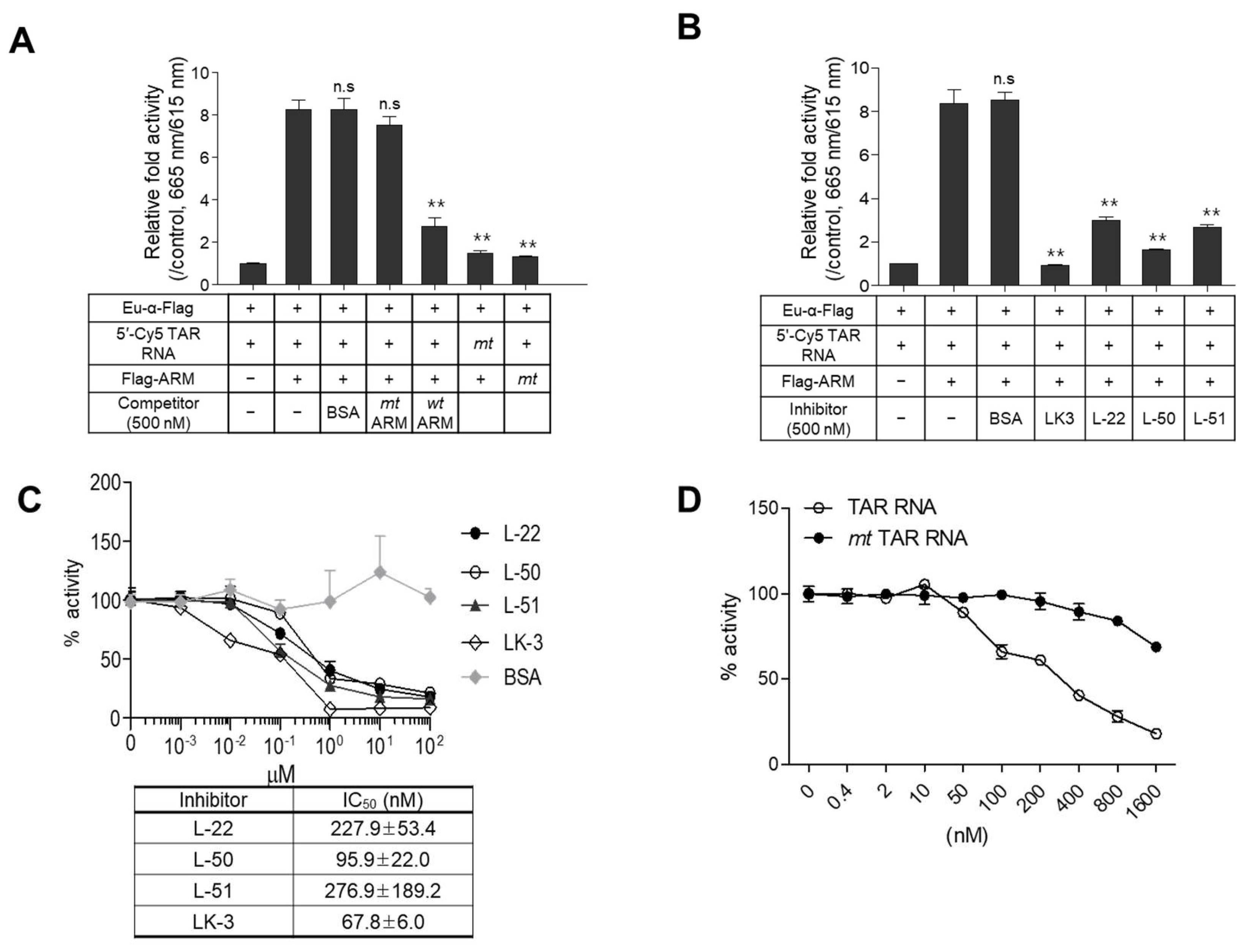
2.3. Evaluation of the Assay
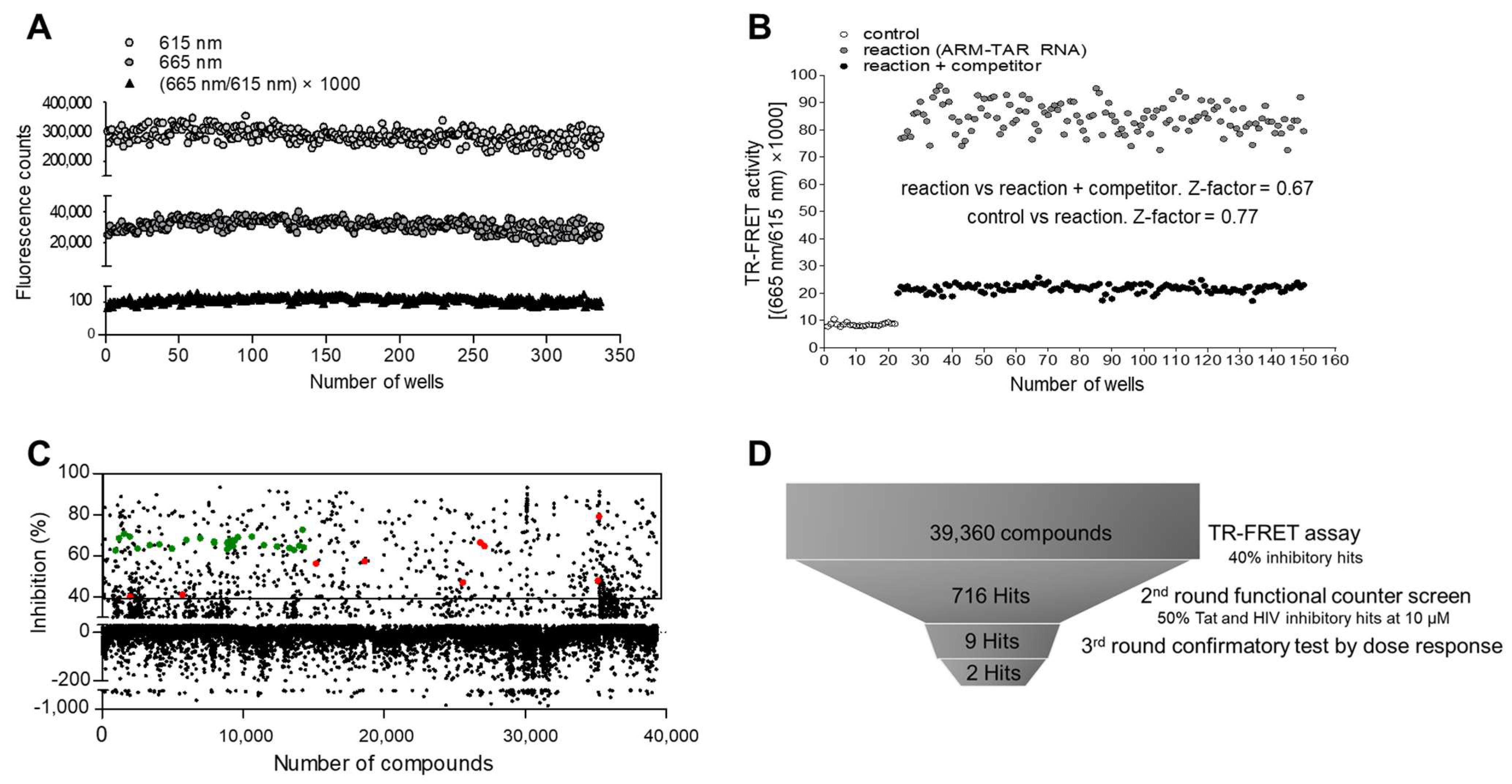
2.4. Suitability of the Assay in HTS
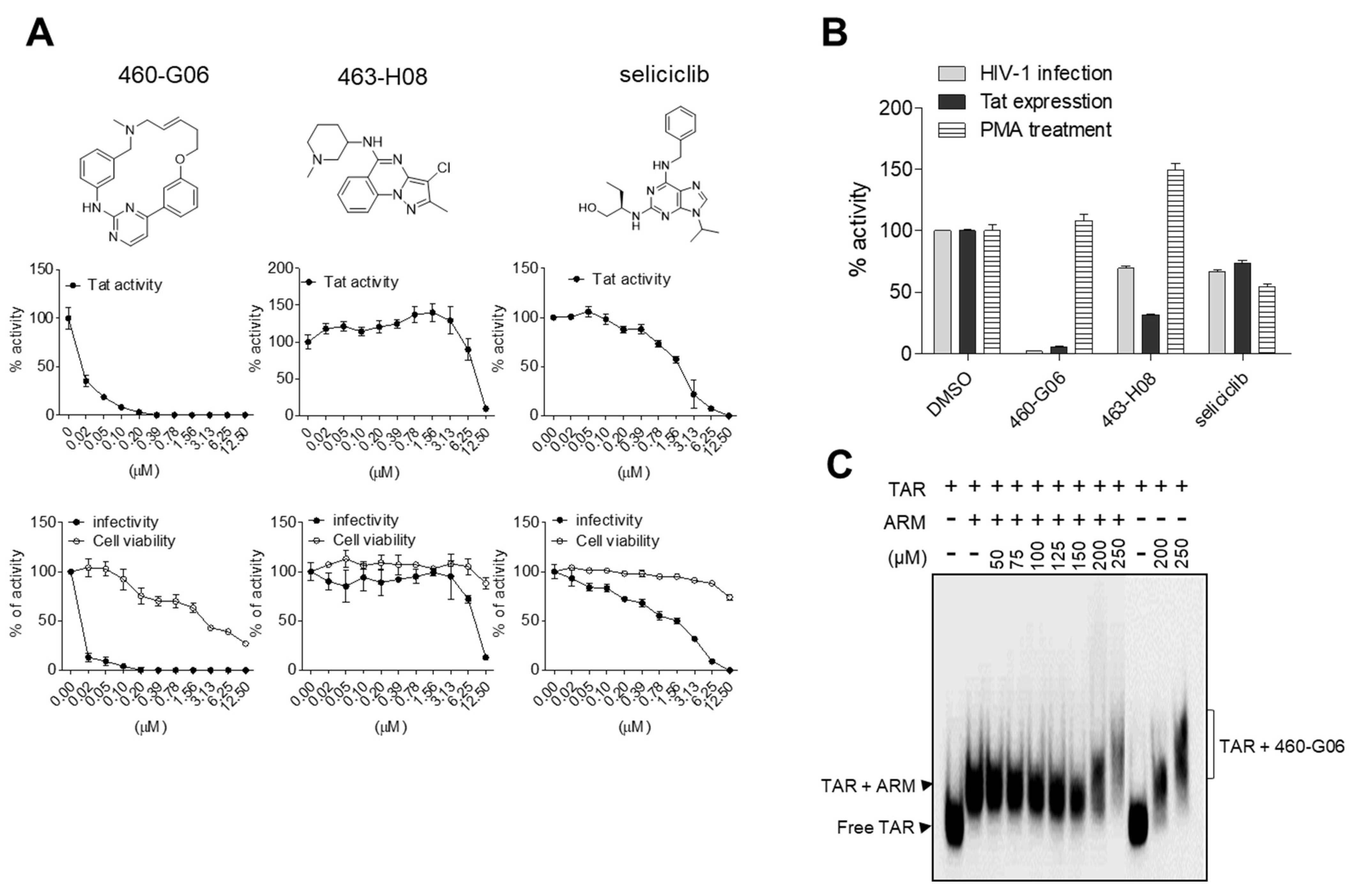
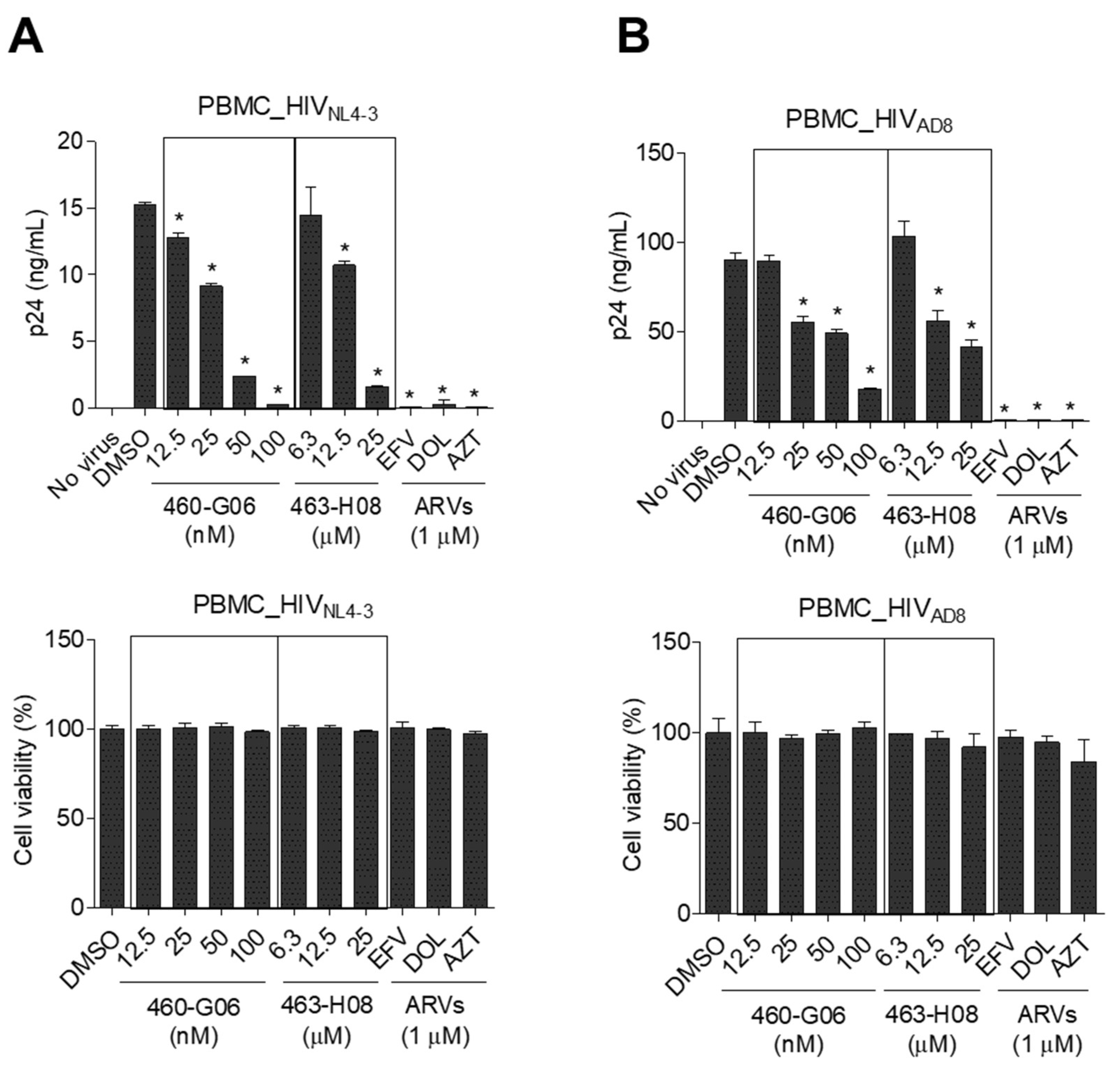
2.5. Pilot Screen
3. Discussion
4. Materials and Methods
4.1. Tat-Driven Peptides and TAR RNA Fragments
4.2. Agents
4.3. TR-FRET Assay
4.3.1. Cy5-TAR RNA:Tat (ARM)-Eu
4.3.2. ULight-TAR RNA:Tat (ARM)-Eu
4.3.3. Eu-TAR RNA:Tat (ARM)-ULight
4.4. Assay for Tat-Mediated HIV-1 Transcription
4.5. Antiviral Effects of the Compounds
4.6. Gel Mobility-Shift Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS: Joint United Nations Programme on HIV/AIDS; UNAIDS: Geneva, Switzerland, 2021; Available online: https://www.unaids.org/en/resources/documents/2021/2021_unaids_data (accessed on 17 May 2023).
- Beck, H.; Harter, M.; Hass, B.; Schmeck, C.; Baerfacker, L. Small molecules and their impact in drug discovery: A perspective on the occasion of the 125th anniversary of the Bayer Chemical Research Laboratory. Drug Discov. Today 2022, 27, 1560–1574. [Google Scholar] [CrossRef]
- Navarro, J.; Punzon, M.C.; Pizarro, A.; Fernandez-Cruz, E.; Fresno, M.; Munoz-Fernandez, M.A. Pentoxifylline inhibits acute HIV-1 replication in human T cells by a mechanism not involving inhibition of tumour necrosis factor synthesis or nuclear factor-kappa B activation. AIDS 1996, 10, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef]
- Shaw, J.P.; Utz, P.J.; Durand, D.B.; Toole, J.J.; Emmel, E.A.; Crabtree, G.R. Identification of a putative regulator of early T cell activation genes. Science 1988, 241, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Dutilleul, A.; Rodari, A.; Van Lint, C. Depicting HIV-1 Transcriptional Mechanisms: A Summary of What We Know. Viruses 2020, 12, 1385. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- Fujinaga, K. P-TEFb as A Promising Therapeutic Target. Molecules 2020, 25, 838. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M.; Jeang, K.T. Biochemical and functional interactions between HIV-1 Tat protein and TAR RNA. Arch. Biochem. Biophys. 1999, 365, 175–185. [Google Scholar] [CrossRef]
- Kim, J.M.; Chun, H. TAR RNA Mediated Folding of a Single-Arginine-Mutant HIV-1 Tat Protein within HeLa Cells Experiencing Intracellular Crowding. Int. J. Mol. Sci. 2021, 22, 9998. [Google Scholar] [CrossRef]
- Wang, Z.; Rana, T.M. RNA conformation in the Tat-TAR complex determined by site-specific photo-cross-linking. Biochemistry 1996, 35, 6491–6499. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.S.; Bonn-Breach, R.; Wedekind, J.E. Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J. Biol. Chem. 2019, 294, 9326–9341. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.W.; Blakeley, B.D.; Chen, P.H.; Sherpa, C.; Le Grice, S.F.; Laird-Offringa, I.A.; McNaughton, B.R. An evolved RNA recognition motif that suppresses HIV-1 Tat/TAR-dependent transcription. ACS Chem. Biol. 2016, 11, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Gahmen, U.; Hurley, J.H. Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex. Proc. Natl. Acad. Sci. USA 2018, 115, 12973–12978. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Echeverria, I.; Stjepanovic, G.; Bai, Y.; Lu, H.; Schneidman-Duhovny, D.; Doudna, J.A.; Zhou, Q.; Sali, A.; Hurley, J.H. Insights into HIV-1 proviral transcription from integrative structure and dynamics of the Tat:AFF4:P-TEFb:TAR complex. eLife 2016, 5, e15910. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.E.; Du, Z.; Fujinaga, K.; Peterlin, B.M.; James, T.L. Structure-based computational database screening, in vitro assay, and NMR assessment of compounds that target TAR RNA. Chem. Biol. 2002, 9, 185–193. [Google Scholar] [CrossRef]
- Renner, S.; Ludwig, V.; Boden, O.; Scheffer, U.; Gobel, M.; Schneider, G. New inhibitors of the Tat-TAR RNA interaction found with a “fuzzy” pharmacophore model. Chembiochem 2005, 6, 1119–1125. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef]
- Alanazi, A.; Ivanov, A.; Kumari, N.; Lin, X.; Wang, S.; Kovalskyy, D.; Nekhai, S. Targeting Tat-TAR RNA Interaction for HIV-1 Inhibition. Viruses 2021, 13, 2004. [Google Scholar] [CrossRef]
- Davidson, A.; Begley, D.W.; Lau, C.; Varani, G. A small-molecule probe induces a conformation in HIV TAR RNA capable of binding drug-like fragments. J. Mol. Biol. 2011, 410, 984–996. [Google Scholar] [CrossRef]
- Mei, H.Y.; Mack, D.P.; Galan, A.A.; Halim, N.S.; Heldsinger, A.; Loo, J.A.; Moreland, D.W.; Sannes-Lowery, K.A.; Sharmeen, L.; Truong, H.N.; et al. Discovery of selective, small-molecule inhibitors of RNA complexes—I. The Tat protein/TAR RNA complexes required for HIV-1 transcription. Bioorg. Med. Chem. 1997, 5, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Hamy, F.; Brondani, V.; Florsheimer, A.; Stark, W.; Blommers, M.J.; Klimkait, T. A new class of HIV-1 Tat antagonist acting through Tat-TAR inhibition. Biochemistry 1998, 37, 5086–5095. [Google Scholar] [CrossRef] [PubMed]
- Sosic, A.; Olivato, G.; Carraro, C.; Gottlich, R.; Fabris, D.; Gatto, B. Bis-3-Chloropiperidines Targeting TAR RNA as A Novel Strategy to Impair the HIV-1 Nucleocapsid Protein. Molecules 2021, 26, 1874. [Google Scholar] [CrossRef]
- Rocchi, C.; Louvat, C.; Miele, A.E.; Batisse, J.; Guillon, C.; Ballut, L.; Lener, D.; Negroni, M.; Ruff, M.; Gouet, P.; et al. The HIV-1 Integrase C-Terminal Domain Induces TAR RNA Structural Changes Promoting Tat Binding. Int. J. Mol. Sci. 2022, 23, 13742. [Google Scholar] [CrossRef]
- Murchie, A.I.; Davis, B.; Isel, C.; Afshar, M.; Drysdale, M.J.; Bower, J.; Potter, A.J.; Starkey, I.D.; Swarbrick, T.M.; Mirza, S.; et al. Structure-based drug design targeting an inactive RNA conformation: Exploiting the flexibility of HIV-1 TAR RNA. J. Mol. Biol. 2004, 336, 625–638. [Google Scholar] [CrossRef]
- Hwang, S.; Tamilarasu, N.; Kibler, K.; Cao, H.; Ali, A.; Ping, Y.H.; Jeang, K.T.; Rana, T.M. Discovery of a small molecule Tat-trans-activation-responsive RNA antagonist that potently inhibits human immunodeficiency virus-1 replication. J. Biol. Chem. 2003, 278, 39092–39103. [Google Scholar] [CrossRef]
- Ma, C.-T.; Sergienko, E.A. Time-Resolved Fluorescence Assays. In High Throughput Screening: Methods and Protocols, 3rd ed.; Janzen, W.P., Ed.; Humana: New York, NY, USA, 2016; pp. 131–142. [Google Scholar]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Interpretation of europium(III) spectra. Coord. Chem. Rev. 2015, 295, 1–45. [Google Scholar] [CrossRef]
- Bazin, H.; Trinquet, E.; Mathis, G. Time resolved amplification of cryptate emission: A versatile technology to trace biomolecular interactions. J. Biotechnol. 2002, 82, 233–250. [Google Scholar] [CrossRef]
- Harada, M.; Nagai, J.; Kurata, R.; Cui, X.; Isagawa, T.; Semba, H.; Yoshida, Y.; Takeda, N.; Maemura, K.; Yonezawa, T. Establishment of Novel Protein Interaction Assays between Sin3 and REST Using Surface Plasmon Resonance and Time-Resolved Fluorescence Energy Transfer. Int. J. Mol. Sci. 2021, 22, 2323. [Google Scholar] [CrossRef]
- Rectenwald, J.M.; Hardy, P.B.; Norris-Drouin, J.L.; Cholensky, S.H.; James, L.I.; Frye, S.V.; Pearce, K.H. A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins. SLAS Discov. 2019, 24, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Appelblom, H.; Nurmi, J.; Soukka, T.; Pasternack, M.; Penttila, K.E.; Lovgren, T.; Niemela, P. Homogeneous TR-FRET high-throughput screening assay for calcium-dependent multimerization of sorcin. J. Biomol. Screen 2007, 12, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Hyun, S.; Kim, S.; Lee, S.; Lee, I.S.; Baba, M.; Lee, Y.; Yu, J. Cell-penetrating, dimeric alpha-helical peptides: Nanomolar inhibitors of HIV-1 transcription. Angew. Chem. Int. Engl. 2014, 53, 10086–10089. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, M.S.; Lobritz, M.A.; Ratcliff, A.; Chamanian, M.; Athanassiou, Z.; Tyagi, M.; Wong, J.; Robinson, J.A.; Karn, J.; Varani, G.; et al. Inhibition of both HIV-1 reverse transcription and gene expression by a cyclic peptide that binds the Tat-transactivating response element (TAR) RNA. PLoS Pathog. 2011, 7, e1002038. [Google Scholar] [CrossRef]
- Lea, W.A.; Simeonov, A. Fluorescence polarization assays in small molecule screening. Expert Opin. Drug Discov. 2011, 6, 17–32. [Google Scholar] [CrossRef]
- Arkin, M.R.; Glicksman, M.A.; Fu, H.; Havel, J.J.; Du, Y. Inhibition of Protein-Protein Interactions: Non-Cellular Assay Formats. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; AGM: Bethesda, MD, USA, 2004. [Google Scholar]
- Weeks, K.M.; Crothers, D.M. RNA recognition by Tat-derived peptides: Interaction in the major groove? Cell 1991, 66, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Churcher, M.J.; Lamont, C.; Hamy, F.; Dingwall, C.; Green, S.M.; Lowe, A.D.; Butler, J.G.; Gait, M.J.; Karn, J. High affinity binding of TAR RNA by the human immunodeficiency virus type-1 tat protein requires base-pairs in the RNA stem and amino acid residues flanking the basic region. J. Mol. Biol. 1993, 230, 90–110. [Google Scholar] [CrossRef]
- Bayer, P.; Kraft, M.; Ejchart, A.; Westendorp, M.; Frank, R.; Rosch, P. Structural studies of HIV-1 Tat protein. J. Mol. Biol. 1995, 247, 529–535. [Google Scholar] [CrossRef]
- Hyun, S.; Na, J.; Lee, S.J.; Park, S.; Yu, J. RNA grooves can accommodate disulfide-bridged bundles of alpha-helical peptides. Chembiochem 2010, 11, 767–770. [Google Scholar] [CrossRef]
- Davidson, A.; Leeper, T.C.; Athanassiou, Z.; Patora-Komisarska, K.; Karn, J.; Robinson, J.A.; Varani, G. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. USA 2009, 106, 11931–11936. [Google Scholar] [CrossRef]
- Poulsen, A.; William, A.; Blanchard, S.; Nagaraj, H.; Williams, M.; Wang, H.; Lee, A.; Sun, E.; Teo, E.L.; Tan, E.; et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J. Mol. Model 2013, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hardeman, E.; Gunning, P.; Eiffe, E. Quinazoline Compounds and the Use Thereof in the Treatment of Cancer. WO/2021/155426, 12 August 2021. [Google Scholar]
- Shin, Y.; Choi, B.S.; Kim, K.C.; Kang, C.; Kim, K.; Yoon, C.H. Development of a dual reporter screening assay for distinguishing the inhibition of HIV Tat-mediated transcription from off-target effects. J. Virol. Methods 2017, 249, 1–9. [Google Scholar] [CrossRef]
- Brodsky, A.S.; Williamson, J.R. Solution structure of the HIV-2 TAR-argininamide complex. J. Mol. Biol. 1997, 267, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Jarman, F.C.; Rickards, W.S.; Hudson, I.L. Late adolescent outcome of early onset anorexia nervosa. J. Paediatr. Child Health 1991, 27, 221–227. [Google Scholar] [CrossRef]
- Shin, Y.; Kim, H.G.; Park, C.M.; Choi, M.S.; Kim, D.E.; Choi, B.S.; Kim, K.; Yoon, C.H. Identification of novel compounds against Tat-mediated human immunodeficiency virus-1 transcription by high-throughput functional screening assay. Biochem. Biophys. Res. Commun. 2020, 523, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Park, C.M.; Kim, H.G.; Kim, D.E.; Choi, M.S.; Kim, J.A.; Choi, B.S.; Yoon, C.H. Identification of Aristolactam Derivatives That Act as Inhibitors of Human Immunodeficiency Virus Type 1 Infection and Replication by Targeting Tat-Mediated Viral Transcription. Virol. Sin. 2021, 36, 254–263. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hit | IC50 a of Tat Activity a (μM) | IC50 of Infectivity (μM) | CC50 b (μM) | SI c |
|---|---|---|---|---|
| 460-G06 | 0.011 ± 0.001 | 0.013 ± 0.003 | 4.940 ± 0.440 | 380.000 |
| 463-H08 | 2.372 ± 0.160 | 8.055 ± 0.488 | N.D. | >1.552 |
| 458-D06 | N.D. | N.D. | N.D. | - |
| 458-F08 | N.D. | 1.286 ± 0.384 | N.D. | >9.720 |
| 459-G07 | 3.929 ± 0.338 | N.D. | N.D. | - |
| 460-F06 | 3.204 ± 0.438 | N.D. | N.D. | - |
| 461-H04 | 10.063 ± 2.730 | N.D. | N.D. | - |
| 463-B11 | 6.816 ± 1.880 | 5.032 ± 0.310 | N.D. | >2.484 |
| 464-A09 | 2.265 ± 0.518 | 2.029 ± 0.132 | 3.364 ± 0.160 | 1.645 |
| Seliciclib | 2.062 ± 0.409 | 2.396 ± 0.327 | N.D. | >5.217 |
| Name | Sequence |
|---|---|
| Biotin-Tat (ARM) (15 aa) | Biotin-GISYGRKKRRQRRRA |
| HA-Tat (ARM) (26 aa) | GAYPYDVPDYA-GISYGRKKRRQRRRA |
| Flag-Tat (ARM) (25 aa) | GADYKDDDDK-GISYGRKKRRQRRRA |
| FITC-Tat (ARM) (15 aa) | FITC-GISYGRKKRRQRRRA |
| Tagged-mutant Tat (ARM) | Tags-GISYGDKKDDQEERA |
| Tat (ARM) (15 aa) | GISYGRKKRRQRRRA |
| Name | Sequence | nt |
|---|---|---|
| TAR (59 nt) | 5′-GGGUCUCUCUGGUUAGACCAGAUCUGAGCCUGGGAGCUCUCUGGCUAACUAGGGAACCC-3′ | 1–59 |
| mt TAR (56 nt) | 5′-GGGUCUCUCUGGUUAGACCAGA***GAGCCUGGGAGCUCUCUGGCUAACUAGGGAACCC-3′ | 1–59 |
| TAR (36 nt) | 5′-UUAGACCAGAUCUGAGCCUGGGAGCUCUCUGGCUAA-3′ | 13–48 |
| mt TAR (33 nt) | 5′-UUAGACCAGA***GAGCCUGGGAGCUCUCUGGCUAA-3′ | 13–48 |
| TAR (31 nt) | 5′-GGCCAGAUCUGAGCCUGGGAGCUCUCUGGCC-3′ | 16–46 |
| mt TAR (28 nt) | 5′-GGCCAGA***GAGCCUGGGAGCUCUCUGGCC-3′ | 16–46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, Y.H.; Kim, D.-E.; Yu, K.L.; Park, C.M.; Kim, H.G.; Kim, K.-C.; Bae, S.; Yoon, C.-H. A Novel Time-Resolved Fluorescence Resonance Energy Transfer Assay for the Discovery of Small-Molecule Inhibitors of HIV-1 Tat-Regulated Transcription. Int. J. Mol. Sci. 2023, 24, 9139. https://doi.org/10.3390/ijms24119139
Shin YH, Kim D-E, Yu KL, Park CM, Kim HG, Kim K-C, Bae S, Yoon C-H. A Novel Time-Resolved Fluorescence Resonance Energy Transfer Assay for the Discovery of Small-Molecule Inhibitors of HIV-1 Tat-Regulated Transcription. International Journal of Molecular Sciences. 2023; 24(11):9139. https://doi.org/10.3390/ijms24119139
Chicago/Turabian StyleShin, Young Hyun, Dong-Eun Kim, Kyung Lee Yu, Chul Min Park, Hong Gi Kim, Kyung-Chang Kim, Songmee Bae, and Cheol-Hee Yoon. 2023. "A Novel Time-Resolved Fluorescence Resonance Energy Transfer Assay for the Discovery of Small-Molecule Inhibitors of HIV-1 Tat-Regulated Transcription" International Journal of Molecular Sciences 24, no. 11: 9139. https://doi.org/10.3390/ijms24119139
APA StyleShin, Y. H., Kim, D.-E., Yu, K. L., Park, C. M., Kim, H. G., Kim, K.-C., Bae, S., & Yoon, C.-H. (2023). A Novel Time-Resolved Fluorescence Resonance Energy Transfer Assay for the Discovery of Small-Molecule Inhibitors of HIV-1 Tat-Regulated Transcription. International Journal of Molecular Sciences, 24(11), 9139. https://doi.org/10.3390/ijms24119139