
Pioglitazone-Loaded PLGA Nanoparticles: Towards the Most Reliable Synthesis Method



Abstract
:1. Introduction
2. Results and Discussion
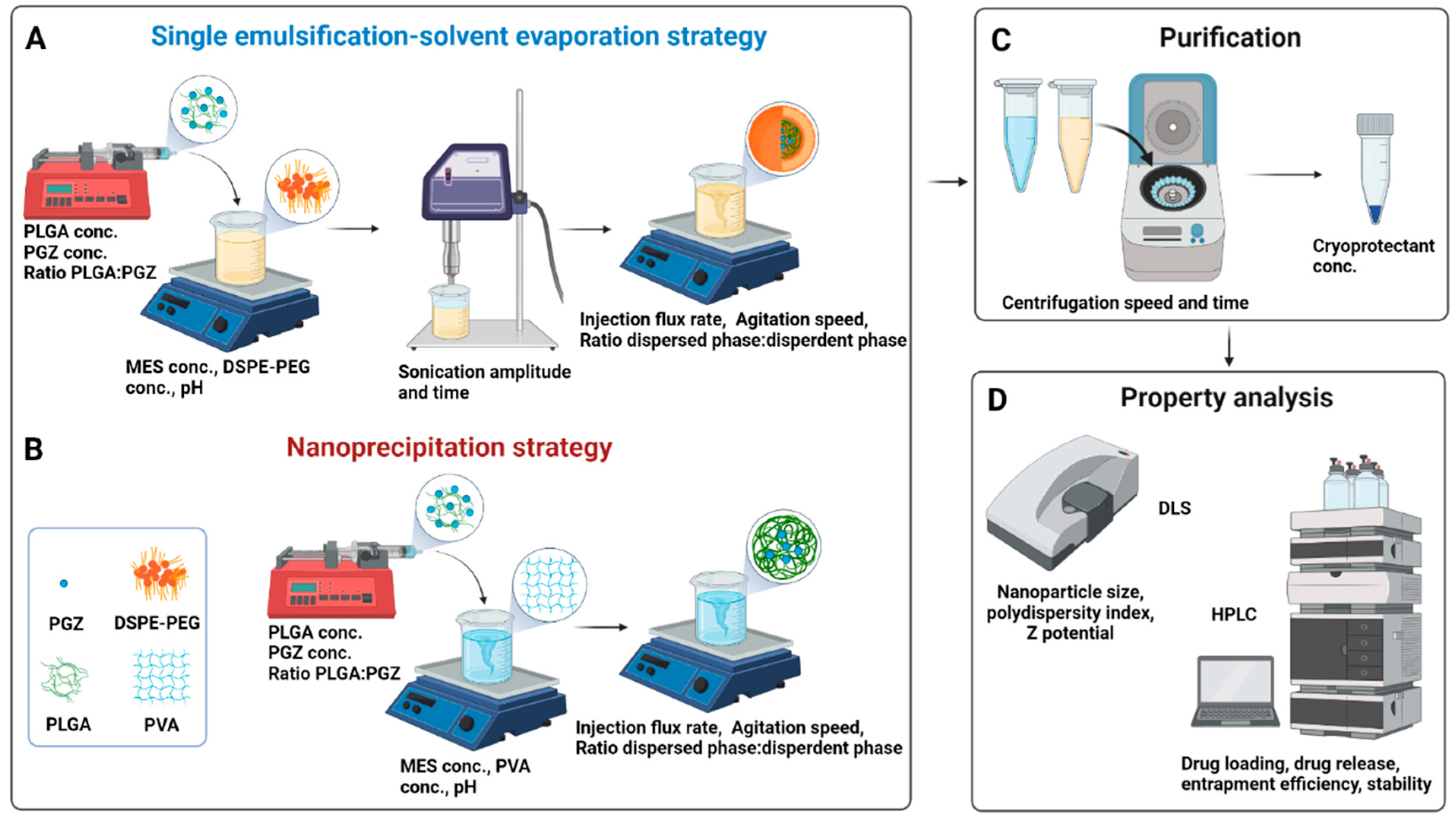
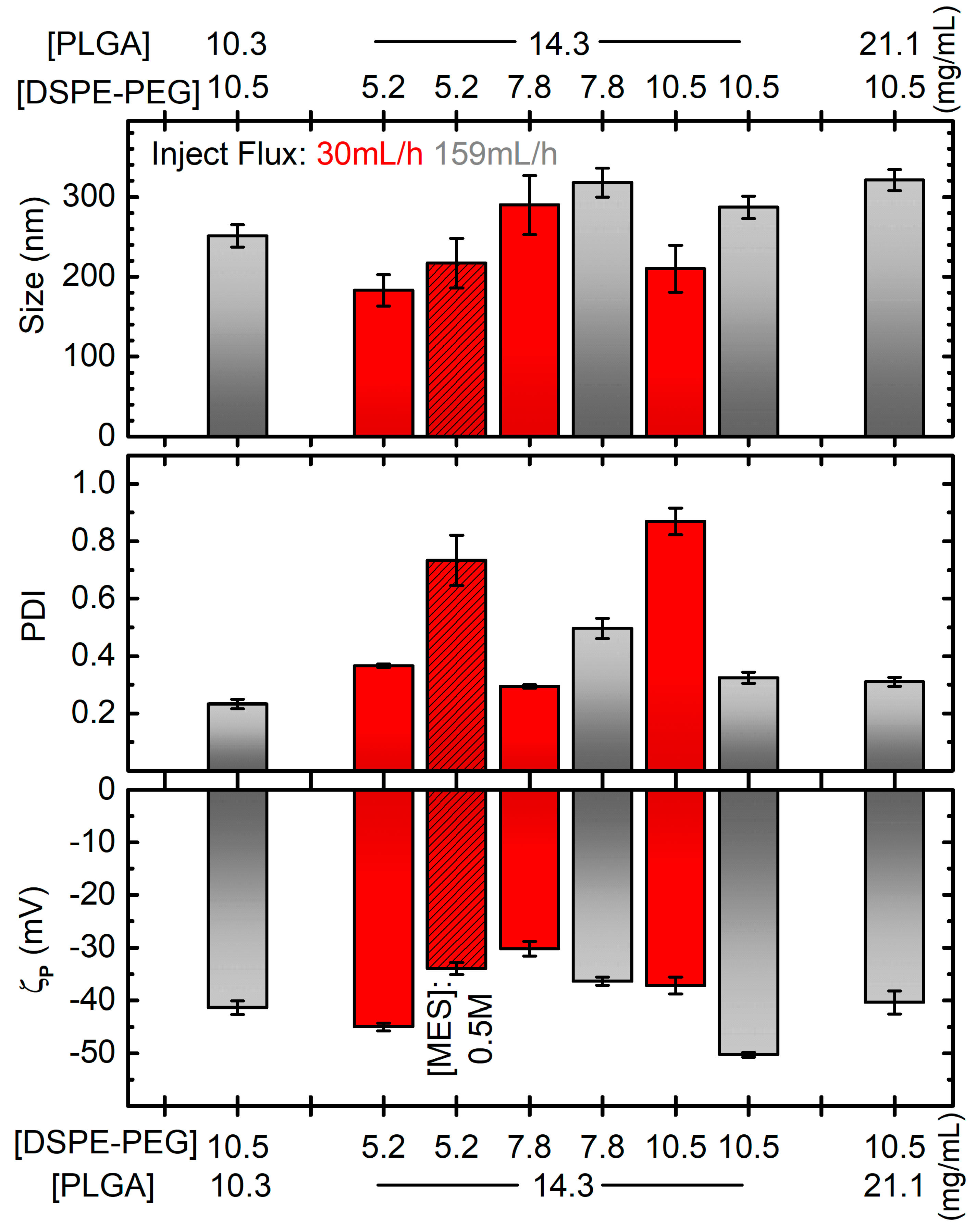
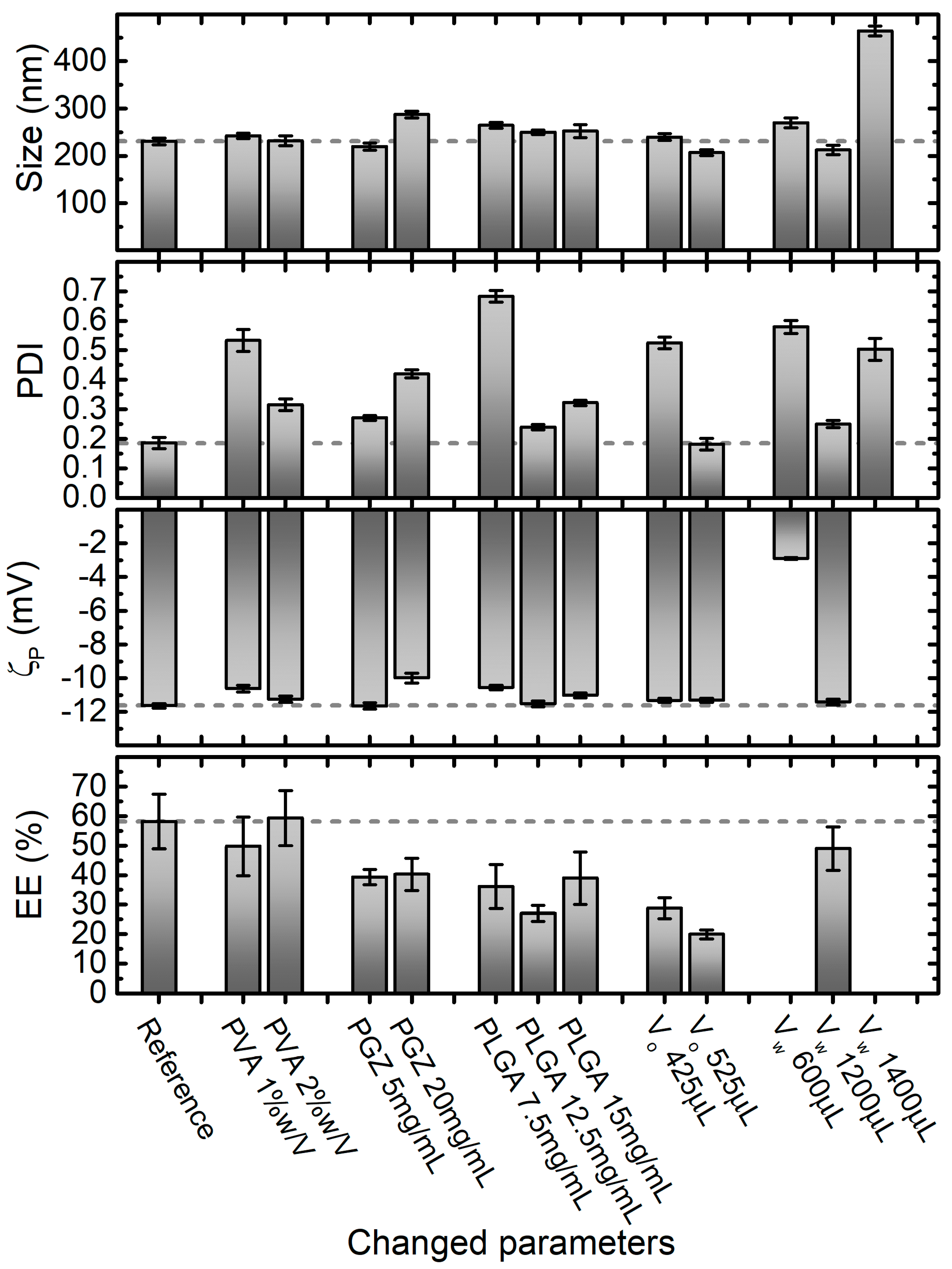
2.1. A Rational Optimization of the Single Emulsification-Solvent Evaporation Formulation Parameters
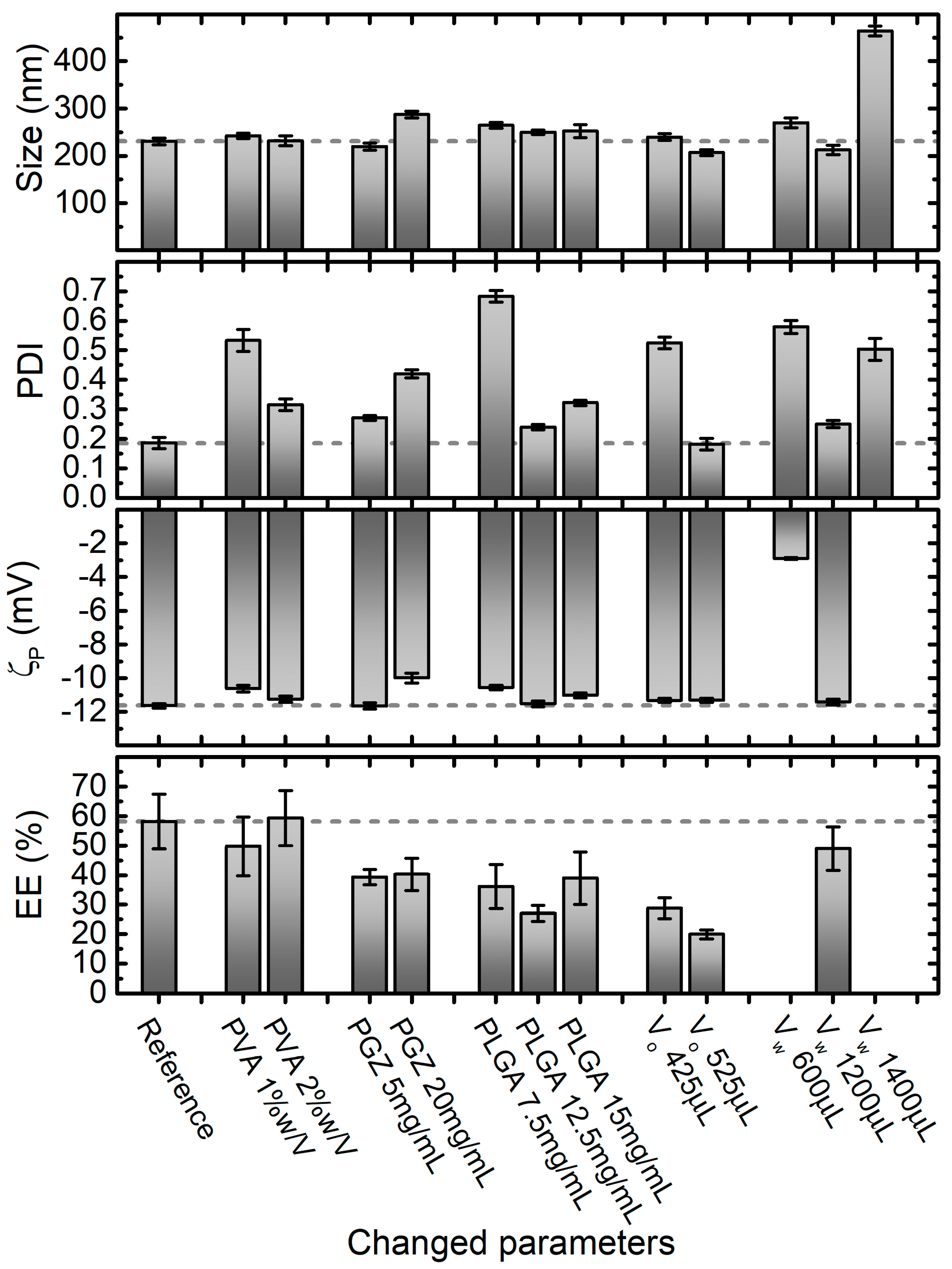
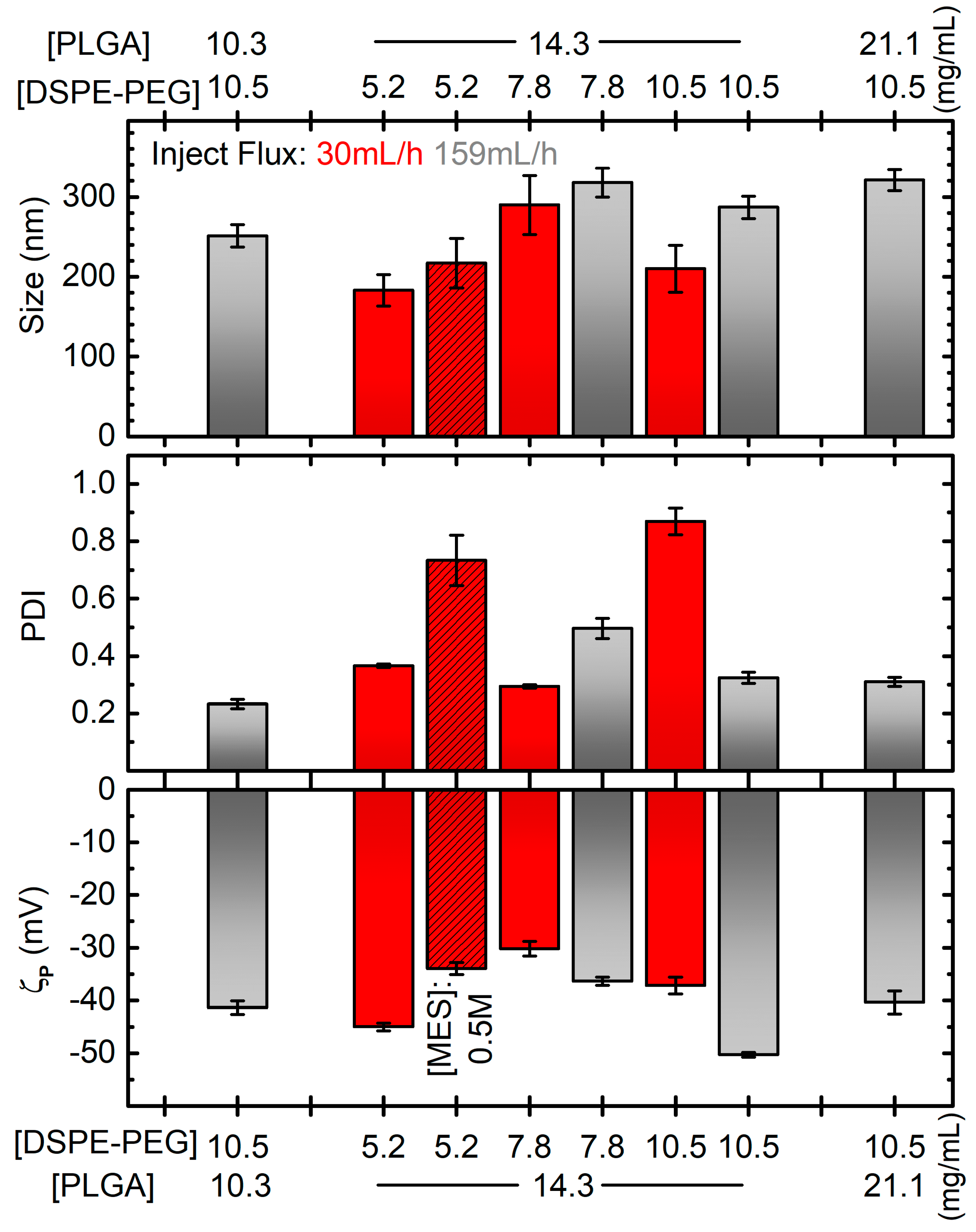
2.2. A Rational Optimization of the Nanoprecipitation Formulation Parameters
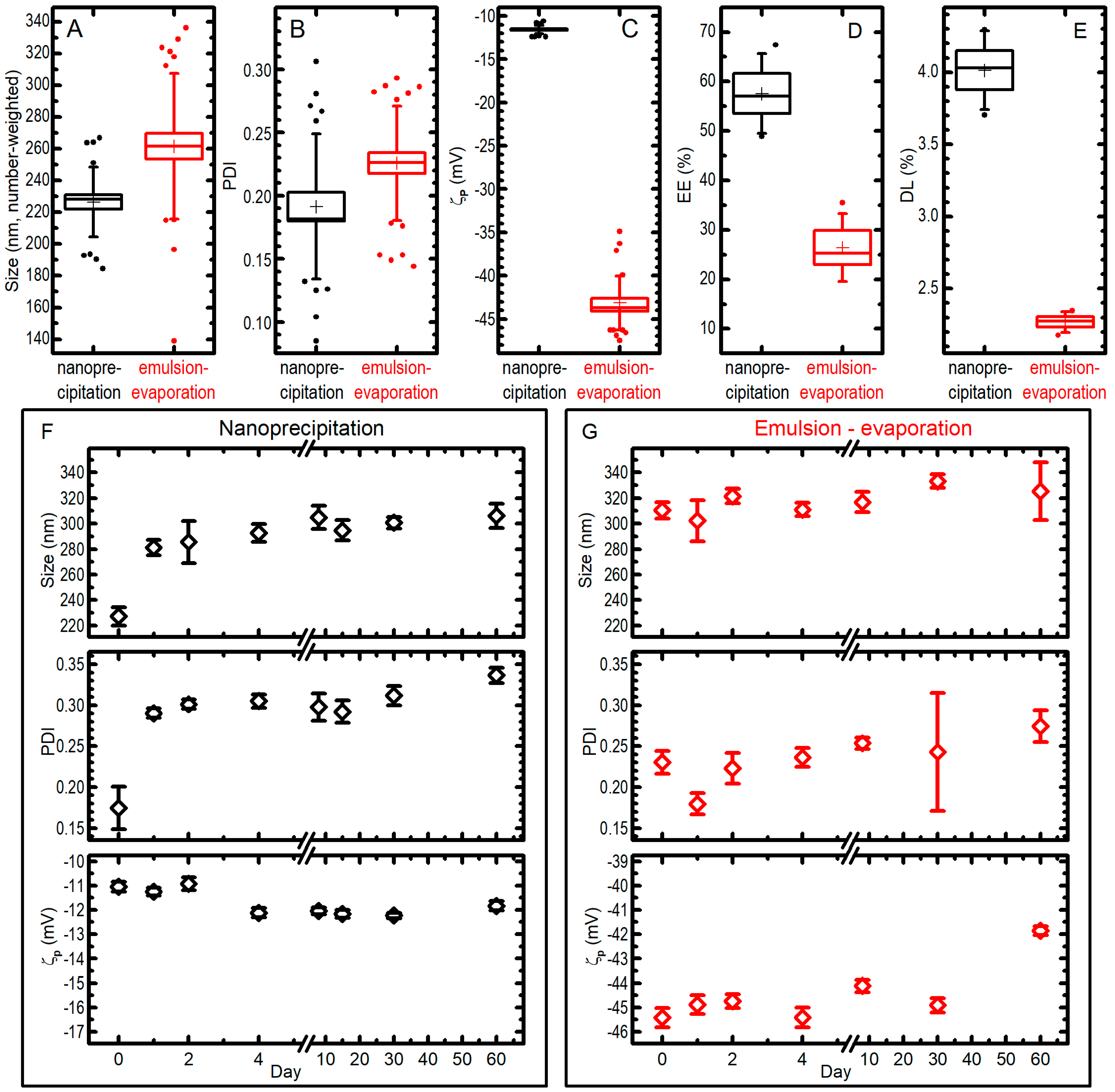
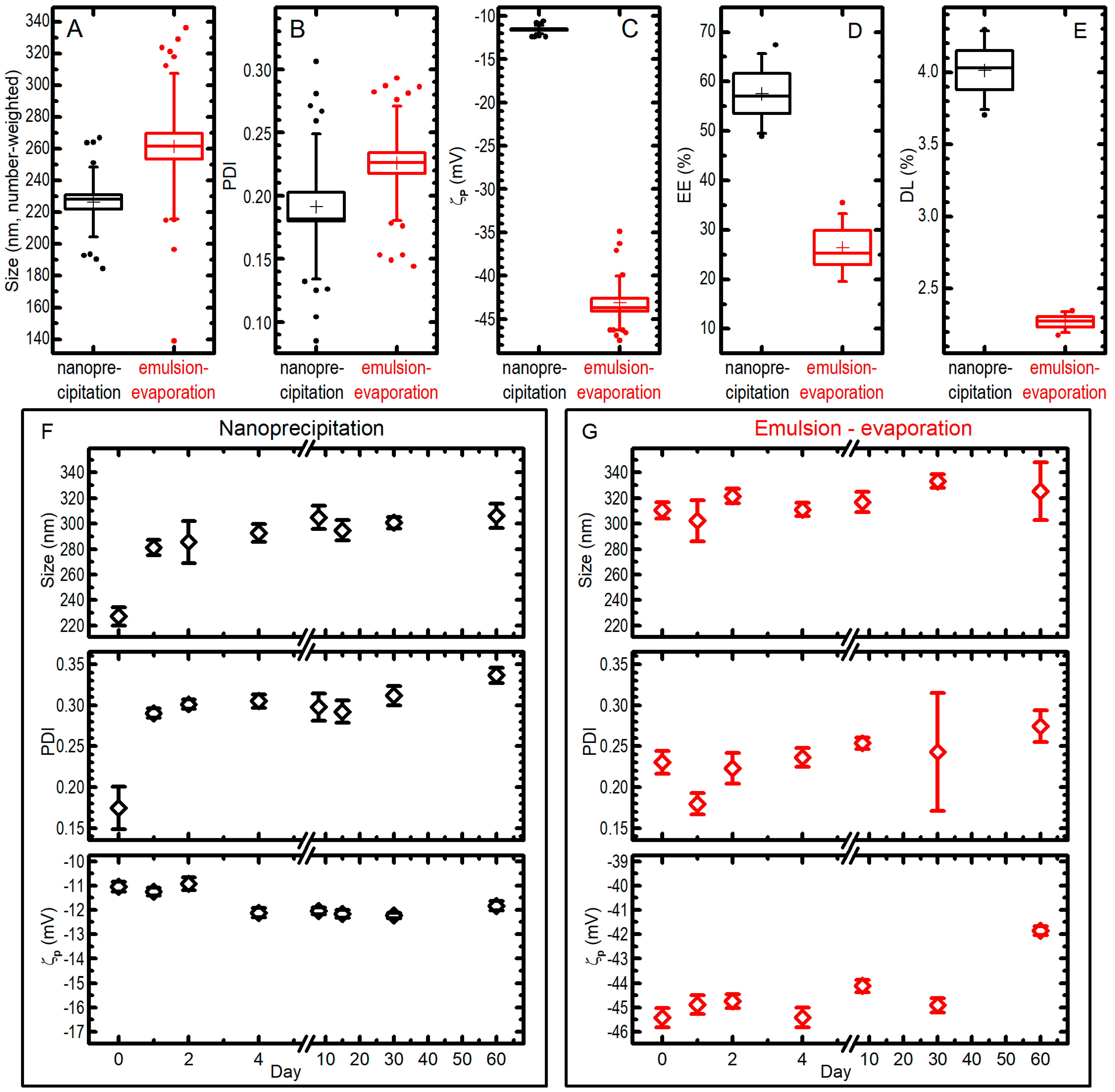
2.3. Comparison between the Two Syntheses
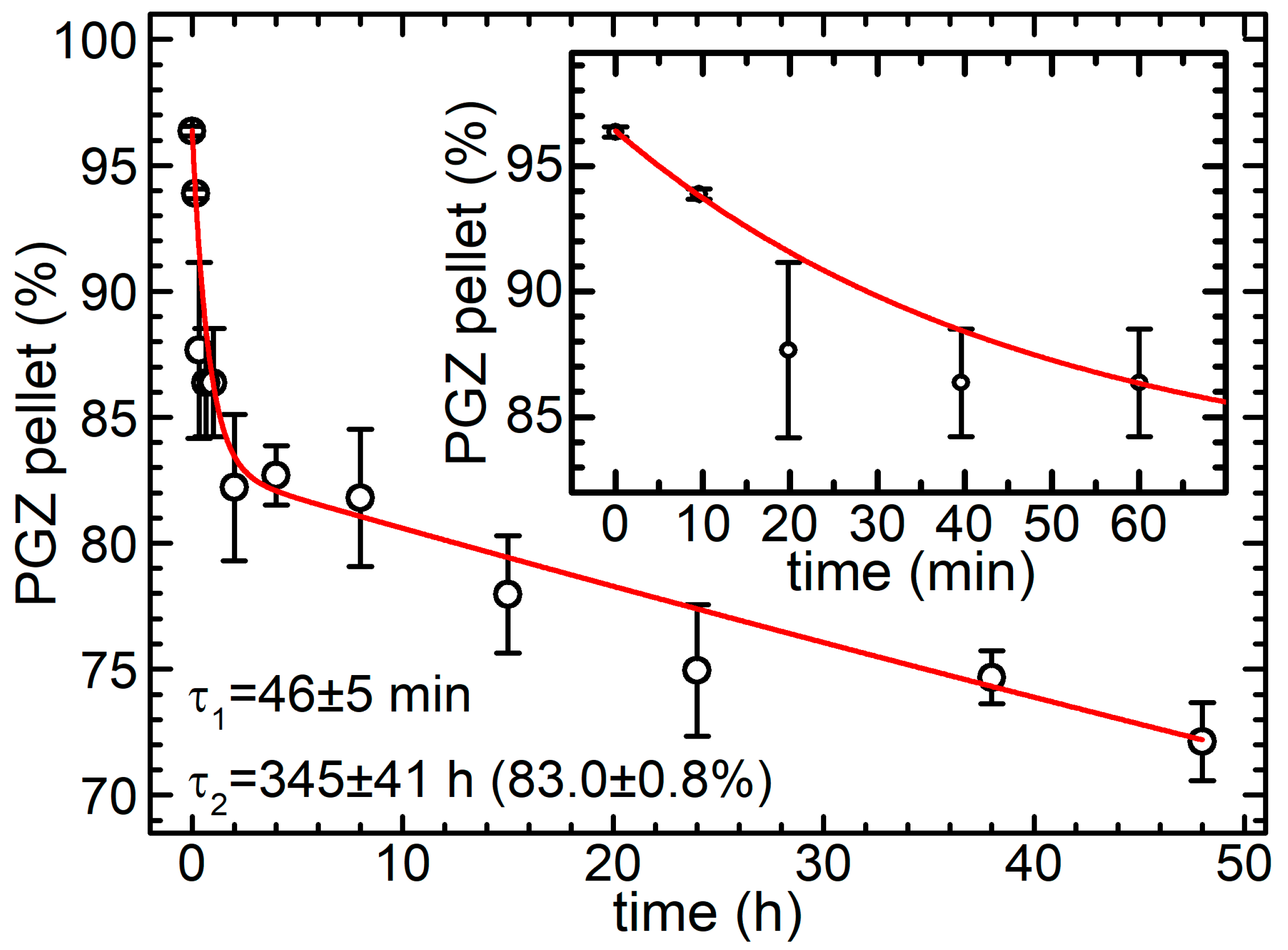
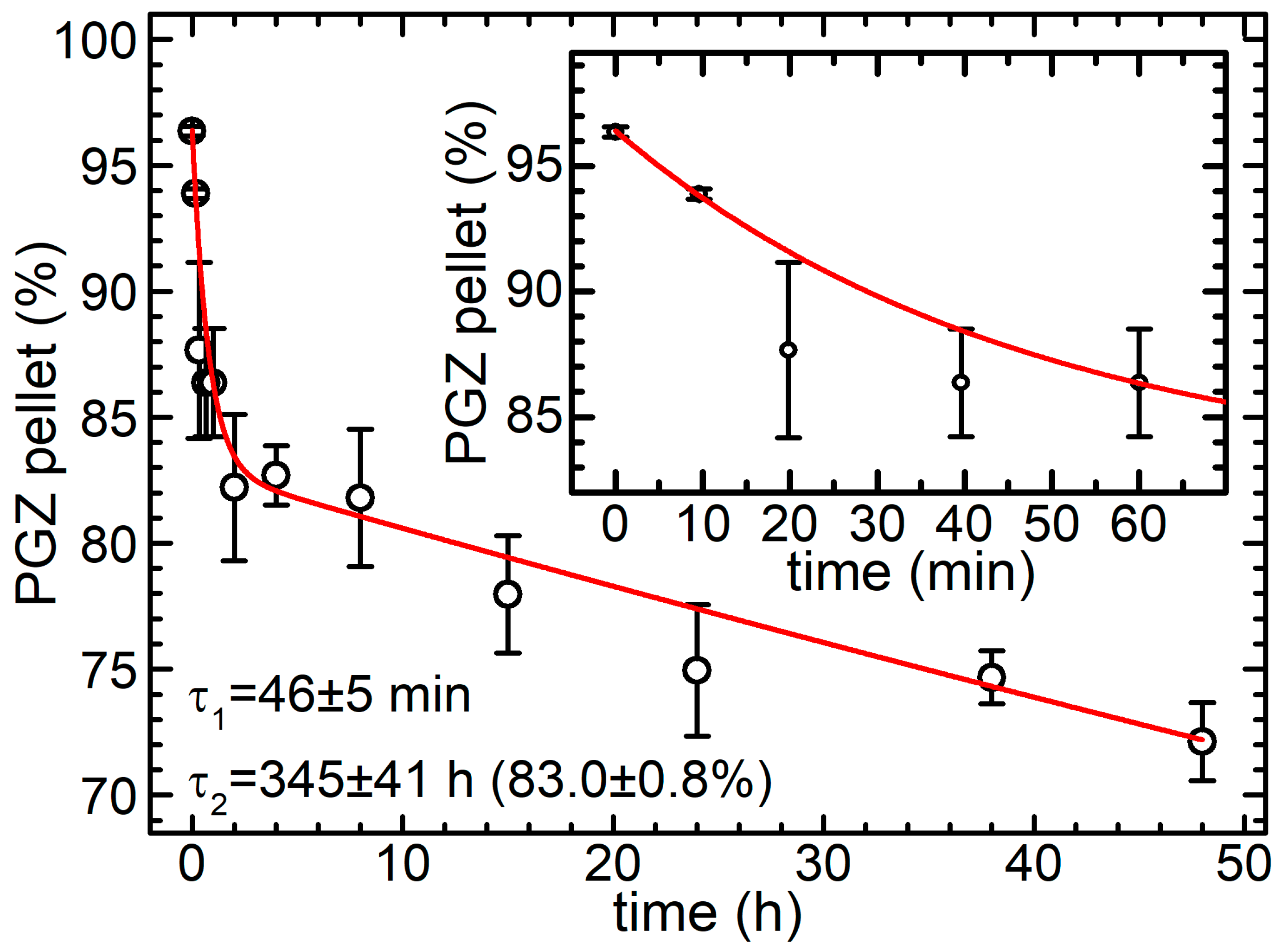
2.4. Release Assay
3. Materials and Methods
3.1. Materials
3.2. Nanoparticles Synthesis via Single Emulsification-Solvent Evaporation Method (A)
3.3. Nanoparticles Synthesis via Nanoprecipitation Method (B)
3.4. High Performance Liquid Chromatography (Hplc) Method
3.5. Nanoparticle Size, Polydispersity Index and Zeta Potential Measurements
3.6. Drug Loading (DL%) and Entrapment Efficiency (EE%) Measurements
3.7. Stability of Frozen Aliquots
3.8. Release Assay
3.9. Data Analysis
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, W.; Schilperoort, M.; Cao, Y.; Shi, J.; Tabas, I.; Tao, W. Macrophage-Targeted Nanomedicine for the Diagnosis and Treatment of Atherosclerosis. Nat. Rev. Cardiol. 2021, 1–22. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Nambi, V.; Chambless, L.; Folsom, A.R.; He, M.; Hu, Y.; Mosley, T.; Volcik, K.; Boerwinkle, E.; Ballantyne, C.M. Carotid Intima-Media Thickness and Presence or Absence of Plaque Improves Prediction of Coronary Heart Disease Risk. J. Am. Coll. Cardiol. 2010, 55, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Song, J.; Watase, H.; Hippe, D.S.; Zhao, X.; Canton, G.; Tian, F.; Du, R.; Ji, S.; Yuan, C.; et al. Differences in Carotid Plaques between Symptomatic Patients With and Without Diabetes Mellitus: A CARE-II Study. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1234–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saremi, A.; Schwenke, D.C.; Buchanan, T.A.; Hodis, H.N.; Mack, W.J.; Banerji, M.; Bray, G.A.; Clement, S.C.; Henry, R.R.; Kitabchi, A.E.; et al. Pioglitazone Slows Progression of Atherosclerosis in Prediabetes Independent of Changes in Cardiovascular Risk Factors. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami-Nishida, S.; Matsumura, T.; Senokuchi, T.; Ishii, N.; Kinoshita, H.; Yamada, S.; Morita, Y.; Nishida, S.; Motoshima, H.; Kondo, T.; et al. Pioglitazone Suppresses Macrophage Proliferation in Apolipoprotein-E Deficient Mice by Activating PPARγ. Atherosclerosis 2019, 286, 30–39. [Google Scholar] [CrossRef]
- Chang, K.; Francis, S.A.; Aikawa, E.; Kohler, R.H.; McCarthy, J.R.; Weissleder, R.; Plutzky, J.; Jaffer, F.A. Pioglitazone Suppresses Inflammation In Vivo In Murine Carotid Atherosclerosis: Novel Detection by Dual-Target Fluorescence Molecular Imaging. Arterioscler. Thromb. Vasc. Biol. 2011, 30, 1933–1939. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.L.; Meibohm, B.; Capparelli, E.V.; Velasquez-Mieyer, P.; Burghen, G.A.; Tamborlane, W.V. Single- and Multiple-Dose Pharmacokinetics of Pioglitazone in Adolescents With Type 2 Diabetes. J. Clin. Pharmacol. 2005, 45, 1137–1144. [Google Scholar] [CrossRef]
- Nakashiro, S.; Matoba, T.; Umezu, R.; Koga, J.; Tokutome, M.; Katsuki, S.; Nakano, K.; Sunagawa, K.; Egashira, K. Pioglitazone-Incorporated Nanoparticles Prevent Plaque Destabilization and Rupture by Regulating Monocyte/Macrophage Differentiation in ApoE−/− Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Cervadoro, A.; Palomba, R.; Vergaro, G.; Cecchi, R.; Menichetti, L.; Decuzzi, P.; Emdin, M.; Luin, S. Targeting Inflammation With Nanosized Drug Delivery Platforms in Cardiovascular Diseases: Immune Cell Modulation in Atherosclerosis. Front. Bioeng. Biotechnol. 2018, 6, 177. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-Based Targeted Drug Delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santi, M.; Maccari, G.; Mereghetti, P.; Voliani, V.; Rocchiccioli, S.; Ucciferri, N.; Luin, S.; Signore, G. Rational Design of a Transferrin-Binding Peptide Sequence Tailored to Targeted Nanoparticle Internalization. Bioconjug. Chem. 2017, 28, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Tapeinos, C.; Battaglini, M.; Ciofani, G. Advances in the Design of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Targeting Brain Diseases. J. Control. Release 2017, 264, 306–332. [Google Scholar] [CrossRef]
- Ghitman, J.; Biru, E.I.; Stan, R.; Iovu, H. Review of Hybrid PLGA Nanoparticles: Future of Smart Drug Delivery and Theranostics Medicine. Mater. Des. 2020, 193, 108805. [Google Scholar] [CrossRef]
- Ahlawat, J.; Henriquez, G.; Narayan, M. Enhancing the Delivery of Chemotherapeutics: Role of Biodegradable Polymeric Nanoparticles. Molecules 2018, 23, 2157. [Google Scholar] [CrossRef] [Green Version]
- Astete, C.E.; Sabliov, C.M. Synthesis and Characterization of PLGA Nanoparticles. J. Biomater. Sci. Polym. Ed. 2006, 17, 247–289. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P. An Overview of Poly(Lactic-Co-Glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Silva-Abreu, M.; Miralles, E.; Kamma-Lorger, C.S.; Espina, M.; García, M.L.; Calpena, A.C. Stabilization by Nano Spray Dryer of Pioglitazone Polymeric Nanosystems: Development, In Vivo, Ex Vivo and Synchrotron Analysis. Pharmaceutics 2021, 13, 1751. [Google Scholar] [CrossRef] [PubMed]
- Zhi, K.; Raji, B.; Nookala, A.R.; Khan, M.M.; Nguyen, X.H.; Sakshi, S.; Pourmotabbed, T.; Yallapu, M.M.; Kochat, H.; Tadrous, E.; et al. PLGA Nanoparticle-Based Formulations to Cross the Blood–Brain Barrier for Drug Delivery: From R&D to CGMP. Pharmaceutics 2021, 13, 500. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, Y.; Sun, Q.; Zhou, C.; Hu, S.; Lenahan, C.; Xu, W.; Deng, Y.; Li, G.; Tao, S. Update on Nanoparticle-Based Drug Delivery System for Anti-Inflammatory Treatment. Front. Bioeng. Biotechnol. 2021, 9, 630352. [Google Scholar] [CrossRef] [PubMed]
- Matoba, T.; Koga, J.; Nakano, K.; Egashira, K.; Tsutsui, H. Nanoparticle-Mediated Drug Delivery System for Atherosclerotic Cardiovascular Disease. J. Cardiol. 2017, 70, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliwal, R.; Babu, R.J.; Palakurthi, S. Nanomedicine Scale-up Technologies: Feasibilities and Challenges. AAPS PharmSciTech 2014, 15, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, E.; Sah, H. Recent Trends in Preparation of Poly(Lactide-co-glycolide) Nanoparticles by Mixing Polymeric Organic Solution with Antisolvent. J. Nanomater. 2015, 2015, 794601. [Google Scholar] [CrossRef] [Green Version]
- Vanderhoff, J.; El-Aasser, M.; Ugelstad, J. Polymer Emulsification Process. U.S. Patent 4177177A, 4 December 1979. [Google Scholar]
- Nava-Arzaluz, M.G.; Pinon-Segundo, E.; Ganem-Rondero, A.; Lechuga-Ballesteros, D. Single Emulsion-Solvent Evaporation Technique and Modifications for the Preparation of Pharmaceutical Polymeric Nanoparticles. Recent Pat. Drug Deliv. Formul. 2012, 6, 209–223. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, J.; Zhao, M.; Tang, S.; Cheng, X.; Zhang, W.; Li, W.; Liu, X.; Peng, H.; Wang, Q. Effects of Polyethylene Glycol on the Surface of Nanoparticles for Targeted Drug Delivery. Nanoscale 2021, 13, 10748–10764. [Google Scholar] [CrossRef]
- Zeng, N.; Hu, Q.; Liu, Z.; Gao, X.; Hu, R.; Song, Q.; Gu, G.; Xia, H.; Yao, L.; Pang, Z.; et al. Preparation and Characterization of Paclitaxel-Loaded DSPE-PEG-Liquid Crystalline Nanoparticles (LCNPs) for Improved Bioavailability. Int. J. Pharm. 2012, 424, 58–66. [Google Scholar] [CrossRef]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule Formation by Interfacial Polymer Deposition Following Solvent Displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Hernández-Giottonini, K.Y.; Rodríguez-Córdova, R.J.; Gutiérrez-Valenzuela, C.A.; Peñuñuri-Miranda, O.; Zavala-Rivera, P.; Guerrero-Germán, P.; Lucero-Acuña, A. PLGA Nanoparticle Preparations by Emulsification and Nanoprecipitation Techniques: Effects of Formulation Parameters. RSC Adv. 2020, 10, 4218–4231. [Google Scholar] [CrossRef] [Green Version]
- Kanemaru, M.; Asai, J.; Jo, J.-I.; Arita, T.; Kawai-Ohnishi, M.; Tsutsumi, M.; Wada, M.; Tabata, Y.; Katoh, N. Nanoparticle-Mediated Local Delivery of Pioglitazone Attenuates Bleomycin-Induced Skin Fibrosis. J. Dermatol. Sci. 2019, 93, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.J.; Kim, J.S.; Huh, K.M.; Cho, K.J.; Lee, Y.K. Preparation and Characterization of Pioglitazone Loaded PLGA Nanospheres for the Treatment of Type 2 Diabetes. Polymer (Korea) 2010, 34, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Laddha, U.D.; Kshirsagar, S.J. Formulation of PPAR-Gamma Agonist as Surface Modified PLGA Nanoparticles for Non-Invasive Treatment of Diabetic Retinopathy: In Vitro and in Vivo Evidences. Heliyon 2020, 6, e04589. [Google Scholar] [CrossRef]
- Lewis, D.R.; Petersen, L.K.; York, A.W.; Zablocki, K.R.; Joseph, L.B.; Kholodovych, V.; Prud’homme, R.K.; Uhrich, K.E.; Moghe, P.V. Sugar-Based Amphiphilic Nanoparticles Arrest Atherosclerosis in Vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 2693–2698. [Google Scholar] [CrossRef] [Green Version]
- Silva-Abreu, M.; Calpena, A.C.; Espina, M.; Silva, A.M.; Gimeno, A.; Egea, M.A.; García, M.L. Optimization, Biopharmaceutical Profile and Therapeutic Efficacy of Pioglitazone-Loaded PLGA-PEG Nanospheres as a Novel Strategy for Ocular Inflammatory Disorders. Pharm Res. 2018, 35, 11. [Google Scholar] [CrossRef] [Green Version]
- Silva-Abreu, M.; Calpena, A.C.; Andrés-Benito, P.; Aso, E.; Romero, I.A.; Roig-Carles, D.; Gromnicova, R.; Espina, M.; Ferrer, I.; García, M.L.; et al. PPARγ Agonist-Loaded PLGA-PEG Nanocarriers as a Potential Treatment for Alzheimer’s Disease: In Vitro and in Vivo Studies. Int. J. Nanomed. 2018, 13, 5577–5590. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-D.; Huang, L. Nanoparticles Evading the Reticuloendothelial System: Role of the Supported Bilayer. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2259–2266. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Rho, S.; Stiles, W.R.; Hu, S.; Baek, Y.; Hwang, D.W.; Kashiwagi, S.; Kim, M.S.; Choi, H.S. Size-Dependent EPR Effect of Polymeric Nanoparticles on Tumor Targeting. Adv. Healthc. Mater. 2020, 9, 1901223. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, C. Tuning the Size of Poly(Lactic-co-glycolic Acid) (PLGA) Nanoparticles Fabricated by Nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Teply, B.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.; Levynissenbaum, E.; Radovicmoreno, A.; Langer, R.; Farokhzad, O. Formulation of Functionalized PLGA–PEG Nanoparticles for in Vivo Targeted Drug Delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Majed, A.; Bakheit, A.H.H.; Abdel Aziz, H.A.; Alharbi, H.; Al-Jenoobi, F.I. Pioglitazone. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 41, pp. 379–438. ISBN 978-0-12-804784-2. [Google Scholar]
- Lee, A.; Di Mascolo, D.; Francardi, M.; Piccardi, F.; Bandiera, T.; Decuzzi, P. Spherical Polymeric Nanoconstructs for Combined Chemotherapeutic and Anti-Inflammatory Therapies. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Galliani, M.; Tremolanti, C.; Signore, G. Nanocarriers for Protein Delivery to the Cytosol: Assessing the Endosomal Escape of Poly(Lactide-Co-Glycolide)-Poly(Ethylene Imine) Nanoparticles. Nanomaterials 2019, 9, 652. [Google Scholar] [CrossRef] [Green Version]
- Galliani, M.; Signore, G. Poly(Lactide-co-glycolide) Nanoparticles Co-Loaded with Chlorophyllin and Quantum Dots as Photodynamic Therapy Agents. ChemPlusChem 2019, 84, 1653–1658. [Google Scholar] [CrossRef]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Haloperidol-Loaded PLGA Nanoparticles: Systematic Study of Particle Size and Drug Content. Int. J. Pharm. 2007, 336, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Martínez Rivas, C.J.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Galindo Rodríguez, S.A.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation Process: From Encapsulation to Drug Delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Lee, J.H.; Yeo, Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic Light Scattering: A Practical Guide and Applications in Biomedical Sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-Drying of Nanoparticles: Formulation, Process and Storage Considerations. Adv. Drug Deliv. Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef]
- Fili, N.; Toseland, C.P. Fluorescence and Labelling: How to Choose and What to Do. In Fluorescent Methods for Molecular Motors; Toseland, C.P., Fili, N., Eds.; Experientia Supplementum; Springer: Basel, Switzerland, 2014; Volume 105, pp. 1–24. ISBN 978-3-0348-0855-2. [Google Scholar]
- Reisch, A.; Klymchenko, A.S. Fluorescent Polymer Nanoparticles Based on Dyes: Seeking Brighter Tools for Bioimaging. Small 2016, 12, 1968–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liet, B.; Laigre, E.; Goyard, D.; Todaro, B.; Tiertant, C.; Boturyn, D.; Berthet, N.; Renaudet, O. Multifunctional Glycoconjugates for Recruiting Natural Antibodies against Cancer Cells. Chem. Eur. J. 2019, 25, 15508–15515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todaro, B.; Achilli, S.; Liet, B.; Laigre, E.; Tiertant, C.; Goyard, D.; Berthet, N.; Renaudet, O. Structural Influence of Antibody Recruiting Glycodendrimers (ARGs) on Antitumoral Cytotoxicity. Biomater. Sci. 2021, 9, 4076–4085. [Google Scholar] [CrossRef] [PubMed]
- Gdowski, A.; Johnson, K.; Shah, S.; Gryczynski, I.; Vishwanatha, J.; Ranjan, A. Optimization and Scale up of Microfluidic Nanolipomer Production Method for Preclinical and Potential Clinical Trials. J. Nanobiotechnol. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Single Emulsification-Solvent Evaporation Method (A) | Nanoprecipitation Method (B) | |
|---|---|---|
| PLGA concentration (mg/mL) | --- | 7.5, 10, 12.5, 15 |
| PLGA* concentration (mg/mL) | 10.3, 14.3, 21.1 | --- |
| PGZ concentration (mg/mL) | 10 | 5, 10, 20 |
| Volume ratio PLGA:PGZ | 7.7:1 | 8:1 |
| Dispersed phase volume (μL) | 228.7 | 225, 425, 525 |
| Dispersant phase volume (μL) | 2062.5 | 600, 800, 1200, 1400 |
| MES concentration (mg/mL) | 97.6, 19.5 | 195.2 |
| pH of dispersant phase | 6.2 | 6.0 |
| PVA concentration (% w/V) | --- | 1, 2, 4 |
| DSPE-PEG(2000) Carboxylic Acid concentration (mg/mL) | 5.2, 7.8, 10.5 | --- |
| Dispersed:dispersant phases Ratio | 1:9 | 1:1.5, 1:1.9, 1:2.66, 1:3.5, 1:5.3, 1:6.2 |
| Stirring rate (rpm) | 400 | 400 |
| Stirring time (min) | 10 | 10 |
| Injection flux rate (mL/h) | 30, 159 | 30 |
| Sonication amplitude (%) | 80, 100 | --- |
| Sonication time (min) | 4.8 | --- |
| Centrifugation speed [rpm (× g)] | 9100 (8000) | 9100 (8000) |
| Centrifugation time (min) | 5 | 5 |
| Cryoprotectant concentration (mg/mL) | 10 | 10 |
| Nanoprecipitation | Emulsion–Solvent Evaporation | |||||||
|---|---|---|---|---|---|---|---|---|
| Reagent | Cost/Unit | Quantities Used for the Synthesis | Costs (€) | Quantities Used for the Synthesis | Costs (€) | |||
| DMF | 155.- | €/1 L | 25.0 | µL | 0.004 | 55.1 | µL | 0.0085 |
| MilliQ water | 13.6 | €/1 L | 2.60 | mL | 0.0354 | 11.9 | mL | 0.0054 |
| PVA (4%) | 211.- | €/500 g | 48.0 | mg | 0.020 | |||
| MES (1 M) | 501.- | €/500 g | 234 | mg | 0.235 | |||
| MES (0.1 M) | 501.- | €/500 g | 224 | mg | 0.225 | |||
| PGZ | 34.1 | €/50 g | 0.250 | mg | 0.0002 | 0.551 | mg | 0.00038 |
| Trehalose (10 mg/mL) | 472.- | €/250 g | 2.00 | mg | 0.004 | 3.84 | mg | 0.0072 |
| PLGA | 238.- | €/5 g | 2.00 | mg | 0.095 | |||
| PLGA* | 225.- | €/5 g | 3.95 | mg | 0.178 | |||
| Acetone | 47.4 | €/1 L | 200 | µL | 0.0095 | |||
| Chloroform | 107.- | €/1 L | 384 | µL | 0.041 | |||
| DSPE-PEG | 268.- | €/50 mg | 1.26 | mg | 6.75 | |||
| Ethanol | 39.6 | €/500 mL | 120 | µL | 0.0095 | |||
| Sum (in Euro) | 0.403 | Sum (in Euro) | 7.22 | |||||
| Method | Dispersed Phase Conditions | Dispersant Phase Conditions | Other Synthetic Parameters | Size (nm) | PDI | ζp (mV) | EE (%) | DL (%) | Cost for Encapsulating 0.145 mg PGZ (€) |
|---|---|---|---|---|---|---|---|---|---|
| Single emulsification-solvent evaporation (A) | 200.0 μL PLGA* (10.3 mg/mL) + 28.7 μL Pio (10.0 mg/mL) | 62.5 μL DSPE-PEG (10.5 mg/mL) + 2.0 mL MES (0.1 M, pH: 6.2) | 159.0 mL/h injection flux rate, sonication time of 4.8 min with 80.0% of amplitude | 262 ± 46 | 0.23 ± 0.05 | −43.2 ± 3.1 | 26 ± 7 | 2.27 ± 0.07 | 7.22 |
| Nanoprecipitation (B) | 200.0 μL PLGA (10.0 mg/mL) + 25.0 μL Pio (10.0 mg/mL) | 400.0 μL PVA (4.0%) + 400.0 μL MES (1.0 M, pH: 6.0) | 30.0 mL/h injection flux rate | 226 ± 22 | 0.19 ± 0.06 | −11.6 ± 0.5 | 58 ± 8 | 4.01 ± 0.27 | 0.403 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todaro, B.; Moscardini, A.; Luin, S. Pioglitazone-Loaded PLGA Nanoparticles: Towards the Most Reliable Synthesis Method. Int. J. Mol. Sci. 2022, 23, 2522. https://doi.org/10.3390/ijms23052522
Todaro B, Moscardini A, Luin S. Pioglitazone-Loaded PLGA Nanoparticles: Towards the Most Reliable Synthesis Method. International Journal of Molecular Sciences. 2022; 23(5):2522. https://doi.org/10.3390/ijms23052522
Chicago/Turabian StyleTodaro, Biagio, Aldo Moscardini, and Stefano Luin. 2022. "Pioglitazone-Loaded PLGA Nanoparticles: Towards the Most Reliable Synthesis Method" International Journal of Molecular Sciences 23, no. 5: 2522. https://doi.org/10.3390/ijms23052522
APA StyleTodaro, B., Moscardini, A., & Luin, S. (2022). Pioglitazone-Loaded PLGA Nanoparticles: Towards the Most Reliable Synthesis Method. International Journal of Molecular Sciences, 23(5), 2522. https://doi.org/10.3390/ijms23052522