
Role of the Ubiquitin System in Stress Granule Metabolism


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Presence of Ubiquitin and Ub-like Modifiers at SGs
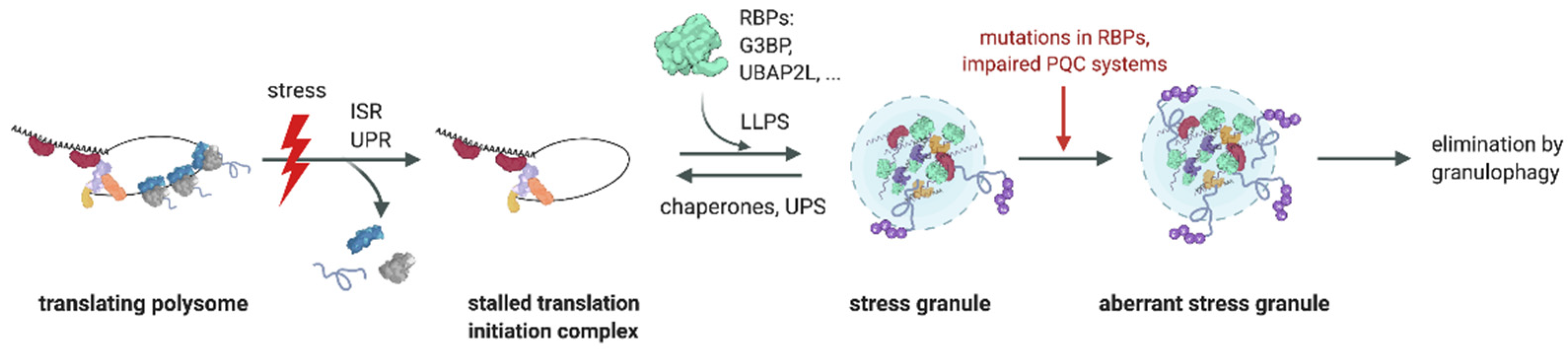
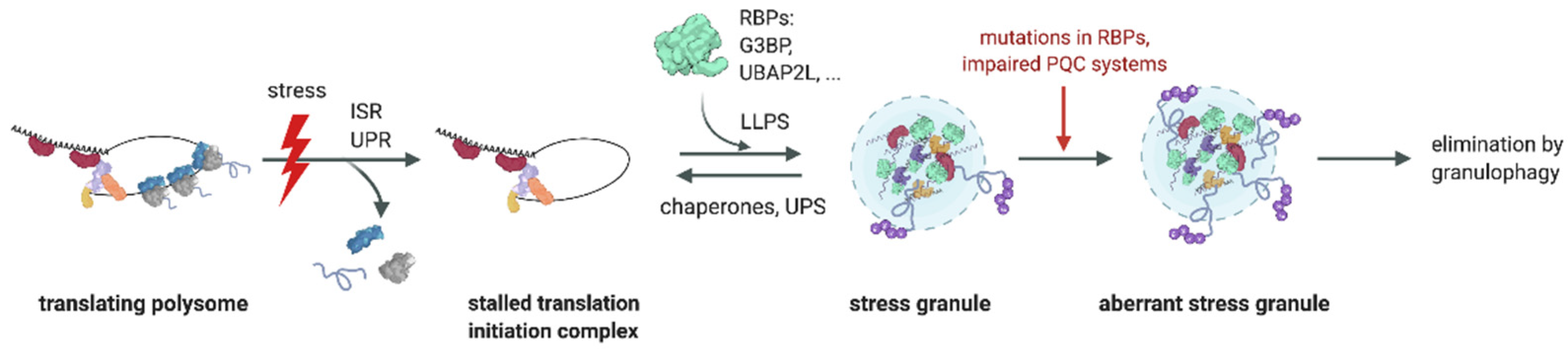
3. Role of the Ub System in SG Formation
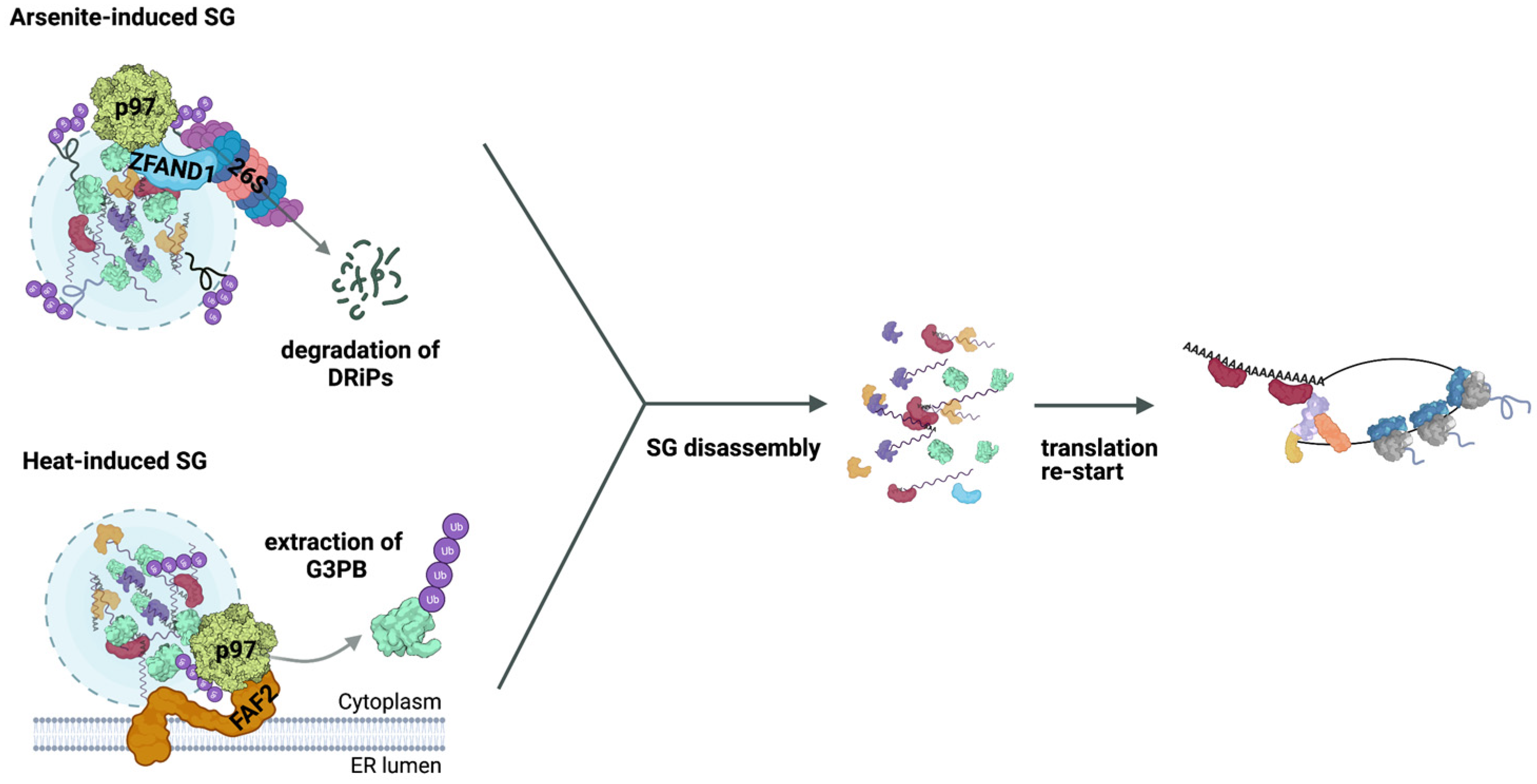
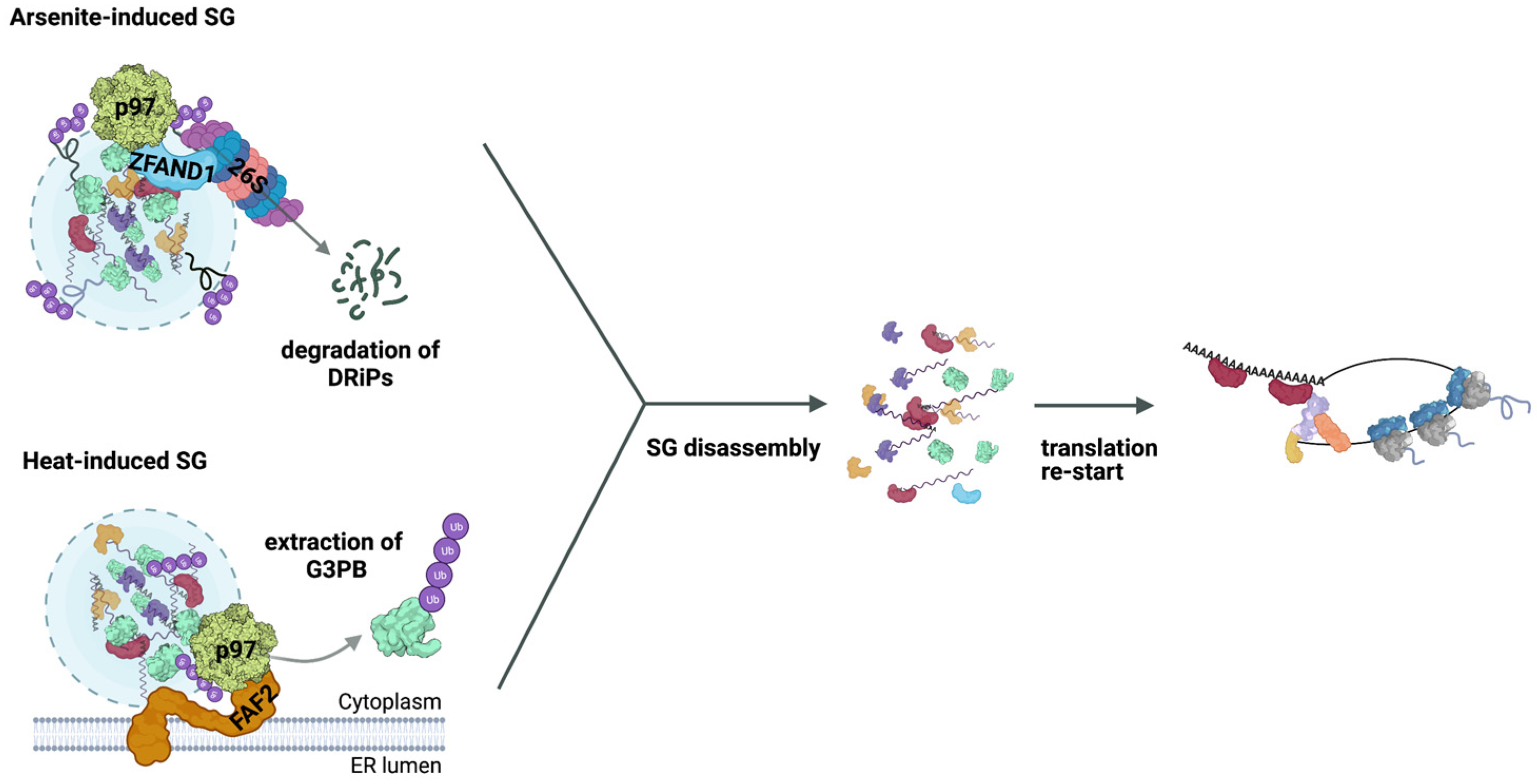
4. Role of the Ub System in SG Clearance
5. Perturbed Granulostasis Is Linked to Neurodegenerative Disorders
6. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress Granules and Cell Signaling: More Than Just a Passing Phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef]
- Hyman, A.; Weber, C.A.; Julicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittag, T.; Parker, R. Multiple Modes of Protein–Protein Interactions Promote RNP Granule Assembly. J. Mol. Biol. 2018, 430, 4636–4649. [Google Scholar] [CrossRef] [PubMed]
- Ries, R.J.; Zaccara, S.; Klein, P.; Olarerin-George, A.; Namkoong, S.; Pickering, B.F.; Patil, D.P.; Kwak, H.; Lee, J.H.; Jaffrey, S.R. m6A enhances the phase separation potential of mRNA. Nature 2019, 571, 424–428. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Namkoong, S.; Ho, A.; Woo, Y.M.; Kwak, H.; Lee, J.H. Systematic Characterization of Stress-Induced RNA Granulation. Mol. Cell 2018, 70, 175–187.e8. [Google Scholar] [CrossRef] [Green Version]
- Padrón, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA Labeling by APEX-Seq Reveals the Organization of Translation Initiation Complexes and Repressive RNA Granules. Mol. Cell 2019, 75, 875–887.e5. [Google Scholar] [CrossRef]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell 2017, 68, 808–820.e5. [Google Scholar] [CrossRef]
- Marcelo, A.; Koppenol, R.; de Almeida, L.P.; Matos, C.A.; Nóbrega, C. Stress Granules, RNA-Binding Proteins and Polyglutamine Diseases: Too Much Aggregation? Cell Death Dis. 2021, 12, 592. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic Stress Granules: The Ins and Outs of Translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasekharan, S.P.; Zhang, F.; Saxena, N.; Ni Huang, J.; Kuo, I.-C.; Low, C.; Bell, R.; Adomat, H.; Stoynov, N.; Foster, L.; et al. G3BP1-linked mRNA Partitioning Supports Selective Protein Synthesis in Response to Oxidative Stress. Nucleic Acids Res. 2020, 48, 6855–6873. [Google Scholar] [CrossRef] [PubMed]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual Specificity Kinase DYRK3 Couples Stress Granule Condensation/Dissolution to mTORC1 Signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Kläsener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.-N.; Kremmer, E.; et al. Inhibition of mTORC1 by Astrin and Stress Granules Prevents Apoptosis in Cancer Cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of Stress Granules Inhibits Apoptosis by Suppressing Stress-Responsive MAPK pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Park, Y.-J.; Choi, D.W.; Cho, S.W.; Han, J.; Yang, S.; Choi, C.Y. Stress Granule Formation Attenuates RACK1-Mediated Apoptotic Cell Death Induced by Morusin. Int. J. Mol. Sci. 2020, 21, 5360. [Google Scholar] [CrossRef]
- Tsai, N.-P.; Wei, L.-N. RhoA/ROCK1 Signaling Regulates Stress Granule Formation and Apoptosis. Cell. Signal. 2010, 22, 668–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical Role of an Antiviral Stress Granule Containing Rig-I and Pkr in Viral Detection and Innate Immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; van Kuppeveld, F.J.M.; Lloyd, R.E. Stress Granules Regulate Double-Stranded RNA-Dependent Protein Kinase Activation through a Complex Containing G3BP1 and Caprin1. mBio 2015, 6, e02486. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.-Y.; Sze, L.; Lam, K.-P. The Stress Granule Protein G3BP1 Binds Viral dsRNA and RIG-I to Enhance Interferon-β Response. J. Biol. Chem. 2019, 294, 6430–6438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Sun, H.; Yin, L.; Li, J.; Mei, S.; Xu, F.; Wu, C.; Liu, X.; Zhao, F.; Zhang, D.; et al. PKR-Dependent Cytosolic cGAS Foci Are Necessary for Intracellular DNA sensing. Sci. Signal. 2019, 12, eaav7934. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral Innate Immunity and Stress Granule Responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An Aberrant Phase Transition of Stress Granules Triggered by Misfolded Protein and Prevented by Chaperone Function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/Vcp Function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein Quality Control of RNP Granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitiprolu, M.; Jagow, C.; Tremblay, V.; Bondy-Chorney, E.; Paris, G.; Savard, A.; Palidwor, G.; Barry, F.A.; Zinman, L.; Keith, J.; et al. A Complex of C9ORF72 and p62 Uses Arginine Methylation to Eliminate Stress Granules by Autophagy. Nat. Commun. 2018, 9, 2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Chitiprolu, M.; Gagnon, D.; Meng, L.; Perez-Iratxeta, C.; Lagace, D.; Gibbings, D. Autophagy Supports Genomic Stability by Degrading Retrotransposon RNA. Nat. Commun. 2014, 5, 5276. [Google Scholar] [CrossRef]
- Ohn, T.; Anderson, P. The Role of Posttranslational Modifications in the Assembly of Stress Granules. Wiley Interdiscip. Rev. RNA 2010, 1, 486–493. [Google Scholar] [CrossRef]
- Hofweber, M.; Dormann, D. Friend or foe—Post-Translational Modifications as Regulators of Phase Separation and RNP Granule Dynamics. J. Biol. Chem. 2019, 294, 7137–7150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Ubiquitin: Same Molecule, Different Degradation Pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the Chains: Deubiquitylating Enzyme Specificity Begets Function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef]
- van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchberger, A. Unfolding by Cdc48/P97: Different Strokes for Different Folks. Trends Cell Biol. 2022, 32, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.-E. Inhibition of the Ubiquitin-Proteasome System Induces Stress Granule Formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.; Zhang, Y.; Matthias, P. The deacetylase HDAC6 Is a novel Critical Component of Stress Granules Involved in the Stress Response. Genes Dev. 2007, 21, 3381–3394. [Google Scholar] [CrossRef] [Green Version]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Böhm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol. Cell 2018, 70, 906–919.e7. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Matsumoto, S.; Endo, A.; Fukushima, T.; Kawahara, H.; Saeki, Y.; Komada, M. Deubiquitylases Usp5 and Usp13 Are Recruited to and Regulate Heat-Induced Stress Granules through Their Deubiquitylating Activities. J. Cell Sci. 2018, 131, jcs210856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolay, N.; Buchberger, A. Comparative Profiling of Stress Granule Clearance Reveals Differential Contributions of the Ubiquitin system. Life Sci. Alliance 2021, 4, e202000927. [Google Scholar] [CrossRef]
- Yu, Y.; Zheng, Q.; Erramilli, S.K.; Pan, M.; Park, S.; Xie, Y.; Li, J.; Fei, J.; Kossiakoff, A.A.; Liu, L.; et al. K29-Linked Ubiquitin Signaling Regulates Proteotoxic Stress Response and Cell Cycle. Nat. Chem. Biol. 2021, 17, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Gwon, Y.; Maxwell, B.A.; Kolaitis, R.-M.; Zhang, P.; Kim, H.J.; Taylor, J.P. Ubiquitination of G3BP1 Mediates Stress Granule Disassembly in a Context-Specific Manner. Science 2021, 372, 6548. [Google Scholar] [CrossRef]
- Maxwell, B.A.; Gwon, Y.; Mishra, A.; Peng, J.; Nakamura, H.; Zhang, K.; Kim, H.J.; Taylor, J.P. Ubiquitination Is Essential for Recovery of Cellular Activities after Heat Shock. Science 2021, 372, 3593. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhu, G.; Tang, Y.; Yan, J.; Han, S.; Yin, J.; Peng, B.; He, X.; Liu, W. HDAC6, A Novel Cargo for Autophagic Clearance of Stress Granules, Mediates the Repression of the Type I Interferon Response During Coxsackievirus A16 Infection. Front. Microbiol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Markmiller, S.; Fulzele, A.; Higgins, R.; Leonard, M.; Yeo, G.W.; Bennett, E.J. Active Protein Neddylation or Ubiquitylation Is Dispensable for Stress Granule Dynamics. Cell Rep. 2019, 27, 1356–1363.e3. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-S.; Bollinger, S.A.; Prada, L.F.; Scavone, F.; Yao, T.; Cohen, R.E. High-affinity free ubiquitin sensors for quantifying ubiquitin homeostasis and deubiquitination. Nat. Methods 2019, 16, 771–777. [Google Scholar] [CrossRef]
- Zhang, P.; Fan, B.; Yang, P.; Temirov, J.; Messing, J.; Kim, H.J.; Taylor, J.P. Chronic Optogenetic Induction of Stress Granules Is Cytotoxic and Reveals the Evolution of ALS-FTD Pathology. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A Distinct Role of Riplet-Mediated K63-Linked Polyubiquitination of the RIG-I Repressor Domain in Human Antiviral Innate Immune Responses. PLOS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal Role of RNA-Binding E3 Ubiquitin Ligase MEX3C in RIG-I–Mediated Antiviral Innate Immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651. [Google Scholar] [CrossRef] [Green Version]
- Marmor-Kollet, H.; Siany, A.; Kedersha, N.; Knafo, N.; Rivkin, N.; Danino, Y.M.; Moens, T.G.; Olender, T.; Sheban, D.; Cohen, N.; et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 2020, 80, 876–891.e6. [Google Scholar] [CrossRef] [PubMed]
- Jongjitwimol, J.; Baldock, R.; Morley, S.J.; Watts, F.Z. Sumoylation of eIF4A2 Affects Stress Granule Formation. J. Cell Sci. 2016, 129, 2407–2415. [Google Scholar] [CrossRef] [Green Version]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Müller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e7. [Google Scholar] [CrossRef]
- Jayabalan, A.K.; Sanchez, A.; Park, R.Y.; Yoon, S.P.; Kang, G.-Y.; Baek, G.-Y.K.J.-H.; Anderson, P.; Kee, A.S.Y.; Ohn, T. NEDDylation Promotes Stress Granule Assembly. Nat. Commun. 2016, 7, 12125. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.; Ivanov, P.; et al. G3BP–Caprin1–USP10 Complexes Mediate Stress Granule Condensation and Associate with 40S Subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Higuchi, M.; Matsuki, H.; Yoshita, M.; Ohsawa, T.; Oie, M.; Fujii, M. Stress Granules Inhibit Apoptosis by Reducing Reactive Oxygen Species Production. Mol. Cell. Biol. 2013, 33, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatnitskaia, S.; Takahashi, M.; Kitaura, H.; Katsuragi, Y.; Kakihana, T.; Zhang, L.; Kakita, A.; Iwakura, Y.; Nawa, H.; Miura, T.; et al. USP10 Is a Critical Factor for Tau-Positive Stress Granule Formation in Neuronal Cells. Sci. Rep. 2019, 9, 10591. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Garzia, A.; Morozov, P.; Molina, H.; Tuschl, T. The G3BP1-Family-USP10 Deubiquitinase Complex Rescues Ubiquitinated 40S Subunits of Ribosomes Stalled in Translation from Lysosomal Degradation. Mol. Cell 2020, 77, 1193–1205.e5. [Google Scholar] [CrossRef] [PubMed]
- Garshott, D.M.; An, H.; Sundaramoorthy, E.; Leonard, M.; Vicary, A.; Harper, J.W.; Bennett, E.J. iRQC, a Surveillance Pathway for 40S Ribosomal Quality Control during mRNA Translation Initiation. Cell Rep. 2021, 36, 109642. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Hospenthal, M.K.; Geurink, P.P.; Elliott, P.R.; Akutsu, M.; Arnaudo, N.; Ekkebus, R.; Kulathu, Y.; Wauer, T.; El Oualid, F.; et al. OTU Deubiquitinases Reveal Mechanisms of Linkage Specificity and Enable Ubiquitin Chain Restriction Analysis. Cell 2013, 154, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Schwintzer, L.; Vinopal, S.; Roca, E.A.; Sylvester, M.; Oprisoreanu, A.-M.; Schoch, S.; Bradke, F.; Broemer, M. New Roles for the De-Ubiquitylating Enzyme OTUD4 in an RNA-Protein Network and RNA Granules. J. Cell Sci. 2019, 132, jcs229252S. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Chen, Y.; Dai, H.; Zhang, H.; Xie, M.; Zhang, H.; Chen, F.; Kang, X.; Bai, X.; Chen, Z. UBAP2L Arginine Methylation by PRMT1 Modulates Stress Granule Assembly. Cell Death Differ. 2020, 27, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Wilde, I.B.; Brack, M.; Winget, J.M.; Mayor, T. Proteomic Characterization of Aggregating Proteins after the Inhibition of the Ubiquitin Proteasome System. J. Proteome Res. 2011, 10, 1062–1072. [Google Scholar] [CrossRef]
- Cirillo, L.; Cieren, A.; Barbieri, S.; Khong, A.; Schwager, F.; Parker, R.; Gotta, M. UBAP2L Forms Distinct Cores that Act in Nucleating Stress Granules Upstream of G3BP. Curr. Biol. 2020, 30, 698–707.e6. [Google Scholar] [CrossRef]
- Youn, J.Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of Mrna-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, C.; Srinivasan, D.; Mah, L.; Kaushik, S.; Peterhoff, C.M.; Ugolino, J.; Fang, S.; Cuervo, A.M.; Nixon, R.A.; Monteiro, M.J. Ubiquilin Functions in Autophagy and Is Degraded by Chaperone-Mediated Autophagy. Hum. Mol. Genet. 2010, 19, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Alexander, E.J.; Niaki, A.G.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 Modulates ALS/FTD-Linked FUS–RNA Complex Dynamics and Stress Granule Formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Gu, A.; Niu, H.; Chen, L.; Chen, Y.; Zhou, M.; Zhang, Y.; Liu, J.; Cai, L.; Liang, D.; et al. Amyotrophic Lateral Sclerosis (ALS) Linked Mutation in Ubiquilin 2 Affects Stress Granule Assembly via TIA-1. CNS Neurosci. Ther. 2021, 28, 105–115. [Google Scholar] [CrossRef]
- Dao, T.P.; Kolaitis, R.-M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castañeda, C.A. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 2018, 69, 965–978.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jones, H.B.; Dao, T.P.; Castañeda, C.A. Single Amino Acid Substitutions in Stickers, but Not Spacers, Substantially Alter UBQLN2 Phase Transitions and Dense Phase Material Properties. J. Phys. Chem. B 2019, 123, 3618–3629. [Google Scholar] [CrossRef]
- Wang, B.; Maxwell, B.A.; Joo, J.H.; Gwon, Y.; Messing, J.; Mishra, A.; Shaw, T.I.; Ward, A.L.; Quan, H.; Sakurada, S.M.; et al. ULK1 and ULK2 Regulate Stress Granule Disassembly through Phosphorylation and Activation of VCP/p97. Mol. Cell 2019, 74, 742–757.e8. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.K.; Xia, D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016, 3, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihl, C.C.; Temiz, P.; Miller, S.E.; Watts, G.D.J.; Smith, C.D.; Forman, M.S.; Hanson, P.I.; Kimonis, V.E.; Pestronk, A. TDP-43 Accumulation in Inclusion Body Myopathy Muscle Suggests a Common Pathogenic Mechanism with Frontotemporal Dementia. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1186–1189. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Ortiz, C.J.; Flores, J.C.; Valenzuela, J.A.; Rodriguez, G.J.; Zumkehr, J.; Tran, D.N.; Kimonis, V.E.; Kitazawa, M. The Myoblast C2C12 Transfected with Mutant Valosin-Containing Protein Exhibits Delayed Stress Granule Resolution on Oxidative Stress. Am. J. Pathol. 2016, 186, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, B.; Klemm, E.J.; Spooner, E.; Claessen, J.H.; Ploegh, H.L. SEL1L Nucleates a Protein Complex Required for Dislocation of Misfolded Glycoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12325–12330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Otsuka, T.; Ohsaki, Y.; Cheng, J.; Taniguchi, T.; Hashimoto, H.; Taniguchi, H.; Fujimoto, T. Derlin-1 and UBXD8 Are Engaged in Dislocation and Degradation of Lipidated ApoB-100 at Lipid Droplets. Mol. Biol. Cell 2012, 23, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.; Alberti, S. Ubiquitin protein helps cells to recover from stress. Nature 2021, 597, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.-B.; Princiotta, M.F.; Bennink, J.R.; Yewdell, J.W. Characterization of Rapidly Degraded Polypeptides in Mammalian Cells Reveals a Novel Layer of Nascent Protein Quality Control. J. Biol. Chem. 2006, 281, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Oania, R.S.; Kolawa, N.J.; Deshaies, R.J. Cdc48/P97 Promotes Degradation of Aberrant Nascent Polypeptides Bound to the Ribosome. eLife 2013, 2, e00308. [Google Scholar] [CrossRef] [PubMed]
- Advani, V.M.; Ivanov, P. Stress Granule Subtypes: An Emerging Link to Neurodegeneration. Cell. Mol. Life Sci. 2020, 77, 4827–4845. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress Granule Mediated Protein Aggregation and Underlying Gene Defects in the FTD-ALS Spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Ripin, N.; Parker, R. Are Stress Granules the RNA Analogs of Misfolded Protein Aggregates? RNA 2021, 28, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechler, M.C.; Crawford, E.D.; Groh, N.; Widmaier, K.; Jung, R.; Kirstein, J.; Trinidad, J.C.; Burlingame, A.L.; David, D.C. Reduced Insulin/IGF-1 Signaling Restores the Dynamic Properties of Key Stress Granule Proteins during Aging. Cell Rep. 2017, 18, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Tan, C.-F.; Eguchi, H.; Tagawa, A.; Onodera, O.; Iwasaki, T.; Tsujino, A.; Nishizawa, M.; Kakita, A.; Takahashi, H. TDP-43 Immunoreactivity in Neuronal Inclusions in Familial Amyotrophic Lateral Sclerosis with or without SOD1 Gene Mutation. Acta Neuropathol. 2007, 113, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA Binding Protein-43 (TDP-43) Associates with Stress Granules: Analysis of Cultured Cells and Pathological Brain Tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Chaplin, J.; Morris, M.J.; Hilliard, M.A.; Wolvetang, E.; Ng, D.C.H.; Noakes, P.G. TDP-43 Mutation Affects Stress Granule Dynamics in Differentiated NSC-34 Motoneuron-Like Cells. Front. Cell Dev. Biol. 2021, 9, 611601. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cohen, T.J. Aggregation of the Nucleic Acid–Binding Protein TDP-43 Occurs via Distinct Routes That Are Coordinated with Stress Granule Formation. J. Biol. Chem. 2019, 294, 3696–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, J.; Zhang, J.; Kwinter, D.M.; Zhai, J.; Jia, H.; Jia, J.; Zhu, H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 2011, 32, 2323.e27–2323.e40. [Google Scholar] [CrossRef] [Green Version]
- Notaro, A.; Messina, A.; La Bella, V. A Deletion of the Nuclear Localization Signal Domain in the Fus Protein Induces Stable Post-stress Cytoplasmic Inclusions in SH-SY5Y Cells. Front. Neurosci. 2021, 15, 759659. [Google Scholar] [CrossRef]
- Mikhaleva, S.; Lemke, E.A. Beyond the Transport Function of Import Receptors: What’s All the FUS about? Cell 2018, 173, 549–553. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yu, J.-T.; Zong, Y.; Zhou, J.; Tan, L. C9ORF72 Mutations in Neurodegenerative Diseases. Mol. Neurobiol. 2014, 49, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Smikle, R.; Reid, M.J.; Mizielinska, S. Altered Phase Separation and Cellular Impact in C9orf72-Linked ALS/FTD. Front. Cell. Neurosci. 2021, 15, 664151. [Google Scholar] [CrossRef]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendredy, L.; Adriaenssens, E.; Timmerman, V. Small Heat Shock Proteins in Neurodegenerative Diseases. Cell Stress Chaperones 2020, 25, 679–699. [Google Scholar] [CrossRef] [Green Version]
- Shy, M.E.; Rebelo, A.P.; Freely, S.M.; Abreu, L.A.; Tao, F.; Swenson, A.; Bacon, C.; Zuchner, S. Mutations in BAG3 cause adult-onset Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Korb, M.; Peck, A.; Alfano, L.N.; Berger, K.I.; James, M.K.; Ghoshal, N.; Healzer, E.; Henchcliffe, C.; Khan, S.; Mammen, P.P.A.; et al. Development of a Standard of Care for Patients with Valosin-Containing Protein Associated Multisystem Proteinopathy. Orphanet J. Rare Dis. 2022, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in Ubqln2 Cause Dominant X-Linked Juvenile and Adult-Onset Als and Als/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key Role of UBQLN2 in Pathogenesis of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef]
- Kim, S.J.; Nam, S.H.; Kanwal, S.; Nam, D.E.; Yoo, D.H.; Chae, J.-H.; Suh, Y.-L.; Chung, K.W.; Choi, B.-O. BAG3 Mutation in a Patient with Atypical Phenotypes of Myofibrillar Myopathy and Charcot–Marie–Tooth disease. Genes Genom. 2018, 40, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I. SQSTM1Mutations in French Patients with Frontotemporal Dementia or Frontotemporal Dementia with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, P.; Komatsu, M. P62/Sqstm1-Steering the Cell through Health and Disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, F.; Hu, Y.; Chen, R.; Meng, D.; Guo, L.; Lv, H.; Guan, J.; Jia, Y. In Vivo Stress Granule Misprocessing Evidenced in a FUS Knock-in ALS Mouse Model. Brain 2020, 143, 1350–1367. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolay, N.; Buchberger, A. Role of the Ubiquitin System in Stress Granule Metabolism. Int. J. Mol. Sci. 2022, 23, 3624. https://doi.org/10.3390/ijms23073624
Tolay N, Buchberger A. Role of the Ubiquitin System in Stress Granule Metabolism. International Journal of Molecular Sciences. 2022; 23(7):3624. https://doi.org/10.3390/ijms23073624
Chicago/Turabian StyleTolay, Nazife, and Alexander Buchberger. 2022. "Role of the Ubiquitin System in Stress Granule Metabolism" International Journal of Molecular Sciences 23, no. 7: 3624. https://doi.org/10.3390/ijms23073624
APA StyleTolay, N., & Buchberger, A. (2022). Role of the Ubiquitin System in Stress Granule Metabolism. International Journal of Molecular Sciences, 23(7), 3624. https://doi.org/10.3390/ijms23073624