
iPSC-Derived MSCs Are a Distinct Entity of MSCs with Higher Therapeutic Potential than Their Donor-Matched Parental MSCs
 , , ,
, , ,  ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
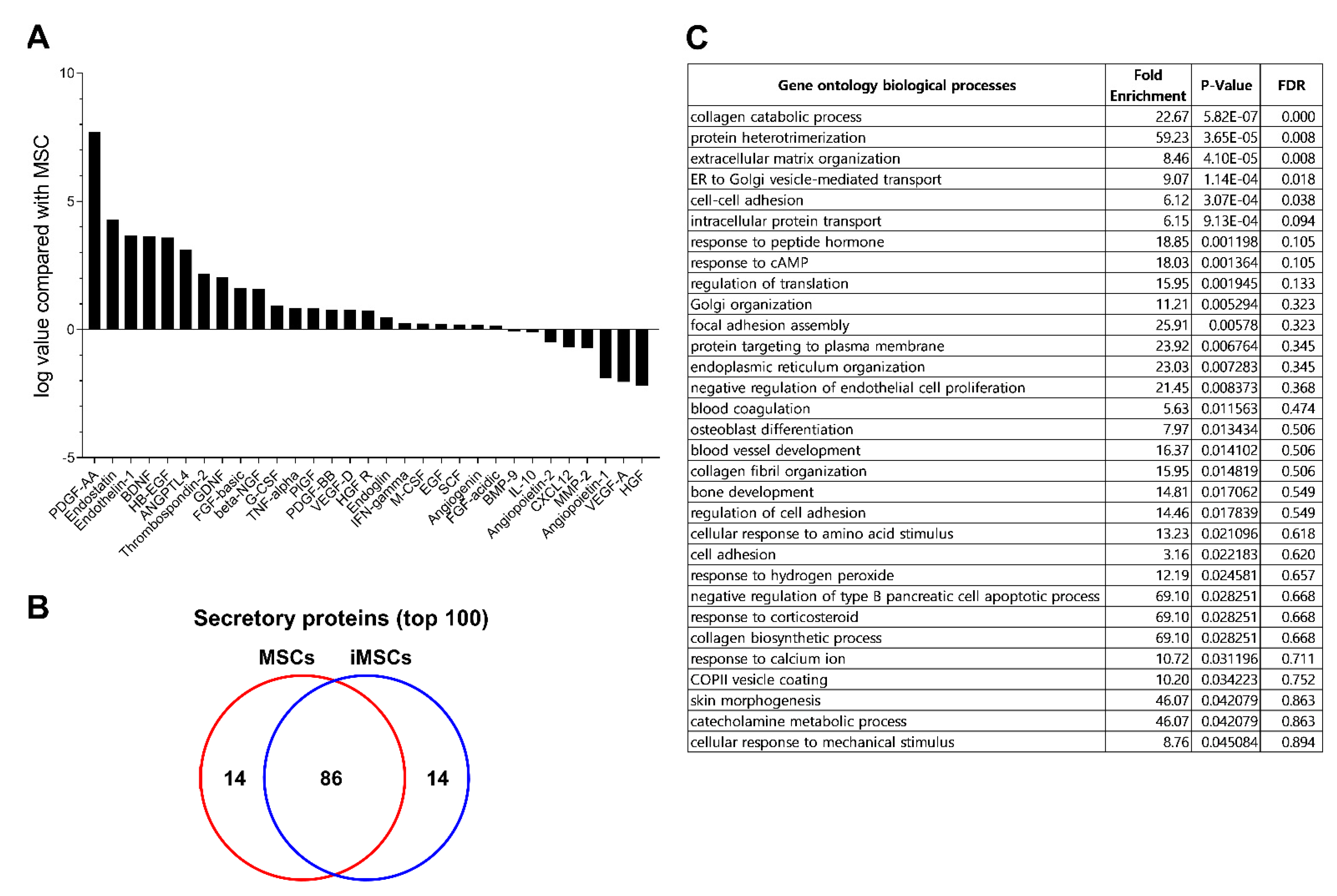
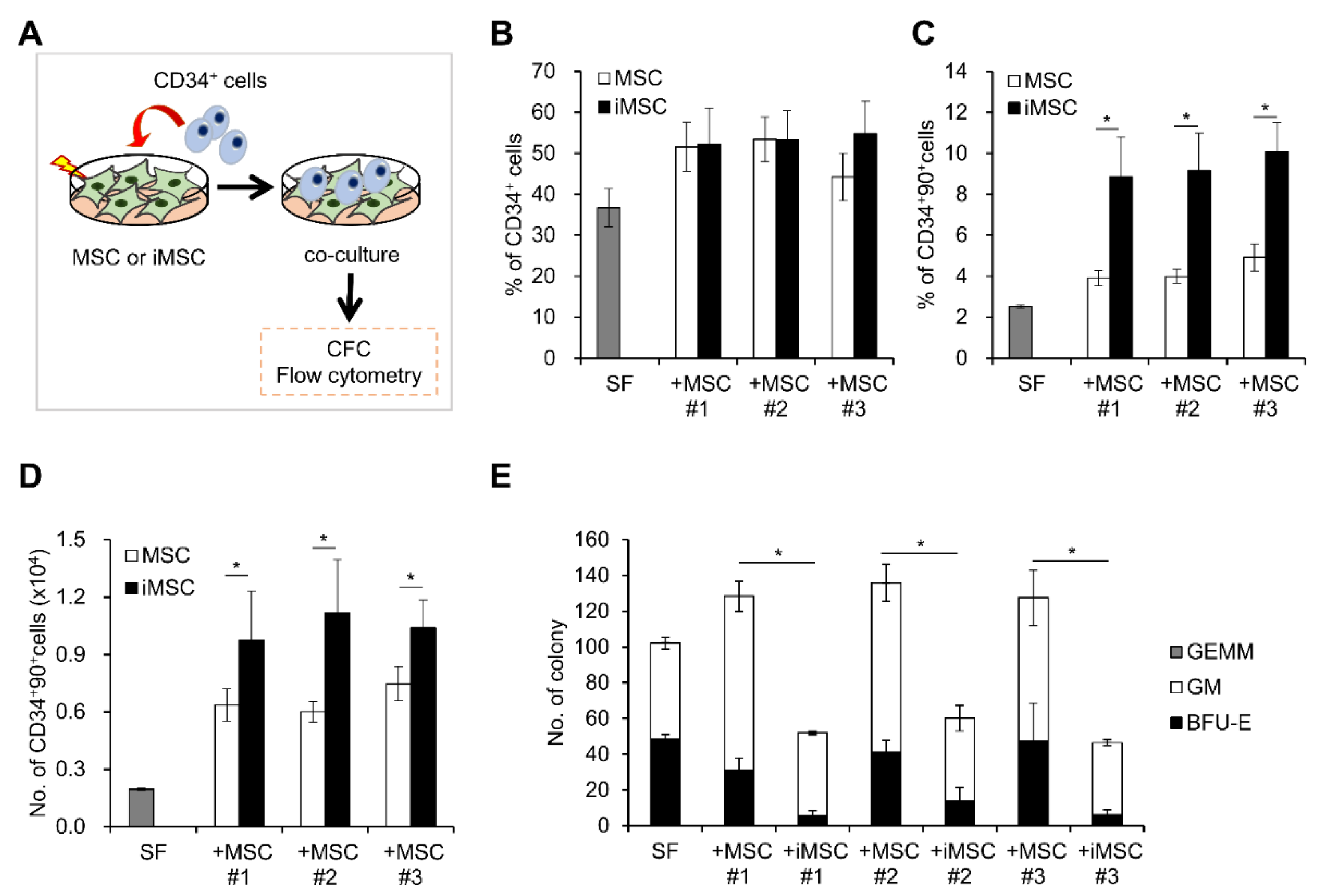
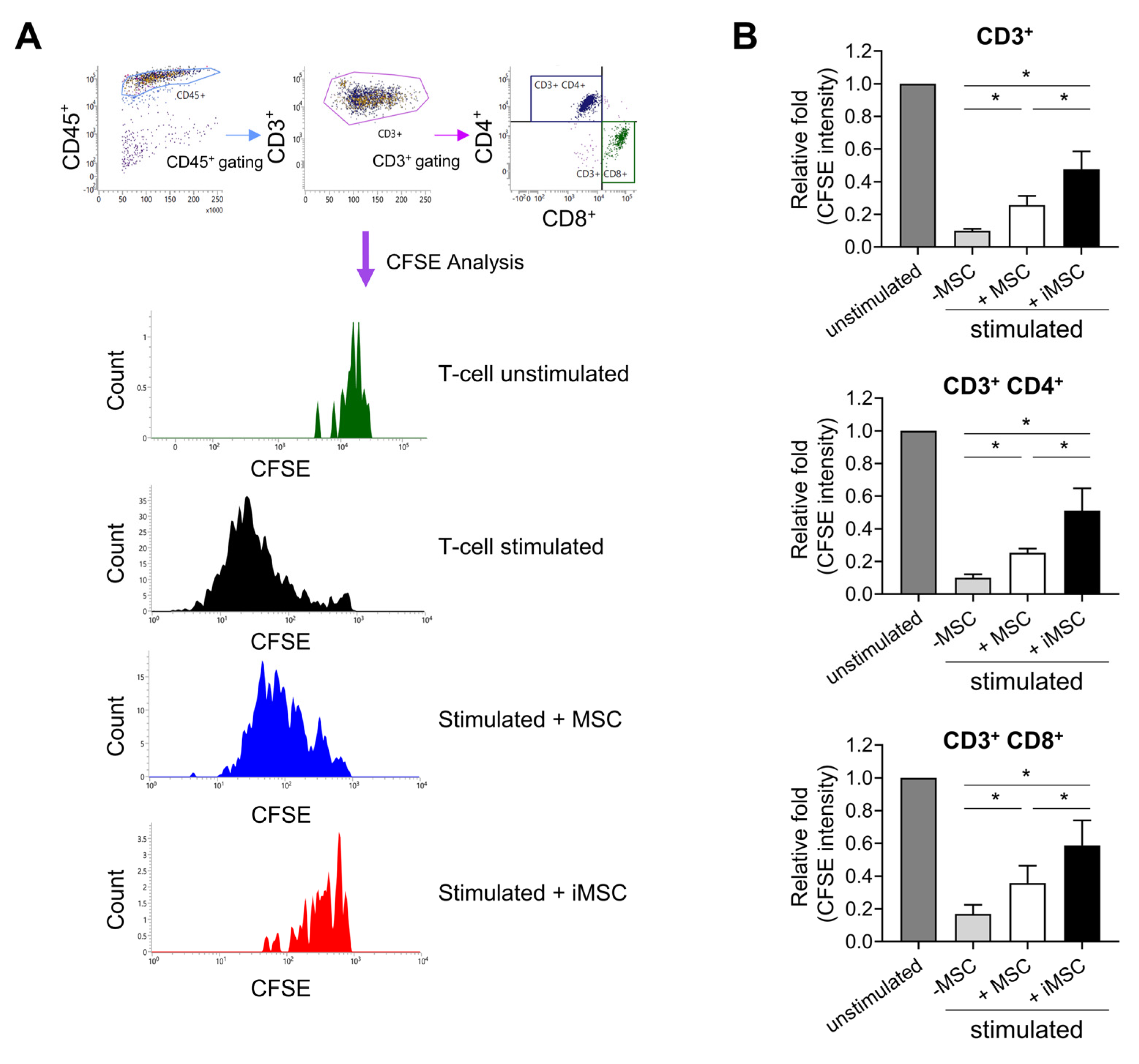
2. Result
3. Discussion
4. Materials and methods
4.1. Human Cells
4.2. Human iPSC Generation and Culture
4.3. In Vitro Differentiation of iPSC into Specific Lineages
4.4. Alkaline Phosphatase and Immunofluorescence Staining
4.5. Karyotype Analysis
4.6. iPSC-Derived MSC Generation and Culture
4.7. Co-Culture
4.8. Flow Cytometry
4.9. Telomere Length Analysis
4.10. CFU-F Assay and Differentiation of MSCs
4.11. Magnetic Bead-Based Multiplex Assay for Cytokine/Growth Factor
4.12. Western Blot
4.13. CFSE (Carboxyfluorescein Succinimidyl Ester) Proliferation Assay
4.14. Real Time-PCR
4.15. mRNA-seq Data
4.16. Preparation of the Secretome
4.17. Mass Spectrometric (MS) Analysis of Secreted Proteins and Bioinformatics Analysis of MS Data
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keating, A. Mesenchymal stromal cells: New directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276, 71–74. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The MSC: An Injury Drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.B.; Moncivais, K.; Caplan, A.I. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp. Mol. Med. 2013, 45. [Google Scholar] [CrossRef] [PubMed]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal Stem Cell: Keystone of the Hematopoietic Stem Cell Niche and a Stepping-Stone for Regenerative Medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef]
- Bruder, S.P.; Jaiswal, N.; Haynesworth, S.E. Growth kinetics, self-renewal, and the osteogenic potential of purified human mesenchymal stem cells during extensive subcultivation and following cryopreservation. J. Cell. Biochem. 1997, 64, 278–294. [Google Scholar] [CrossRef]
- Costa, L.A.; Eiro, N.; Fraile, M.; Gonzalez, L.O.; Saa, J.; Garcia-Portabella, P.; Vega, B.; Schneider, J.; Vizoso, F.J. Functional heterogeneity of mesenchymal stem cells from natural niches to culture conditions: Implications for further clinical uses. Cell. Mol. Life Sci. 2021, 78, 447–467. [Google Scholar] [CrossRef]
- Mitchell, J.B.; McIntosh, K.; Zvonic, S.; Garretta, S.; Floyd, Z.E.; Kloster, A.; Di Halvorsen, Y.; Storms, R.W.; Goh, B.; Kilroy, G.; et al. Immunophenotype of human adipose-derived cells: Temporal changes in stromal-associated and stem cell-associated markers. Stem Cells 2006, 24, 376–385. [Google Scholar] [CrossRef]
- Wagner, W.; Horn, P.; Castoldi, M.; Diehlmann, A.; Bork, S.; Saffrich, R.; Benes, V.; Blake, J.; Pfister, S.; Eckstein, V.; et al. Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS ONE 2008, 3, e2213. [Google Scholar] [CrossRef] [PubMed]
- Larson, B.L.; Ylostalo, J.; Lee, R.H.; Gregory, C.; Prockop, D.J. Sox11 is expressed in early progenitor human multipotent stromal cells and decreases with extensive expansion of the cells. Tissue Eng. Part A 2010, 16, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Kretlow, J.D.; Jin, Y.Q.; Liu, W.; Zhang, W.J.; Hong, T.H.; Zhou, G.; Baggett, L.S.; Mikos, A.G.; Cao, Y. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 2008, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE 2009, 4, e5846. [Google Scholar] [CrossRef]
- Xin, Y.; Wang, Y.M.; Zhang, H.; Li, J.; Wang, W.; Wei, Y.J.; Hu, S.S. Aging adversely impacts biological properties of human bone marrow-derived mesenchymal stem cells: Implications for tissue engineering heart valve construction. Artif. Organs 2010, 34, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.; Keating, A.; Deans, R.; Hematti, P.; Prockop, D.; Stroncek, D.F.; Stacey, G.; Weiss, D.J.; Mason, C.; Rao, M.S. Soliciting strategies for developing cell-based reference materials to advance mesenchymal stromal cell research and clinical translation. Stem Cells Dev. 2014, 23, 1157–1167. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.Z.; Liao, J.Q.; Chang, C.F.; Liu, Y.Q.; Padhiar, A.A.; Zhou, Y.; Zhou, G.Q. Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Hold Lower Heterogeneity and Great Promise in Biological Research and Clinical Applications. Front. Cell Dev. Biol. 2021, 9, 716907. [Google Scholar]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Sarić, T.; Zenke, M.; Wagner, W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 414–422. [Google Scholar] [CrossRef]
- Moslem, M.; Eberle, I.; Weber, I.; Henschler, R.; Cantz, T. Mesenchymal Stem/Stromal Cells Derived from Induced Pluripotent Stem Cells Support CD34(pos) Hematopoietic Stem Cell Propagation and Suppress Inflammatory Reaction. Stem Cells Int. 2015, 2015. [Google Scholar] [CrossRef]
- Jung, Y.; Bauer, G.; Nolta, J.A. Concise review: Induced pluripotent stem cell-derived mesenchymal stem cells: Progress toward safe clinical products. Stem Cells 2012, 30, 42–47. [Google Scholar] [CrossRef]
- Sheyn, D.; Ben-David, S.; Shapiro, G.; De Mel, S.; Bez, M.; Ornelas, L.; Sahabian, A.; Sareen, D.; Da, X.; Pelled, G.; et al. Human Induced Pluripotent Stem Cells Differentiate into Functional Mesenchymal Stem Cells and Repair Bone Defects. Stem Cells Transl. Med. 2016, 5, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Hynes, K.; Menicanin, D.; Han, J.; Marino, V.; Mrozik, K.; Gronthos, S.; Bartold, P.M. Mesenchymal stem cells from iPS cells facilitate periodontal regeneration. J. Dent. Res. 2013, 92, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Kuroda, Y.; Bartosh, T.J.; Liu, F.; Zhao, Q.; Gregory, C.; Reger, R.; Xu, J.; Lee, R.H.; Prockop, D.J. iPS-derived MSCs from an expandable bank to deliver a prodrug-converting enzyme that limits growth and metastases of human breast cancers. Cell Death Discov. 2017, 3, 16064. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Z.; Han, X.; Ouyang, X.; Fang, J.; Huang, X.; Wei, H. Transplantation of induced pluripotent stem cell-derived mesenchymal stem cells improved erectile dysfunction induced by cavernous nerve injury. Theranostics 2019, 9, 6354–6368. [Google Scholar] [CrossRef]
- Lin, L.; Bolund, L.; Luo, Y. Towards Personalized Regenerative Cell Therapy: Mesenchymal Stem Cells Derived from Human Induced Pluripotent Stem Cells. Curr. Stem Cell Res. Ther. 2016, 11, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Sim, H.; Ahn, H.; Ha, J.; Baek, A.; Jeon, Y.J.; Son, M.Y.; Kim, J. Direct Reprogramming to Human Induced Neuronal Progenitors from Fibroblasts of Familial and Sporadic Parkinson’s Disease Patients. Int. J. Stem Cells 2019, 12, 474–483. [Google Scholar] [CrossRef]
- Soares, F.A.; Pedersen, R.A.; Vallier, L. Generation of Human Induced Pluripotent Stem Cells from Peripheral Blood Mononuclear Cells Using Sendai Virus. Methods Mol. Biol. 2016, 1357, 23–31. [Google Scholar] [CrossRef]
- Chan, C.K.F.; Chen, C.C.; Luppen, C.A.; Kim, J.B.; DeBoer, A.T.; Wei, K.; Helms, J.A.; Kuo, C.J.; Kraft, D.L.; Weissman, I.L. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature 2009, 457, 490–494. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef]
- Hollmann, J.; Brecht, J.; Goetzke, R.; Franzen, J.; Selich, A.; Schmidt, M.; Eipel, M.; Ostrowska, A.; Hapala, J.; Fernandez-Rebollo, E.; et al. Genetic barcoding reveals clonal dominance in iPSC-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2020, 11, 105. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.M.; Park, T.I.H.; Dragunow, M. Markers for human brain pericytes and smooth muscle cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Meirelles, L.D.; Wagatsuma, V.M.D.; Malta, T.M.; Palma, P.V.B.; Araujo, A.G.; Panepucci, R.A.; Silva, W.A.; Kashima, S.; Covas, D.T. The gene expression profile of non-cultured, highly purified human adipose tissue pericytes: Transcriptomic evidence that pericytes are stem cells in human adipose tissue. Exp. Cell Res. 2016, 349, 239–254. [Google Scholar] [CrossRef]
- Jeon, S.; Lee, H.S.; Lee, G.Y.; Park, G.; Kim, T.M.; Shin, J.; Lee, C.; Oh, I.H. Shift of EMT gradient in 3D spheroid MSCs for activation of mesenchymal niche function. Sci. Rep. 2017, 7, 6859. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Nakahara, F.; Asada, N.; Zhang, D.; Gao, X.; Xu, C.; Alfieri, A.; Brodin, N.P.; Zimmerman, S.E.; Mar, J.C.; et al. Snai2 Maintains Bone Marrow Niche Cells by Repressing Osteopontin Expression. Dev. Cell 2020, 53, 503–513.e505. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Su, P.F.; Huang, Y.F.; Yew, T.L.; Hung, S.C. Oct4 and Nanog Directly Regulate Dnmt1 to Maintain Self-Renewal and Undifferentiated State in Mesenchymal Stem Cells. Mol. Cell 2012, 47, 169–182. [Google Scholar] [CrossRef]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2003, 101, 3722–3729. [Google Scholar] [CrossRef]
- Le Blanc, K.; Tammik, L.; Sundberg, B.; Haynesworth, S.E.; Ringden, O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand. J. Immunol. 2003, 57, 11–20. [Google Scholar] [CrossRef]
- Zaim, M.; Karaman, S.; Cetin, G.; Isik, S. Donor age and long-term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann. Hematol. 2012, 91, 1175–1186. [Google Scholar] [CrossRef]
- Eto, S.; Goto, M.; Soga, M.; Kaneko, Y.; Uehara, Y.; Mizuta, H.; Era, T. Mesenchymal stem cells derived from human iPS cells via mesoderm and neuroepithelium have different features and therapeutic potentials. PLoS ONE 2018, 13, e0200790. [Google Scholar] [CrossRef]
- Ishida, M.; Miyagawa, S.; Saito, A.; Fukushima, S.; Harada, A.; Ito, E.; Ohashi, F.; Watabe, T.; Hatazawa, J.; Matsuura, K.; et al. Transplantation of Human-induced Pluripotent Stem Cell-derived Cardiomyocytes Is Superior to Somatic Stem Cell Therapy for Restoring Cardiac Function and Oxygen Consumption in a Porcine Model of Myocardial Infarction. Transplantation 2019, 103, 291–298. [Google Scholar] [CrossRef]
- Li, F.; Niyibizi, C. Cells derived from murine induced pluripotent stem cells (iPSC) by treatment with members of TGF-beta family give rise to osteoblasts differentiation and form bone in vivo. BMC Cell Biol. 2012, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Lee, Y.W.; Hsu, P.J.; Wang, H.H.; Wang, Y.; Liou, J.Y.; Hsu, S.H.; Wu, K.K.; Yen, B.L. Human pluripotent stem cell (PSC)-derived mesenchymal stem cells (MSCs) show potent neurogenic capacity which is enhanced with cytoskeletal rearrangement. Oncotarget 2016, 7, 43949–43959. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Q.; Zhang, Y.; Li, X.; Deng, M.X.; Gao, W.X.; Yao, Y.; Chiu, S.M.; Liang, X.; Gao, F.; Chan, C.W.; et al. Insensitivity of Human iPS Cells-Derived Mesenchymal Stem Cells to Interferon-γ-induced HLA Expression Potentiates Repair Efficiency of Hind Limb Ischemia in Immune Humanized NOD Scid Gamma Mice. Stem Cells 2015, 33, 3452–3467. [Google Scholar] [CrossRef] [PubMed]
- Vasko, T.; Frobel, J.; Lubberich, R.; Goecke, T.W.; Wagner, W. iPSC-derived mesenchymal stromal cells are less supportive than primary MSCs for co-culture of hematopoietic progenitor cells. J. Hematol. Oncol. 2016, 9, 43. [Google Scholar] [CrossRef][Green Version]
- Wu, H.J.; Yiu, W.H.; Wong, D.W.L.; Li, R.X.; Chan, L.Y.Y.; Leung, J.C.K.; Zhang, Y.; Lian, Q.; Lai, K.N.; Tse, H.F.; et al. Human induced pluripotent stem cell-derived mesenchymal stem cells prevent adriamycin nephropathy in mice. Oncotarget 2017, 8, 103640–103656. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Gregory, C.A.; Lee, R.H.; Reger, R.L.; Qin, L.; Hai, B.; Park, M.S.; Yoon, N.; Clough, B.; McNeill, E.; et al. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc. Natl. Acad. Sci.USA 2015, 112, 530–535. [Google Scholar] [CrossRef]
- Li, F.; Bronson, S.; Niyibizi, C. Derivation of murine induced pluripotent stem cells (iPS) and assessment of their differentiation toward osteogenic lineage. J. Cell. Biochem. 2010, 109, 643–652. [Google Scholar] [CrossRef]
- Diederichs, S.; Tuan, R.S. Functional comparison of human-induced pluripotent stem cell-derived mesenchymal cells and bone marrow-derived mesenchymal stromal cells from the same donor. Stem Cells Dev. 2014, 23, 1594–1610. [Google Scholar] [CrossRef]
- Hynes, K.; Menicanin, D.; Mrozik, K.; Gronthos, S.; Bartold, P.M. Generation of functional mesenchymal stem cells from different induced pluripotent stem cell lines. Stem Cells Dev. 2014, 23, 1084–1096. [Google Scholar] [CrossRef]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef]
- Greenbaum, A.; Hsu, Y.M.S.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is. required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef]
- Pinho, S.; Lacombe, J.; Hanoun, M.; Mizoguchi, T.; Bruns, I.; Kunisaki, Y.; Frenette, P.S. PDGFR alpha and CD51 mark human Nestin(+) sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013, 210, 1351–1367. [Google Scholar] [CrossRef] [PubMed]
- Péault, B.; Weissman, I.L.; Buckle, A.M.; Tsukamoto, A.; Baum, C. Thy-1-expressing CD34+ human cells express multiple hematopoietic potentialities in vitro and in SCID-hu mice. Nouv. Rev. Fr. Hematol. 1978 1993, 35, 91–93. [Google Scholar] [PubMed]
- Danet, G.H.; Lee, H.W.; Luongo, J.L.; Simon, M.C.; Bonnet, D.A. Dissociation between stem cell phenotype and NOD/SCID repopulating activity in human peripheral blood CD34(+) cells after ex vivo expansion. Exp. Hematol. 2001, 29, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- de Wynter, E.A.; Heyworth, C.M.; Mukaida, N.; Matsushima, K.; Testa, N.G. NOD/SCID repopulating cells but not LTC-IC are enriched in human CD34+ cells expressing the CCR1 chemokine receptor. Leukemia 2001, 15, 1092–1101. [Google Scholar] [CrossRef]
- Giuliani, M.; Oudrhiri, N.; Noman, Z.M.; Vernochet, A.; Chouaib, S.; Azzarone, B.; Durrbach, A.; Bennaceur-Griscelli, A. Human mesenchymal stem cells derived from induced pluripotent stem cells down-regulate NK-cell cytolytic machinery. Blood 2011, 118, 3254–3262. [Google Scholar] [CrossRef]
- Son, M.Y.; Kim, H.J.; Kim, M.J.; Cho, Y.S. Physical passaging of embryoid bodies generated from human pluripotent stem cells. PLoS ONE 2011, 6, e19134. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-R.; Kim, S.; Shin, S.; Jeong, S.-Y.; Lee, D.-W.; Lim, S.-U.; Kang, J.Y.; Son, M.-Y.; Lee, C.; Yu, K.-R.; et al. iPSC-Derived MSCs Are a Distinct Entity of MSCs with Higher Therapeutic Potential than Their Donor-Matched Parental MSCs. Int. J. Mol. Sci. 2023, 24, 881. https://doi.org/10.3390/ijms24010881
Lee H-R, Kim S, Shin S, Jeong S-Y, Lee D-W, Lim S-U, Kang JY, Son M-Y, Lee C, Yu K-R, et al. iPSC-Derived MSCs Are a Distinct Entity of MSCs with Higher Therapeutic Potential than Their Donor-Matched Parental MSCs. International Journal of Molecular Sciences. 2023; 24(1):881. https://doi.org/10.3390/ijms24010881
Chicago/Turabian StyleLee, Hae-Ri, Soo Kim, Sungho Shin, Seon-Yeong Jeong, Dae-Won Lee, Sun-Ung Lim, Ji Yeon Kang, Mi-Young Son, Cheolju Lee, Kyung-Rok Yu, and et al. 2023. "iPSC-Derived MSCs Are a Distinct Entity of MSCs with Higher Therapeutic Potential than Their Donor-Matched Parental MSCs" International Journal of Molecular Sciences 24, no. 1: 881. https://doi.org/10.3390/ijms24010881
APA StyleLee, H.-R., Kim, S., Shin, S., Jeong, S.-Y., Lee, D.-W., Lim, S.-U., Kang, J. Y., Son, M.-Y., Lee, C., Yu, K.-R., Kim, M., & Oh, I.-H. (2023). iPSC-Derived MSCs Are a Distinct Entity of MSCs with Higher Therapeutic Potential than Their Donor-Matched Parental MSCs. International Journal of Molecular Sciences, 24(1), 881. https://doi.org/10.3390/ijms24010881