
Pan-Cancer Prediction of Cell-Line Drug Sensitivity Using Network-Based Methods


 , , ,
, , ,
Abstract
:1. Introduction
2. Results
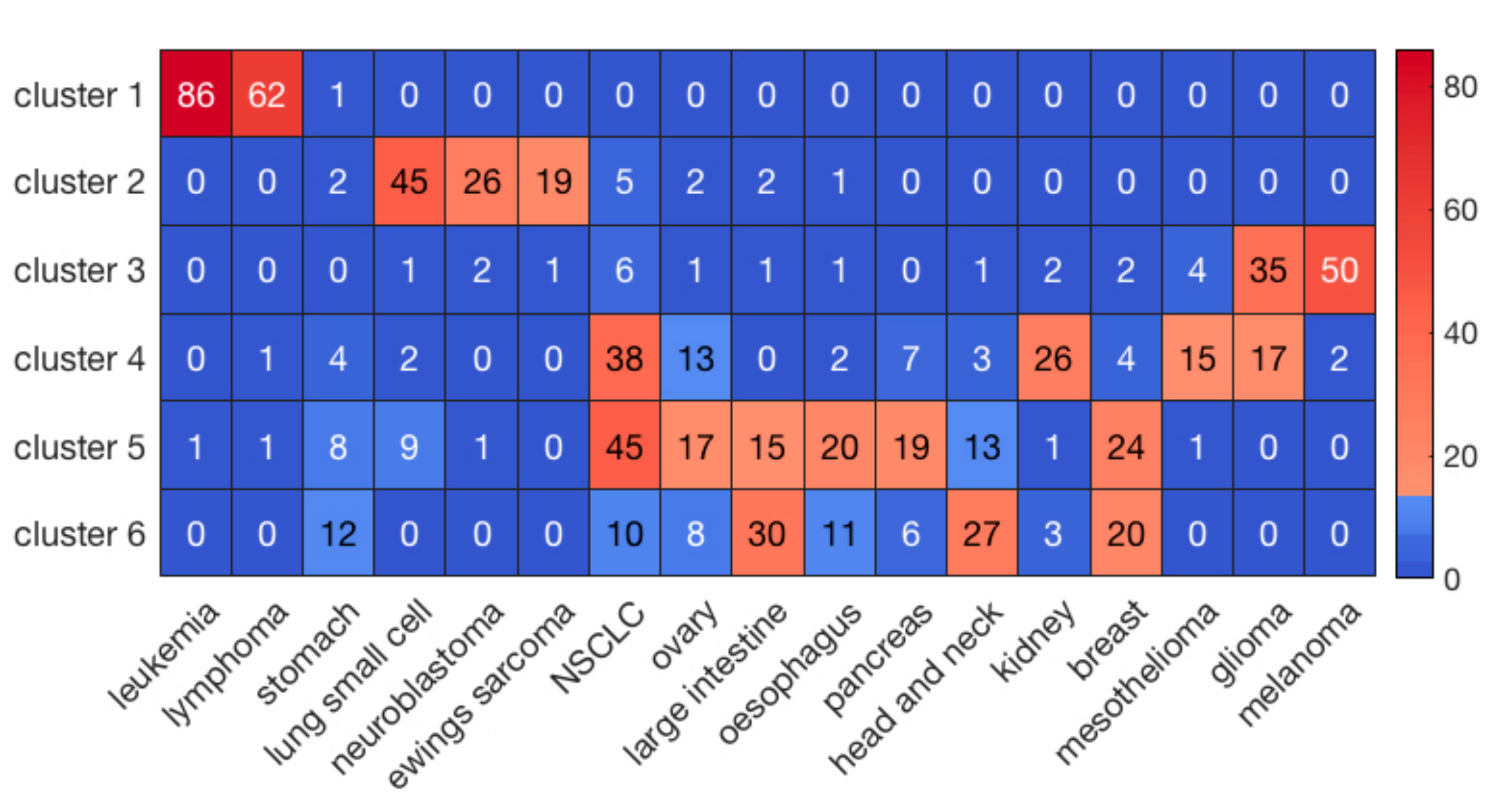
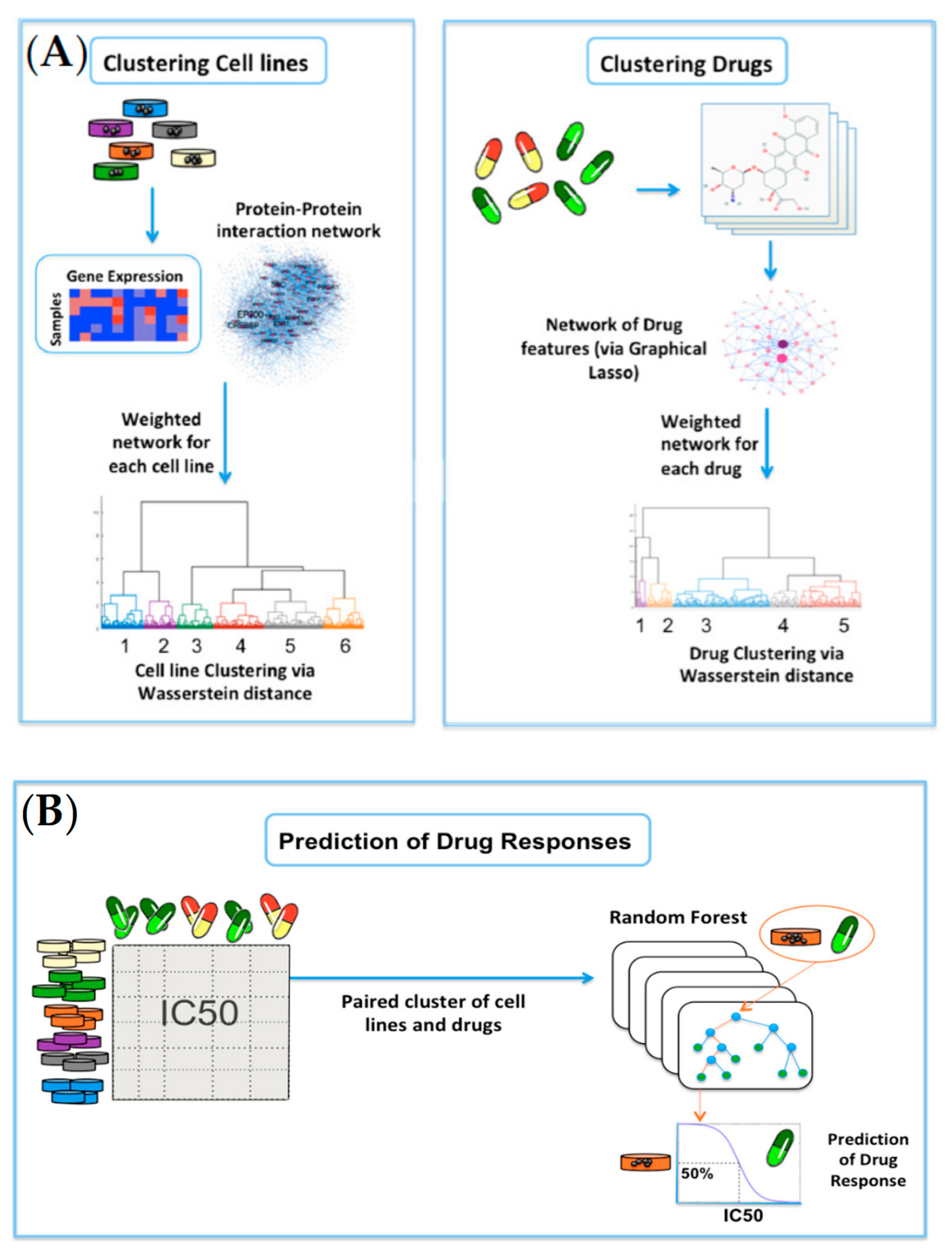
2.1. Clustering of Cell Lines and Drugs
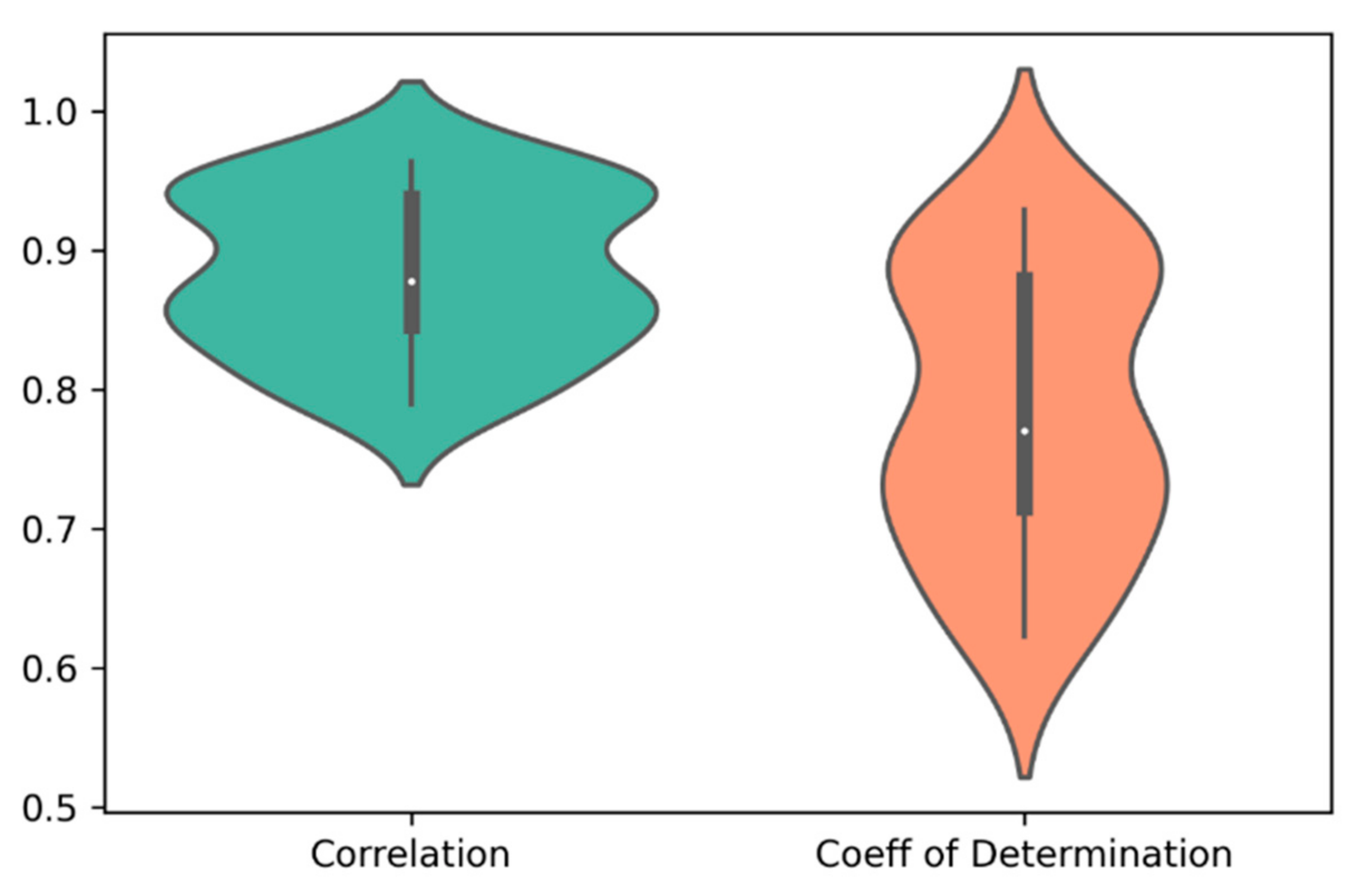
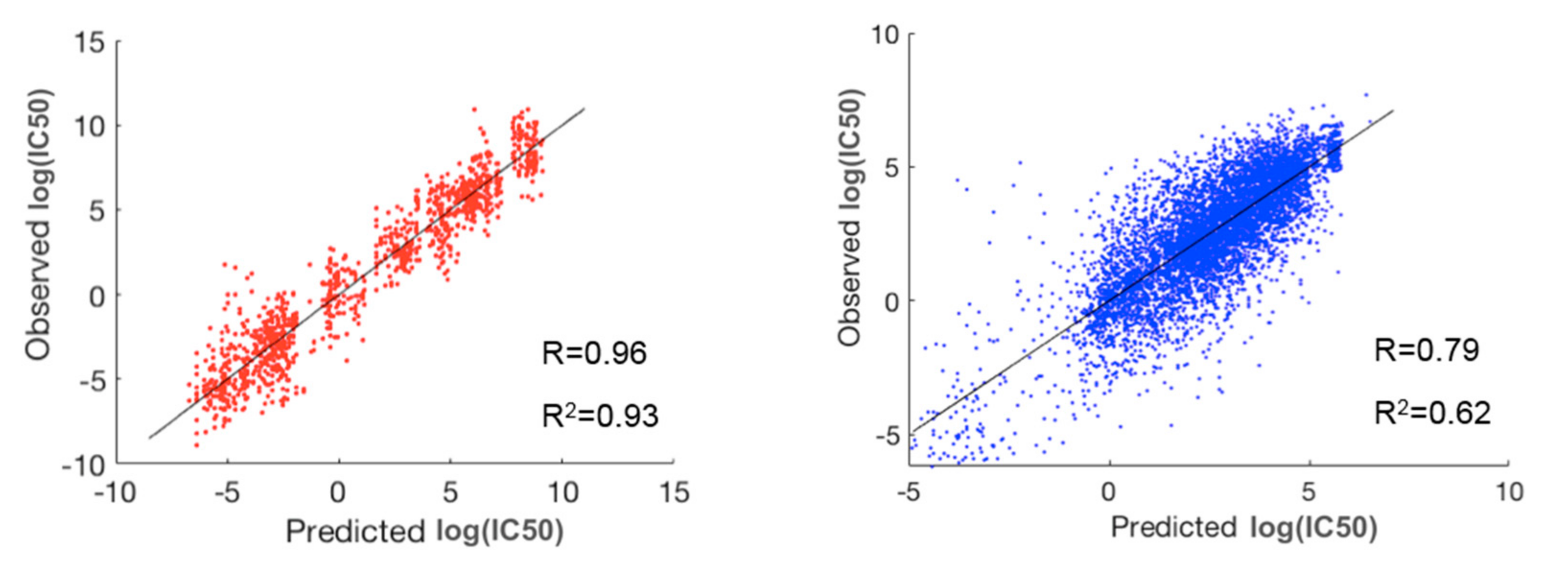
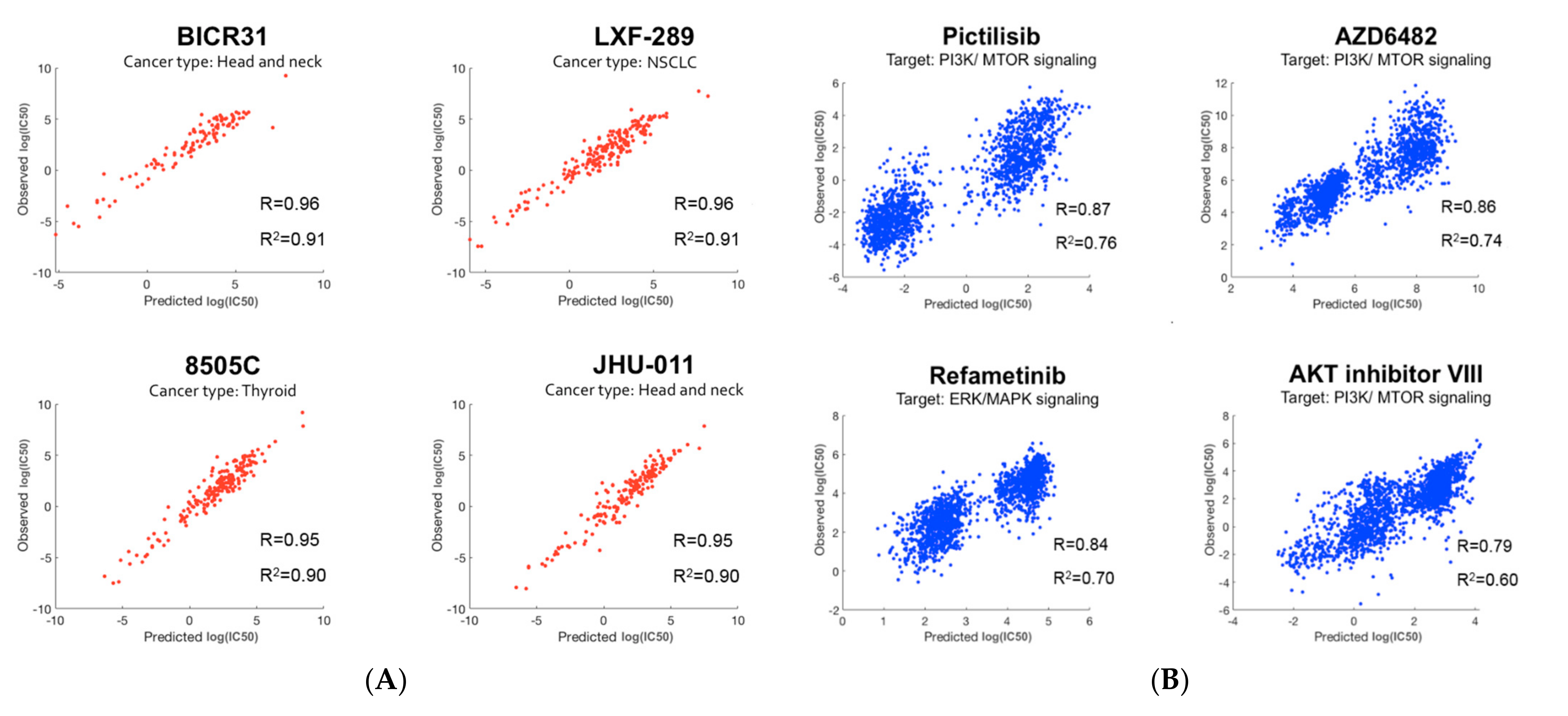
2.2. Prediction of Drug Responses in Paired Cell-Line Drug Clusters
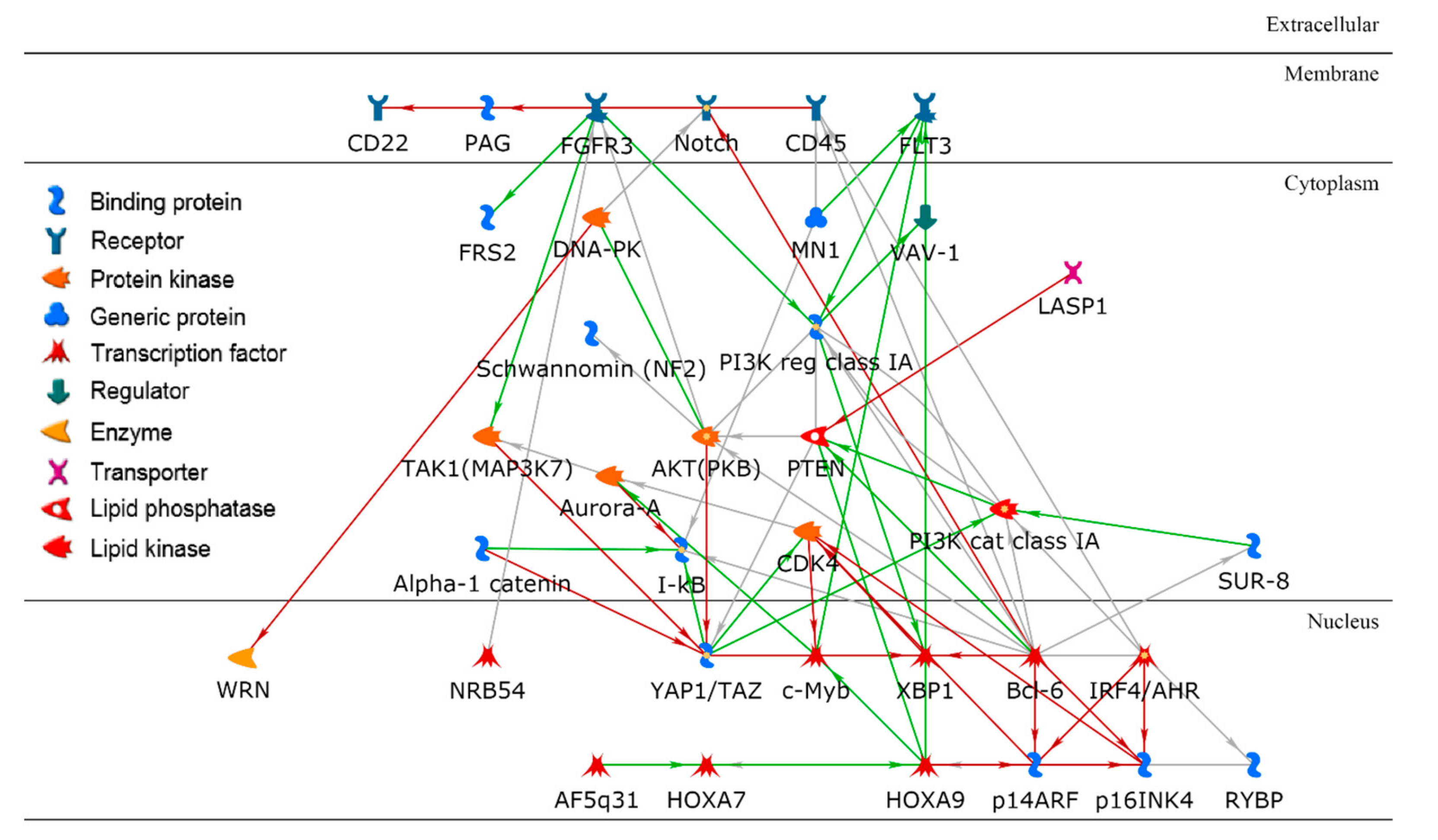
2.3. Biological Analysis
3. Discussion
4. Materials and Methods
4.1. Data and Preprocessing
4.2. The Invariant Measure of Gene Expression in a PPI Network
4.3. Network Construction of Cheminformatic Drug Features via Graphical LASSO
4.4. Network-Based Clustering via the Wasserstein Distance
4.5. Prediction of Drug Responses in Paired Cell-Line Drug Clusters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix B
Appendix C
References
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Chabner, B.A. NCI-60 Cell Line Screening: A Radical Departure in its Time. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Weinstein, J.N. Integromic analysis of the NCI-60 cancer cell lines. Breast Dis. 2004, 19, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Staunton, J.E.; Slonim, D.K.; Coller, H.A.; Tamayo, P.; Angelo, M.J.; Park, J.; Scherf, U.; Lee, J.K.; Reinhold, W.O.; Weinstein, J.N.; et al. Chemosensitivity prediction by transcriptional profiling. Proc. Natl. Acad. Sci. USA 2001, 98, 10787–10792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuaje, F. Computational models for predicting drug responses in cancer research. Brief. Bioinform. 2017, 18, 820–829. [Google Scholar] [CrossRef]
- Yates, J.W.T.; Mistry, H. Clone Wars: Quantitatively Understanding Cancer Drug Resistance. JCO Clin. Cancer Inform. 2020, 4, 938–946. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, N.; Li, C.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X. Anticancer drug sensitivity prediction in cell lines from baseline gene expression through recursive feature selection. BMC Cancer 2015, 15, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daemen, A.; Griffith, O.L.; Heiser, L.M.; Wang, N.J.; Enache, O.M.; Sanborn, Z.; Pepin, F.; Durinck, S.; Korkola, J.E.; Griffith, M.; et al. Modeling precision treatment of breast cancer. Genome Biol. 2013, 14, R110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddick, G.; Song, H.; Ahn, S.; Walling, J.; Borges-Rivera, D.; Zhang, W.; Fine, H.A. Predicting in vitro drug sensitivity using Random Forests. Bioinformatics 2011, 27, 220–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef]
- Wang, W.; Yang, S.; Li, J. Drug target predictions based on heterogeneous graph inference. In Biocomputing; World Scientific: Singapore, 2013; pp. 53–64. [Google Scholar]
- Zhang, N.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X.; Liu, X.S. Predicting Anticancer Drug Responses Using a Dual-Layer Integrated Cell Line-Drug Network Model. PLoS Comput. Biol. 2015, 11, e1004498. [Google Scholar] [CrossRef]
- Stanfield, Z.; Coskun, M.; Koyuturk, M. Drug Response Prediction as a Link Prediction Problem. Sci. Rep. 2017, 7, 40321. [Google Scholar] [CrossRef]
- Ahmed, K.T.; Park, S.; Jiang, Q.; Yeu, Y.; Hwang, T.; Zhang, W. Network-based drug sensitivity prediction. BMC Med. Genom. 2020, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [Green Version]
- Villani, C. Optimal Transport; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Villani, C. Topics in Optimal Transportation (Graduate Studies in Mathematics); American Mathematical Society: Providence, RI, USA, 2003. [Google Scholar]
- Evans, L.C. Partial differential equations and Monge-Kantorovich mass transfer. Curr. Dev. Math. 1997, 1997, 65–126. [Google Scholar] [CrossRef]
- Wei, D.; Liu, C.; Zheng, X.; Li, Y. Comprehensive anticancer drug response prediction based on a simple cell line-drug complex network model. BMC Bioinform. 2019, 20, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouryahya, M.; Oh, J.H.; Mathews, J.C.; Deasy, J.O.; Tannenbaum, A.R. Characterizing Cancer Drug Response and Biological Correlates: A Geometric Network Approach. Sci. Rep. 2018, 8, 6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, S.; Pardos, Z.A.; Heffernan, N.T. The utility of clustering in prediction tasks. arXiv 2015, arXiv:1509.06163. [Google Scholar]
- Ye, B.H.; Lista, F.; Lo Coco, F.; Knowles, D.M.; Offit, K.; Chaganti, R.S.; Dalla-Favera, R. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 1993, 262, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, M.G.; Oswald, E.; Yu, W.; Xue, F.; MacKerell, A.D.; Melnick, A.M. The Expanding Role of the BCL6 Oncoprotein as a Cancer Therapeutic Target. Clin. Cancer Res. 2017, 23, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, T.; Fukuda, T.; Miki, T.; Miura, O. BCL6 overexpression prevents increase in reactive oxygen species and inhibits apoptosis induced by chemotherapeutic reagents in B-cell lymphoma cells. Oncogene 2003, 22, 4459–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhu, Q.; Chen, M.; Huang, Q.; Wang, W.; Li, Q.; Huang, Y.; Di, W. The changing 50% inhibitory concentration (IC50) of cisplatin: A pilot study on the artifacts of the MTT assay and the precise measurement of density-dependent chemoresistance in ovarian cancer. Oncotarget 2016, 7, 70803–70821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Cruz, F.D.; Sandhu, R.; Kung, A.L.; Mundi, P.; Deasy, J.O.; Tannenbaum, A. Pediatric Sarcoma Data Forms a Unique Cluster Measured via the Earth Mover’s Distance. Sci. Rep. 2017, 7, 7035. [Google Scholar] [CrossRef] [Green Version]
- Pouryahya, M.; Oh, J.H.; Javanmard, P.; Mathews, J.C.; Belkhatir, Z.; Deasy, J.O.; Tannenbaum, A.R. aWCluster: A Novel integrative Network-based Clustering of Multiomics Breast Cancer Data. bioRxiv 2019, 558700. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Banerji, C.R.; Severini, S.; Kuehn, R.; Sollich, P. Increased signaling entropy in cancer requires the scale-free property of protein interaction networks. Sci. Rep. 2015, 5, 9646. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 2008, 9, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.H.; Deasy, J.O. Inference of radio-responsive gene regulatory networks using the graphical lasso algorithm. BMC Bioinform. 2014, 15 (Suppl. S7), S5. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, R.; Hastie, T. The graphical lasso: New insights and alternatives. Electron. J. Stat. 2012, 6, 2125–2149. [Google Scholar] [CrossRef] [PubMed]
- Monge, G. Mémoire sur la Théorie des Déblais et des Remblais. In Histoire de l’Académie Royale des Sciences de Paris; De l’Imprimerie Royale: Paris, France, 1781. [Google Scholar]
- Kantorovitch, L. On the translocation of masses. Manag. Sci. 1958, 5, 1–4. [Google Scholar] [CrossRef]
- Rubner, Y. The earth mover’s distance as a metric for image retrieval. Int. J. Comput. Vis. 2000, 40, 99–121. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | R | R2 |
|---|---|---|
| a. Random forest using prior WD-based clustering | 0.89 | 0.79 |
| b. CDCN model with WD | 0.86 | 0.59 |
| c. Random forest on the whole data | 0.77 | 0.60 |
| d. CDCN model with Pearson correlation | 0.74 | 0.53 |
| Ranking | Biological Processes | FDR | Number of Input Genes |
|---|---|---|---|
| 1 | Regulation of apoptotic process | 2.55 × 10−20 | 40 |
| 2 | Regulation of programmed cell death | 2.55 × 10−20 | 40 |
| 3 | Regulation of cell death | 4.94 × 10−20 | 41 |
| 4 | System development | 1.93 × 10−18 | 56 |
| 5 | Positive regulation of nitrogen compound metabolic process | 5.35 × 10−18 | 48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pouryahya, M.; Oh, J.H.; Mathews, J.C.; Belkhatir, Z.; Moosmüller, C.; Deasy, J.O.; Tannenbaum, A.R. Pan-Cancer Prediction of Cell-Line Drug Sensitivity Using Network-Based Methods. Int. J. Mol. Sci. 2022, 23, 1074. https://doi.org/10.3390/ijms23031074
Pouryahya M, Oh JH, Mathews JC, Belkhatir Z, Moosmüller C, Deasy JO, Tannenbaum AR. Pan-Cancer Prediction of Cell-Line Drug Sensitivity Using Network-Based Methods. International Journal of Molecular Sciences. 2022; 23(3):1074. https://doi.org/10.3390/ijms23031074
Chicago/Turabian StylePouryahya, Maryam, Jung Hun Oh, James C. Mathews, Zehor Belkhatir, Caroline Moosmüller, Joseph O. Deasy, and Allen R. Tannenbaum. 2022. "Pan-Cancer Prediction of Cell-Line Drug Sensitivity Using Network-Based Methods" International Journal of Molecular Sciences 23, no. 3: 1074. https://doi.org/10.3390/ijms23031074
APA StylePouryahya, M., Oh, J. H., Mathews, J. C., Belkhatir, Z., Moosmüller, C., Deasy, J. O., & Tannenbaum, A. R. (2022). Pan-Cancer Prediction of Cell-Line Drug Sensitivity Using Network-Based Methods. International Journal of Molecular Sciences, 23(3), 1074. https://doi.org/10.3390/ijms23031074