
Challenges of CRISPR-Based Gene Editing in Primary T Cells




Abstract
:1. Introduction
2. CRISPR/Cas System Origin, Structure, and Molecular Mechanisms of Genome Editing
3. Practical Aspects of CRISPR/Cas9 Gene Editing on T Cells
3.1. Culturing of T Cells for Genome Editing
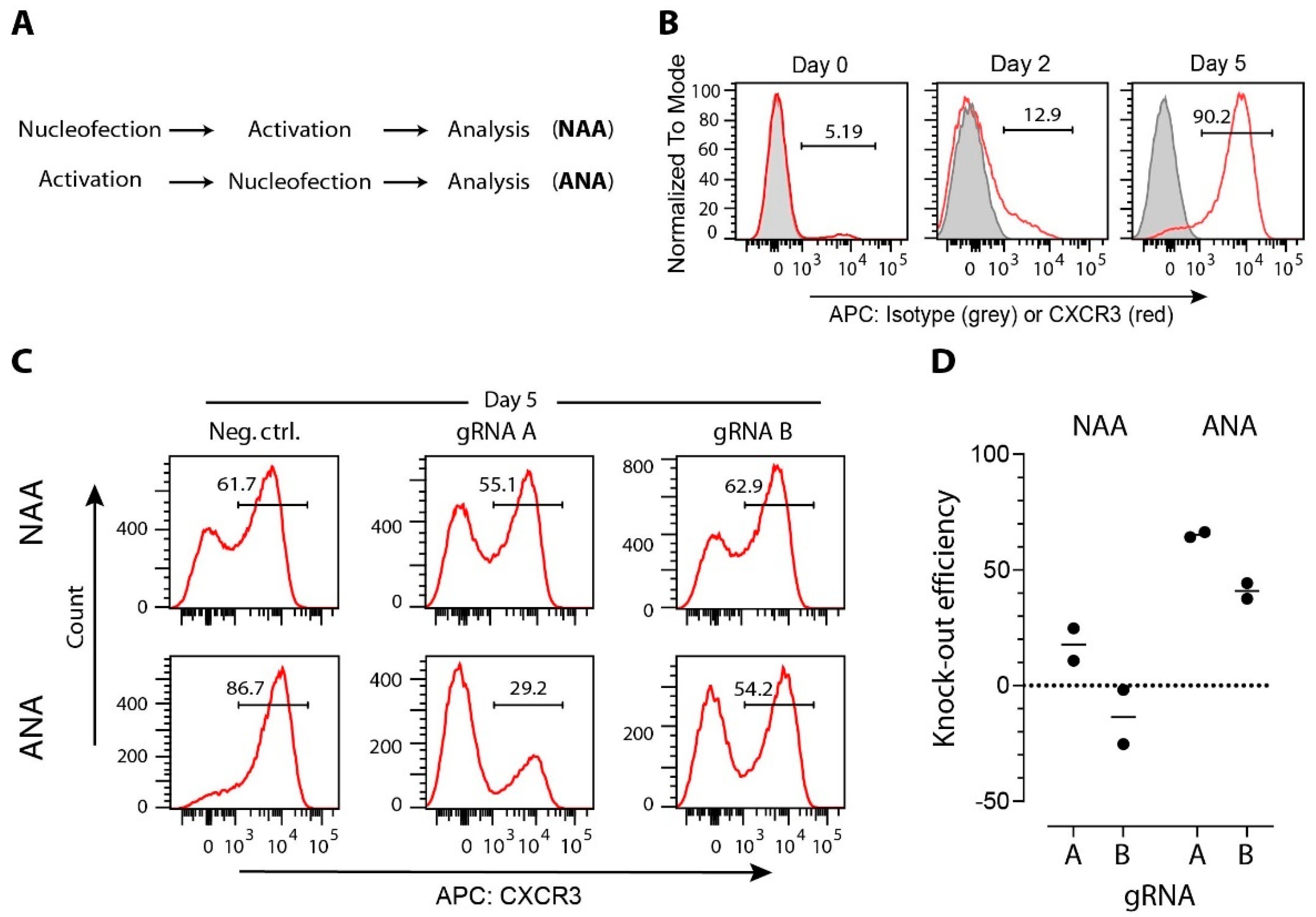
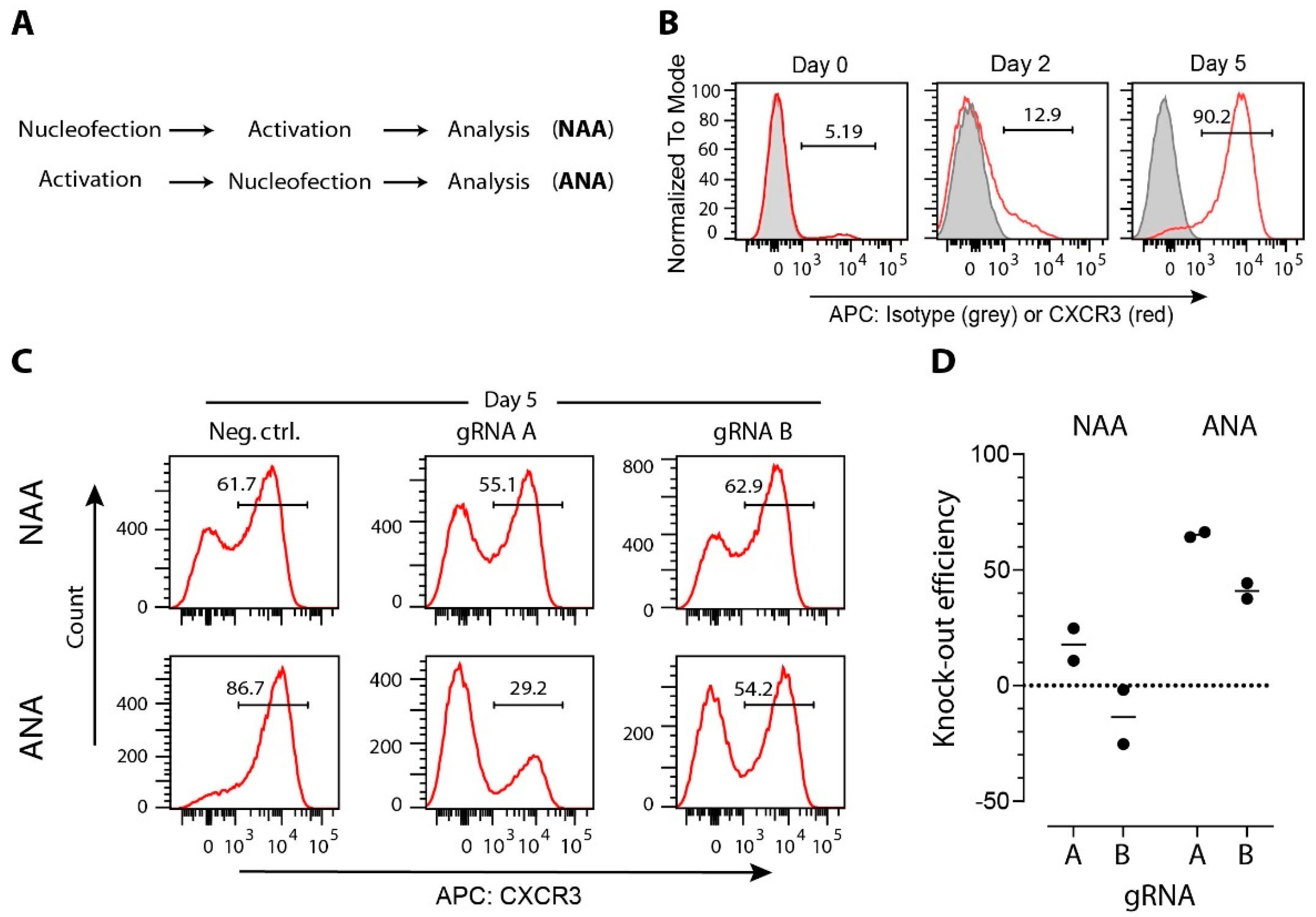
3.2. gRNA Selection
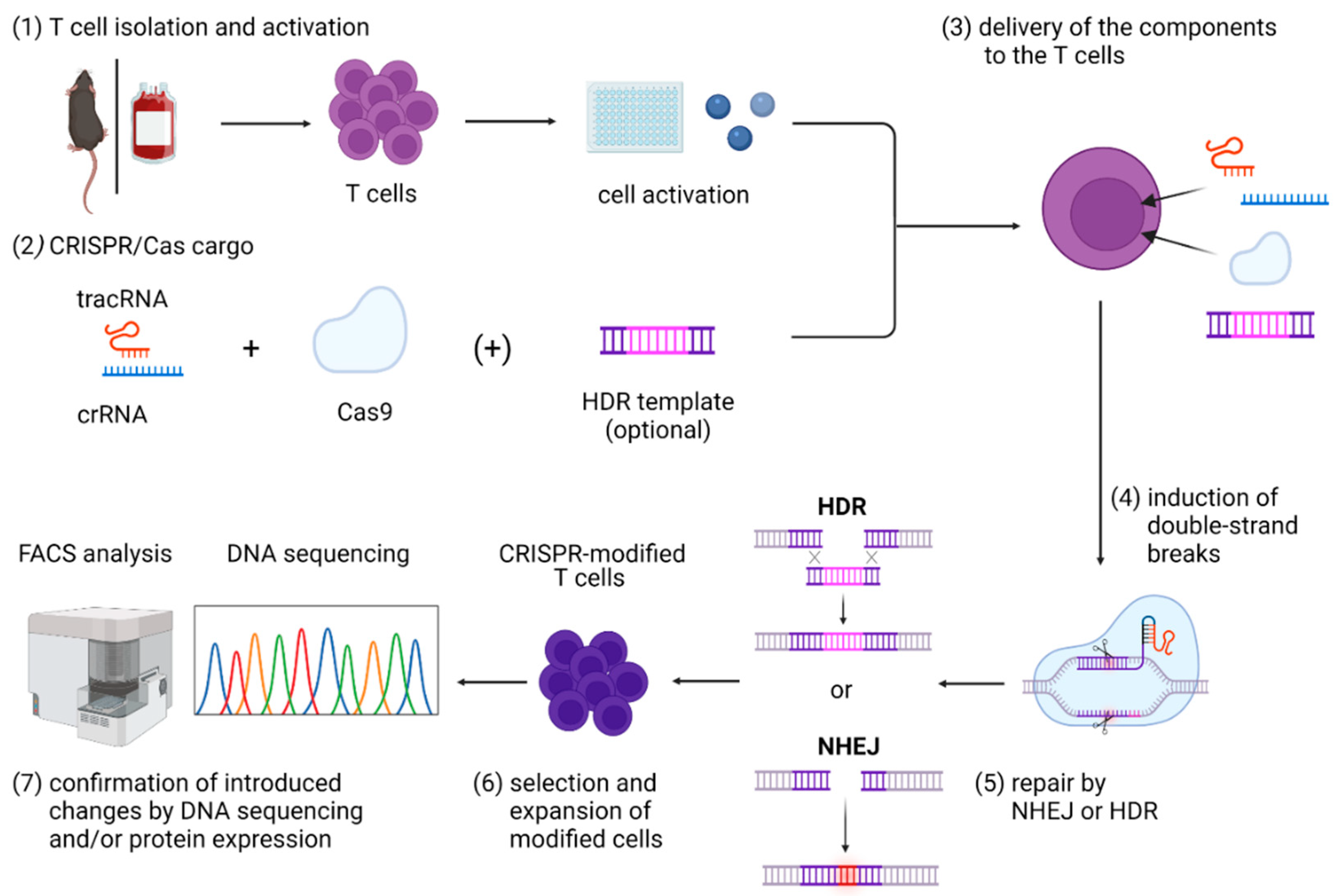
3.3. CRISPR/Cas Cargo
3.4. Strategies for Delivery of CRISPR/Cas Cargo to T Cells
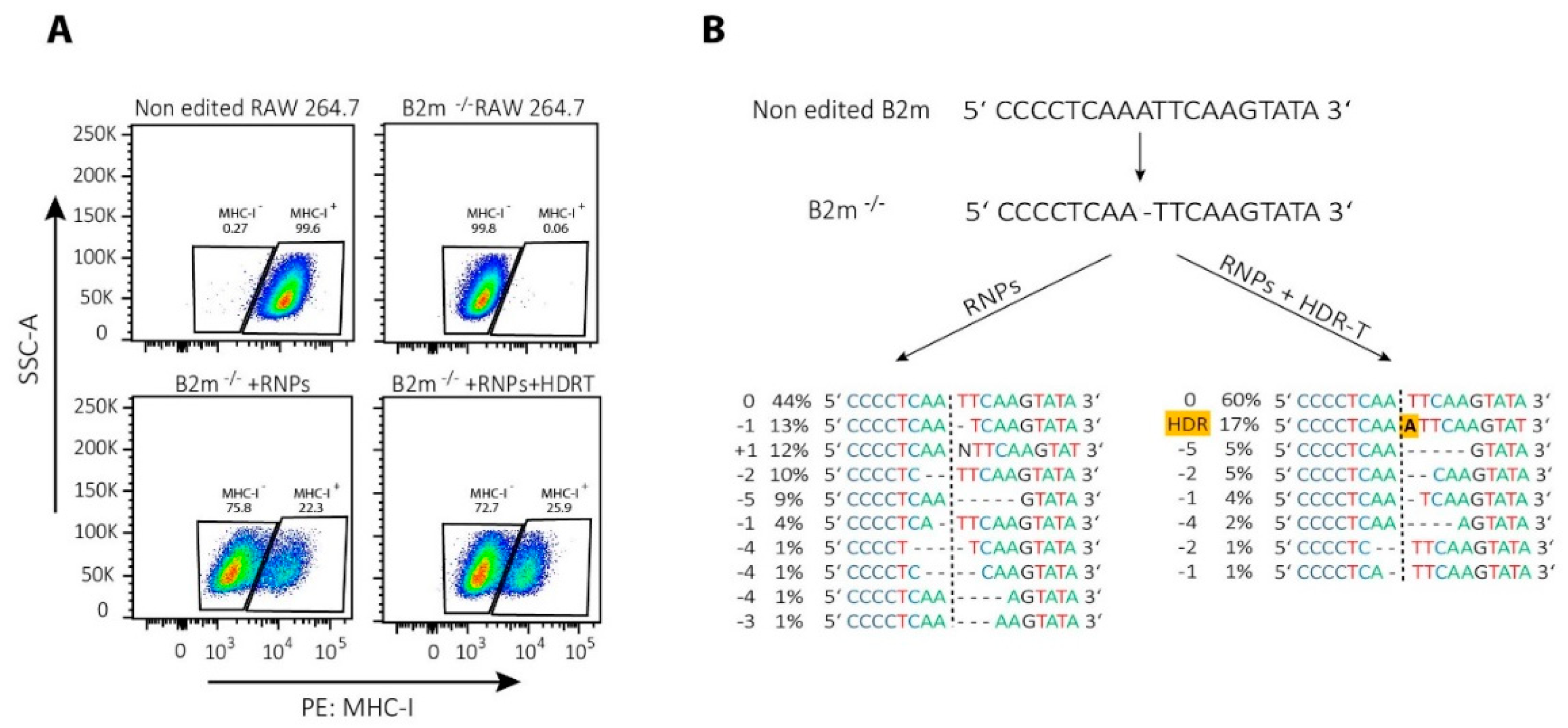
4. Gene Modifications in T Cells by NHEJ-Mediated Repair of CRISPR/Cas9-Induced DSBs
5. Gene Knock-in Strategies in T Cells, Using HDR-Mediated Repair of CRISPR/Cas9-Induced DSBs
5.1. Selection of Appropriate gRNAs for HDR-Mediated Repair
5.2. HDR Template Type
5.3. Design of HDR Template “Homology Arms”
5.4. Inhibition of HDR Template Toxic Effects
5.5. Promoting HDR over NHEJ
5.6. Practical Considerations during Performing HDR-Mediated CRISPR/Cas9 Gene-Editing Experiments
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Linares, L.; Sanclemente, G.; Cervera, C.; Hoyo, I.; Cofán, F.; Ricart, M.J.; Pérez-Villa, F.; Navasa, M.; Marcos, M.A.; Antón, A.; et al. Influence of cytomegalovirus disease in outcome of solid organ transplant patients. Transplant. Proc. 2011, 43, 2145–2148. [Google Scholar] [CrossRef]
- Beam, E.; Razonable, R.R. Cytomegalovirus in solid organ transplantation: Epidemiology, prevention, and treatment. Curr. Infect. Dis. Rep. 2012, 14, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R.; Humar, A. AST Infectious Diseases Community of Practice Cytomegalovirus in solid organ transplantation. Am. J. Transplant. 2013, 13 (Suppl. 4), 93–106. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, S.; Bayatipoor, H.; Pashangzadeh, S.; Jafarpour, R.; Shojaei, Z.; Motallebnezhad, M. Immune checkpoints and cancer development: Therapeutic implications and future directions. Pathol. Res. Pract. 2021, 223, 153485. [Google Scholar] [CrossRef]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Löffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.-G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef] [Green Version]
- Cobbold, M.; Khan, N.; Pourgheysari, B.; Tauro, S.; McDonald, D.; Osman, H.; Assenmacher, M.; Billingham, L.; Steward, C.; Crawley, C.; et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J. Exp. Med. 2005, 202, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mohty, M.; Or, R.; Maschan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peggs, K.S.; Thomson, K.; Samuel, E.; Dyer, G.; Armoogum, J.; Chakraverty, R.; Pang, K.; Mackinnon, S.; Lowdell, M.W. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin. Infect. Dis. 2011, 52, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.C.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.K.; Bowman, L.C.; Krance, R.A.; Brenner, M.K.; et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood 1998, 92, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.P.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Annesley, C.E.; Summers, C.; Ceppi, F.; Gardner, R.A. The Evolution and Future of CAR T Cells for B-Cell Acute Lymphoblastic Leukemia. Clin. Pharmacol. Ther. 2018, 103, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Lulla, P.D.; Hill, L.C.; Ramos, C.A.; Heslop, H.E. The use of chimeric antigen receptor T cells in patients with non-Hodgkin lymphoma. Clin. Adv. Hematol. Oncol. 2018, 16, 375–386. [Google Scholar]
- Hay, K.A.; Turtle, C.J. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs 2017, 77, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, M.L.; Sadelain, M. Biology and clinical application of CAR T cells for B cell malignancies. Int. J. Hematol. 2016, 104, 6–17. [Google Scholar] [CrossRef]
- Harris, E.; Elmer, J.J. Optimization of electroporation and other non-viral gene delivery strategies for T cells. Biotechnol. Prog. 2021, 37, e3066. [Google Scholar] [CrossRef]
- Xu, C.L.; Ruan, M.Z.C.; Mahajan, V.B.; Tsang, S.H. Viral Delivery Systems for CRISPR. Viruses 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, P.E.; Almeida, J.S.; Murphy, P.N.; Moreira, J.L.; Carrondo, M.J.T. Modeling retrovirus production for gene therapy. 1. Determination Of optimal bioreaction mode and harvest strategy. Biotechnol. Prog. 2000, 16, 213–221. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Verhoeyen, E.; Costa, C.; Cosset, F.-L. Lentiviral vector gene transfer into human T cells. Methods Mol. Biol. 2009, 506, 97–114. [Google Scholar] [CrossRef]
- Gill, S.; June, C.H. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol. Rev. 2015, 263, 68–89. [Google Scholar] [CrossRef]
- Carroll, D. Focus: Genome Editing: Genome Editing: Past, Present, and Future. Yale J. Biol. Med. 2017, 90, 653. [Google Scholar]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.G.; Chandrasegaran, S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.; McLachlan, A.D.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Pabo, C.O.; Peisach, E.; Grant, R.A. Design and selection of novel Cys2His2 zinc finger proteins. Annu. Rev. Biochem. 2001, 70, 313–340. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.H.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Mussolino, C.; Morbitzer, R.; Lütge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.-Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Alzubi, J.; Lock, D.; Rhiel, M.; Schmitz, S.; Wild, S.; Mussolino, C.; Hildenbeutel, M.; Brandes, C.; Rositzka, J.; Lennartz, S.; et al. Automated generation of gene-edited CAR T cells at clinical scale. Mol. Ther. Methods Clin. Dev. 2021, 20, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR—A widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef]
- Van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Nozawa, T.; Furukawa, N.; Aikawa, C.; Watanabe, T.; Haobam, B.; Kurokawa, K.; Maruyama, F.; Nakagawa, I. CRISPR inhibition of prophage acquisition in Streptococcus pyogenes. PLoS ONE 2011, 6, e19543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasiunas, G.; Siksnys, V. RNA-dependent DNA endonuclease Cas9 of the CRISPR system: Holy Grail of genome editing? Trends Microbiol. 2013, 21, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Han, M.; Romanienko, P.J.; Jasin, M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5172–5177. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, W.; Chen, Y.; Guo, W.; Zhang, J.; Tang, H.; Xu, Z.; Zhang, H.; Tao, Y.; Wang, F.; et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 2016, 23, 1765–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Feng, Y.L.; Xiao, J.J.; Liu, Q.; Sun, X.N.; Xiang, J.F.; Kong, N.; Liu, S.C.; Chen, G.Q.; Wang, Y.; et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stope, M.B. Phosphorylation of histone H2A.X as a DNA-associated biomarker (Review). World Acad. Sci. J. 2021, 3, 1–5. [Google Scholar] [CrossRef]
- Marini, F.; Rawal, C.C.; Liberi, G.; Pellicioli, A. Regulation of DNA Double Strand Breaks Processing: Focus on Barriers. Front. Mol. Biosci. 2019, 6, 55. [Google Scholar] [CrossRef]
- Renkawitz, J.; Lademann, C.A.; Jentsch, S. Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 2014, 15, 369–383. [Google Scholar] [CrossRef]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Pastwa, E.; Błasiak, J. Non-homologous DNA end joining. Acta Biochim. Pol. 2003, 50, 891–908. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Carballar, R.; Martínez-Láinez, J.M.; Samper, B.; Bru, S.; Bállega, E.; Mirallas, O.; Ricco, N.; Clotet, J.; Jiménez, J. CDK-mediated Yku80 Phosphorylation Regulates the Balance Between Non-homologous End Joining (NHEJ) and Homologous Directed Recombination (HDR). J. Mol. Biol. 2020, 432, 166715. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef]
- Buis, J.; Stoneham, T.; Spehalski, E.; Ferguson, D.O. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012, 19, 246–253. [Google Scholar] [CrossRef]
- Huertas, P.; Jackason, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Gagné, J.-P.; Genois, M.-M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.-Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 2015, 17, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Farkash, E.A.; Gao, W.; Thompson, C.B. IL-7 Enhances the Survival and Maintains the Size of Naive T Cells. J. Immunol. 2001, 167, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Rutz, S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef]
- Nüssing, S.; House, I.G.; Kearney, C.J.; Chen, A.X.Y.; Vervoort, S.J.; Beavis, P.A.; Oliaro, J.; Johnstone, R.W.; Trapani, J.A.; Parish, I.A. Efficient CRISPR/Cas9 Gene Editing in Uncultured Naive Mouse T Cells for In Vivo Studies. J. Immunol. 2020, 204, 2308–2315. [Google Scholar] [CrossRef]
- Majumder, S.; Jugovic, I.; Saul, D.; Bell, L.; Hundhausen, N.; Seal, R.; Beilhack, A.; Rosenwald, A.; Mougiakakos, D.; Berberich-Siebelt, F. Rapid and Efficient Gene Editing for Direct Transplantation of Naive Murine Cas9+ T Cells. Front. Immunol. 2021, 12, 683631. [Google Scholar] [CrossRef]
- Verkuijl, S.A.; Rots, M.G. The influence of eukaryotic chromatin state on CRISPR–Cas9 editing efficiencies. Curr. Opin. Biotechnol. 2019, 55, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowicz, P.; Chan, H.; Medina, D.; Gumpelmair, S.; Kjelstrup, H.; Spurkland, A. A simple and efficient workflow for generation of knock-in mutations in Jurkat T cells using CRISPR/Cas9. Scand. J. Immunol. 2020, 91, e12862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for improving HDR efficiency. Front. Genet. 2019, 10, 691. [Google Scholar] [CrossRef]
- Ghaffari, S.; Torabi-Rahvar, M.; Omidkhoda, A.; Ahmadbeigi, N. Impact of various culture conditions on ex vivo expansion of polyclonal T cells for adoptive immunotherapy. APMIS 2019, 127, 737–745. [Google Scholar] [CrossRef]
- Bere, A.; Denny, L.; Hanekom, W.; Burgers, W.A.; Passmore, J.A.S. Comparison of polyclonal expansion methods to improve the recovery of cervical cytobrush-derived T cells from the female genital tract of HIV-infected women. J. Immunol. Methods 2010, 354, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.J.; Lin, D.T.S.; Gillies, J.K.; Uday, P.; Pesenacker, A.M.; Kobor, M.S.; Levings, M.K. Optimized CRISPR-mediated gene knockin reveals FOXP3-independent maintenance of human Treg identity. Cell Rep. 2021, 36, 109494. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, N.; Cao, W.; Huang, L.; Zhou, J.; Sheng, L. Influence of various medium environment to in vitro human T cell culture. Vitr. Cell. Dev. Biol.-Anim. 2018, 54, 559–566. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- Fu, Y.W.; Dai, X.Y.; Wang, W.T.; Yang, Z.X.; Zhao, J.J.; Zhang, J.P.; Wen, W.; Zhang, F.; Oberg, K.C.; Zhang, L.; et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021, 49, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Kath, J.; Du, W.; Thommandru, B.; Turk, R.; Amini, L.; Stein, M.; Zittel, T.; Martini, S.; Ostendorf, L.; Wilhelm, A.; et al. Fast, Efficient and Virus-Free Generation of TRAC-Replaced CAR T Cells. SSRN Electron. J. 2021, 47. [Google Scholar] [CrossRef]
- Ghaffari, S.; Torabi-Rahvar, M.; Aghayan, S.; Jabbarpour, Z.; Moradzadeh, K.; Omidkhoda, A.; Ahmadbeigi, N. Optimizing interleukin-2 concentration, seeding density and bead-to-cell ratio of T-cell expansion for adoptive immunotherapy. BMC Immunol. 2021, 22, 43. [Google Scholar] [CrossRef]
- Raulf, M. T cell: Primary culture from peripheral blood. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2019. [Google Scholar]
- Wang, W.; Ai, X. Primary culture of immature, naïve mouse CD4+ T cells. STAR Protoc. 2021, 2, 100756. [Google Scholar] [CrossRef] [PubMed]
- Martens, R.; Permanyer, M.; Werth, K.; Yu, K.; Braun, A.; Halle, O.; Halle, S.; Patzer, G.E.; Bošnjak, B.; Kiefer, F.; et al. Efficient homing of T cells via afferent lymphatics requires mechanical arrest and integrin-supported chemokine guidance. Nat. Commun. 2020, 11, 1114. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Zhang, T. Computational approaches for effective CRISPR guide RNA design and evaluation. Comput. Struct. Biotechnol. J. 2020, 18, 35–44. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, J.; Cheng, M.; Liao, X.; Peng, S. Review of CRISPR/Cas9 sgRNA Design Tools. Interdiscip. Sci. 2018, 10, 455–465. [Google Scholar] [CrossRef]
- Huang, M.C.; Cheong, W.C.; Lim, L.S.; Li, M.-H. A simple, high sensitivity mutation screening using Ampligase mediated T7 endonuclease I and Surveyor nuclease with microfluidic capillary electrophoresis. Electrophoresis 2012, 33, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Vakulskas, C.A.; Behlke, M.A. Evaluation and Reduction of CRISPR Off-Target Cleavage Events. Nucleic Acid Ther. 2019, 29, 167–174. [Google Scholar] [CrossRef]
- DeBruin, K.A.; Krassowska, W. Modeling electroporation in a single cell. I. Effects Of field strength and rest potential. Biophys. J. 1999, 77, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.-D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302–315.e23. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl. 2017, 56, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michieletto, D.; Lusic, M.; Marenduzzo, D.; Orlandini, E. Physical principles of retroviral integration in the human genome. Nat. Commun. 2019, 10, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.N.; Roth, T.L.; Li, P.J.; Chen, P.A.; Apathy, R.; Mamedov, M.R.; Vo, L.T.; Tobin, V.R.; Goodman, D.; Shifrut, E.; et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020, 38, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.S.; Thommandru, B.; Woodley, J.; Turk, R.; Yan, S.; Kurgan, G.; McNeill, M.S.; Rettig, G.R. Optimized design parameters for CRISPR Cas9 and Cas12a homology-directed repair. Sci. Rep. 2021, 11, 19482. [Google Scholar] [CrossRef] [PubMed]
- Kotowski, M.; Sharma, S. CRISPR-Based Editing Techniques for Genetic Manipulation of Primary T Cells. Methods Protoc. 2020, 3, 79. [Google Scholar] [CrossRef]
- Atsavapranee, E.S.; Billingsley, M.M.; Mitchell, M.J. Delivery technologies for T cell gene editing: Applications in cancer immunotherapy. EBioMedicine 2021, 67, 103354. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Arai, Y.; Yamazaki, M.; Morita, S.; Kimura, M.; Itoh, M.; Abe, Y.; Hatada, I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 2014, 4, 4513. [Google Scholar] [CrossRef]
- Raveux, A.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. Optimization of the production of knock-in alleles by CRISPR/Cas9 microinjection into the mouse zygote. Sci. Rep. 2017, 7, 42661. [Google Scholar] [CrossRef]
- Rahimmanesh, I.; Totonchi, M.; Khanahmad, H. The challenging nature of primary T lymphocytes for transfection: Effect of protamine sulfate on the transfection efficiency of chemical transfection reagents. Res. Pharm. Sci. 2020, 15, 437–446. [Google Scholar] [CrossRef]
- Bošnjak, B.; Permanyer, M.; Sethi, M.K.; Galla, M.; Maetzig, T.; Heinemann, D.; Willenzon, S.; Förster, R.; Heisterkamp, A.; Kalies, S. CRISPR/Cas9 Genome Editing Using Gold-Nanoparticle-Mediated Laserporation. Adv. Biosyst. 2018, 2, 1700184. [Google Scholar] [CrossRef]
- Boukany, P.E.; Morss, A.; Liao, W.-C.; Henslee, B.; Jung, H.; Zhang, X.; Yu, B.; Wang, X.; Wu, Y.; Li, L.; et al. Nanochannel electroporation delivers precise amounts of biomolecules into living cells. Nat. Nanotechnol. 2011, 6, 747–754. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Electroporation. Cold Spring Harb. Protoc. 2019, 2019, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Schober, K.; Müller, T.R.; Gökmen, F.; Grassmann, S.; Effenberger, M.; Poltorak, M.; Stemberger, C.; Schumann, K.; Roth, T.L.; Marson, A.; et al. Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019, 3, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Ibarra, G.S.R.; Hubbard, N.; Einhaus, T.; Astrakhan, A.; Rawlings, D.J.; Kiem, H.-P.; Peterson, C.W. Efficient Enrichment of Gene-Modified Primary T Cells via CCR5-Targeted Integration of Mutant Dihydrofolate Reductase. Mol. Ther. Methods Clin. Dev. 2018, 9, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, T.L.; Li, P.J.; Blaeschke, F.; Nies, J.F.; Apathy, R.; Mowery, C.; Yu, R.; Nguyen, M.L.T.; Lee, Y.; Truong, A.; et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 2020, 181, 728–744.e21. [Google Scholar] [CrossRef]
- Durán, V.; Grabski, E.; Hozsa, C.; Becker, J.; Yasar, H.; Monteiro, J.T.; Costa, B.; Koller, N.; Lueder, Y.; Wiegmann, B.; et al. Fucosylated lipid nanocarriers loaded with antibiotics efficiently inhibit mycobacterial propagation in human myeloid cells. J. Control. Release 2021, 334, 201–212. [Google Scholar] [CrossRef]
- Chicaybam, L.; Sodre, A.L.; Curzio, B.A.; Bonamino, M.H. An Efficient Low Cost Method for Gene Transfer to T Lymphocytes. PLoS ONE 2013, 8, e60298. [Google Scholar] [CrossRef]
- Zhang, M.; Ma, Z.; Selliah, N.; Weiss, G.; Genin, A.; Finkel, T.H.; Cron, R.Q. The impact of Nucleofection® on the activation state of primary human CD4 T cells. J. Immunol. Methods 2014, 408, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiTommaso, T.; Cole, J.M.; Cassereau, L.; Buggé, J.A.; Hanson, J.L.S.; Bridgen, D.T.; Stokes, B.D.; Loughhead, S.M.; Beutel, B.A.; Gilbert, J.B.; et al. Cell engineering with microfluidic squeezing preserves functionality of primary immune cells in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E10907–E10914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Astolfo, D.S.; Pagliero, R.J.; Pras, A.; Karthaus, W.R.; Clevers, H.; Prasad, V.; Lebbink, R.J.; Rehmann, H.; Geijsen, N. Efficient intracellular delivery of native proteins. Cell 2015, 161, 674–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholosy, W.M.; Visscher, M.; Ogink, K.; Buttstedt, H.; Griffin, K.; Beier, A.; Gerlach, J.P.; Molenaar, J.J.; Geijsen, N.; de Boer, M.; et al. Simple, fast and efficient iTOP-mediated delivery of CRISPR/Cas9 RNP in difficult-to-transduce human cells including primary T cells. J. Biotechnol. 2021, 338, 71–80. [Google Scholar] [CrossRef]
- Agrotis, A.; Ketteler, R. A new age in functional genomics using CRISPR/Cas9 in arrayed library screening. Front. Genet. 2015, 6, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, W.; Jiang, Y.; Boettcher, M.; Ding, K.; Mollenauer, M.; Liu, Z.; Wen, X.; Liu, C.; Hao, P.; Zhao, S.; et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc. Natl. Acad. Sci. USA 2018, 115, E4051–E4060. [Google Scholar] [CrossRef] [Green Version]
- Cortez, J.T.; Montauti, E.; Shifrut, E.; Gatchalian, J.; Zhang, Y.; Shaked, O.; Xu, Y.; Roth, T.L.; Simeonov, D.R.; Zhang, Y.; et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 2020, 582, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Arai, E.; Khan, O.; Zhang, Z.; Ngiow, S.F.; He, Y.; Huang, H.; Manne, S.; Cao, Z.; Baxter, A.E.; et al. In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 2021, 184, 1262–1280. [Google Scholar] [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef]
- Konishi, J.; Yamazaki, K.; Azuma, M.; Kinoshita, I.; Dosaka-Akita, H.; Nishimura, M. B7-H1 Expression on Non-Small Cell Lung Cancer Cells and Its Relationship with Tumor-Infiltrating Lymphocytes and Their PD-1 Expression. Clin. Cancer Res. 2004, 10, 5094–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Shi, L.; Zhang, W.; Han, J.; Zhang, S.; Fu, Z.; Cai, J. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget 2018, 9, 5208–5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Zou, Z.; Chen, F.; Ding, N.; Du, J.; Shao, J.; Li, L.; Fu, Y.; Hu, B.; Yang, Y.; et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology 2017, 6, e1249558. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Terawaki, S.; Ikegawa, M.; Okazaki, T.; Honjo, T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med. 2003, 198, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Amini, L.; Wagner, D.L.; Rössler, U.; Zarrinrad, G.; Wagner, L.F.; Vollmer, T.; Wendering, D.J.; Kornak, U.; Volk, H.D.; Reinke, P.; et al. CRISPR-Cas9-Edited Tacrolimus-Resistant Antiviral T Cells for Advanced Adoptive Immunotherapy in Transplant Recipients. Mol. Ther. 2021, 29, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.-Y.; Kim, Y.-Y.; Yu, H.-S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Mølck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef] [PubMed]
- Kamali, E.; Rahbarizadeh, F.; Hojati, Z.; Frödin, M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Van Overbeek, M.; Capurso, D.; Carter, M.M.; Thompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B.; et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollanoori, H.; Teimourian, S. Therapeutic applications of CRISPR/Cas9 system in gene therapy. Biotechnol. Lett. 2018, 40, 907–914. [Google Scholar] [CrossRef]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, Y.; Kang, X.; Lin, B.; Yu, Q.; Song, B.; Gao, G.; Chen, Y.; Sun, X.; Li, X.; et al. One-Step Biallelic and Scarless Correction of a β-Thalassemia Mutation in Patient-Specific iPSCs without Drug Selection. Mol. Ther. Nucleic Acids 2017, 6, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Park, C.-Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.-H.; Kim, D.-W.; Kim, J.-S. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Jafari, H.; Hesami, S.; Safi, M.; Ghasemi, F.; Banan, M. Expression and hydroxyurea-triggered induction of EGFP upon CRISPR/Cas9-mediated integration into the γ-globin gene of K562 cells. Biotechnol. Lett. 2019, 41, 691–700. [Google Scholar] [CrossRef]
- Wiebking, V.; Lee, C.M.; Mostrel, N.; Lahiri, P.; Bak, R.; Bao, G.; Roncarolo, M.G.; Bertaina, A.; Porteus, M.H. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica 2021, 106, 847–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatiossian, K.J.; Clark, R.D.E.; Huang, C.; Thornton, M.E.; Grubbs, B.H.; Cannon, P.M. Rational Selection of CRISPR-Cas9 Guide RNAs for Homology-Directed Genome Editing. Mol. Ther. 2021, 29, 1057–1069. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- O’Brien, A.R.; Wilson, L.O.W.; Burgio, G.; Bauer, D.C. Unlocking HDR-mediated nucleotide editing by identifying high-efficiency target sites using machine learning. Sci. Rep. 2019, 9, 2788. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Amaishi, Y.; Maki, I.; Enoki, T.; Mineno, J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019, 9, 4811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, K.I.; Sutrisnoh, N.A.B.; Srinivasan, H.; Zhang, J.; Li, J.; Zhang, F.; Lalith, C.R.J.; Xing, H.; Shanmugam, R.; et al. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biol. 2018, 19, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; Quan, Z.J.; Li, S.A.; Yang, Z.X.; Fu, Y.W.; Zhang, F.; Li, G.H.; Zhao, M.; Yin, M.-D.; Xu, J.; et al. Effective control of large deletions after double-strand breaks by homology-directed repair and dsODN insertion. Genome Biol. 2021, 22, 236. [Google Scholar] [CrossRef]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Mintier, G.; Ma-Edmonds, M.; Storton, D.; Wang, X.; Xiao, X.; Kienzle, B.; Zhao, D.; Feder, J.N. ‘Cold shock’ increases the frequency of homology directed repair gene editing in induced pluripotent stem cells. Sci. Rep. 2018, 8, 2080. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
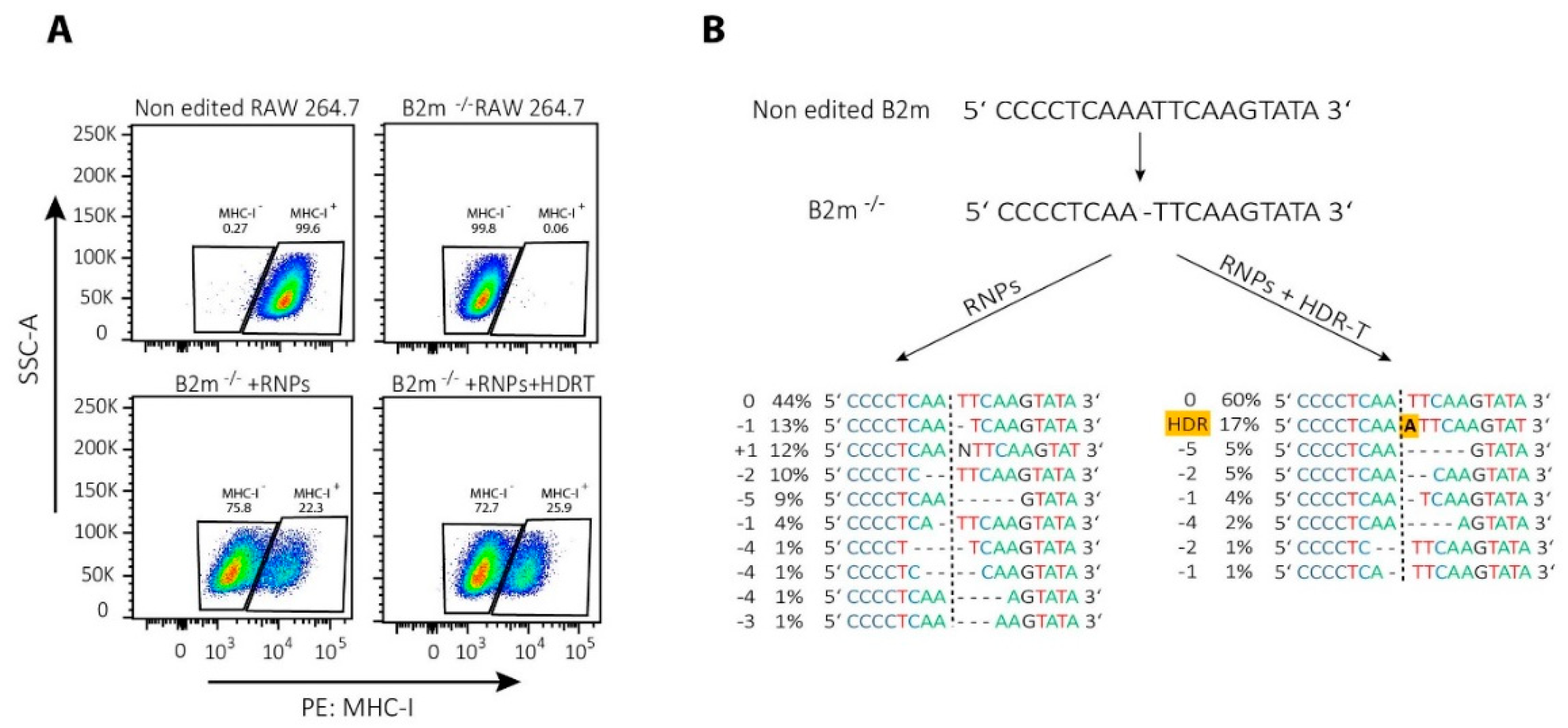{kind=link}
| ClinicalTrials.gov Identifier | Official Study Title | Indication of Treatment | Target of Genome Editing | Study Phase |
|---|---|---|---|---|
| NCT04037566 | “A Safety Study of Autologous T Cells Engineered to Target CD19 and CRISPR Gene Edited to Eliminate Endogenous HPK1 (XYF19 CAR-T Cells) for Relapsed or Refractory Haematopoietic Malignancies” | Relapsed or refractory CD19+ leukemia or lymphoma | XYF19 CAR | Phase 1 |
| NCT03545815 | “Phase I Study to Evaluate Treatment of CRISPR-Cas9 Mediated PD-1 and TCR Gene -knocked Out Chimeric Antigen Receptor (CAR) T Cells in Patients with Mesothelin Positive Multiple Solid Tumors” | Solid tumors | KO of PD-1 and TCR | Phase 1 |
| NCT04502446 | “A Phase 1, Open-Label, Multicenter, Dose Escalation and Cohort Expansion Study of the Safety and Efficacy of Anti-CD70 Allogeneic CRISPR-Cas9-Engineered T Cells (CTX130) in Subjects with Relapsed or Refractory T or B Cell Malignancies” | Relapsed or refractory T- or B-cell malignancies | CTX130 CAR | Phase 1 |
| NCT04637763 | “A Phase 1, Multicenter, Open-Label Study of CB-010, a CRISPR-Edited Allogeneic Anti-CD19 CAR-T Cell Therapy in Patients with Relapsed/Refractory B Cell Non-Hodgkin Lymphoma (ANTLER)” | Relapsed or refractory B-cell non-Hodgkin lymphoma | CB-010 CAR | Phase 1 |
| NCT03398967 | “Phase I/II Study to Evaluate Treatment of Relapsed or Refractory Leukemia and Lymphoma with Universal CRISPR-Cas9 Gene-Editing CAR-T Cells Targeting CD19 and CD20 or CD22” | B-cell leukemia; B-cell lymphoma | CAR (CD19 and CD20 or CD22) | Phase 1 and Phase 2 |
| NCT04438083 | “A Phase 1 Dose Escalation and Cohort Expansion Study of the Safety and Efficacy of Allogeneic CRISPR-Cas9-Engineered T Cells (CTX130) in Subjects with Advanced, Relapsed or Refractory Renal Cell Carcinoma with Clear Cell Differentiation” | Refractory renal cell carcinoma | CTX13 CAR | Phase 1 |
| NCT04244656 | “A Phase 1 Dose Escalation and Cohort Expansion Study of the Safety and Efficacy of Anti-BCMA Allogeneic CRISPR-Cas9-Engineered T Cells (CTX120) in Subjects with Relapsed or Refractory” | Multiple myeloma | CTX120 CAR | Phase 1 |
| NCT04035434 | “A Phase 1 Dose Escalation and Cohort Expansion Study of the Safety and Efficacy of Allogeneic CRISPR-Cas9-Engineered T Cells (CTX110) in Subjects with Relapsed or Refractory B-Cell Malignancies (CARBON)” | B-cell malignancy; non-Hodgkin lymphoma; B-cell lymphoma; adult B-cell acute lymphoblastic leukemia | CTX110 CAR | Phase 1 |
| NCT04557436 | “Phase 1, Open Label Study of CRISPR-CAR Genome Edited T Cells (PBLTT52CAR19) in Relapsed/Refractory B Cell Acute Lymphoblastic Leukaemia” | B acute lymphoblastic leukemia | PBLTT52CAR19 | Phase 1 |
| NCT03166878 | “Phase I/II Study to Determine the Safety, Tolerability, Biological Activity and Efficacy of Universal CRISPR-Cas9 Gene-Editing CAR-T Cells Targeting CD19(UCART019) in Patients with Relapsed or Refractory CD19+ Leukemia and Lymphoma” | B-Cell leukemia; B-Cell lymphoma | UCART019 CAR | Phase 1 and Phase 2 |
| NCT04417764 | “Safety and Effect Assessment of TACE in Combination with Autologous PD-1 Knockout Engineered T Cells by Percutaneous Infusion in the Patients with Advanced Hepatocellular Carcinoma” | Advanced hepatocellular carcinoma | PD-1 KO | Phase 1 |
| NCT05066165 | “Phase 1/2a, Single Dose Study Investigating NTLA-5001 in Subjects with Acute Myeloid Leukemia” | Acute myeloid leukemia | NTLA-5001 CAR | Phase 1 and Phase 2 |
| NCT03044743 | “A Phase I/II Trial of PD-1 Knockout EBV-CTLs for Advanced Stage EBV Associated Malignancies” | Carcinoma; T-Cell lymphoma; adult Hodgkin lymphoma; diffuse large B-Cell lymphoma | PD-1 KO in EBV-specific T cells | Phase 1 and Phase 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezalotfi, A.; Fritz, L.; Förster, R.; Bošnjak, B. Challenges of CRISPR-Based Gene Editing in Primary T Cells. Int. J. Mol. Sci. 2022, 23, 1689. https://doi.org/10.3390/ijms23031689
Rezalotfi A, Fritz L, Förster R, Bošnjak B. Challenges of CRISPR-Based Gene Editing in Primary T Cells. International Journal of Molecular Sciences. 2022; 23(3):1689. https://doi.org/10.3390/ijms23031689
Chicago/Turabian StyleRezalotfi, Alaleh, Lea Fritz, Reinhold Förster, and Berislav Bošnjak. 2022. "Challenges of CRISPR-Based Gene Editing in Primary T Cells" International Journal of Molecular Sciences 23, no. 3: 1689. https://doi.org/10.3390/ijms23031689
APA StyleRezalotfi, A., Fritz, L., Förster, R., & Bošnjak, B. (2022). Challenges of CRISPR-Based Gene Editing in Primary T Cells. International Journal of Molecular Sciences, 23(3), 1689. https://doi.org/10.3390/ijms23031689