
CRISPR Editing Enables Consequential Tag-Activated MicroRNA-Mediated Endogene Deactivation

 , ,
, , 

Abstract
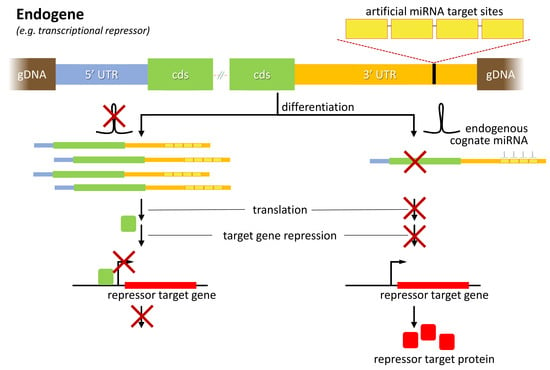
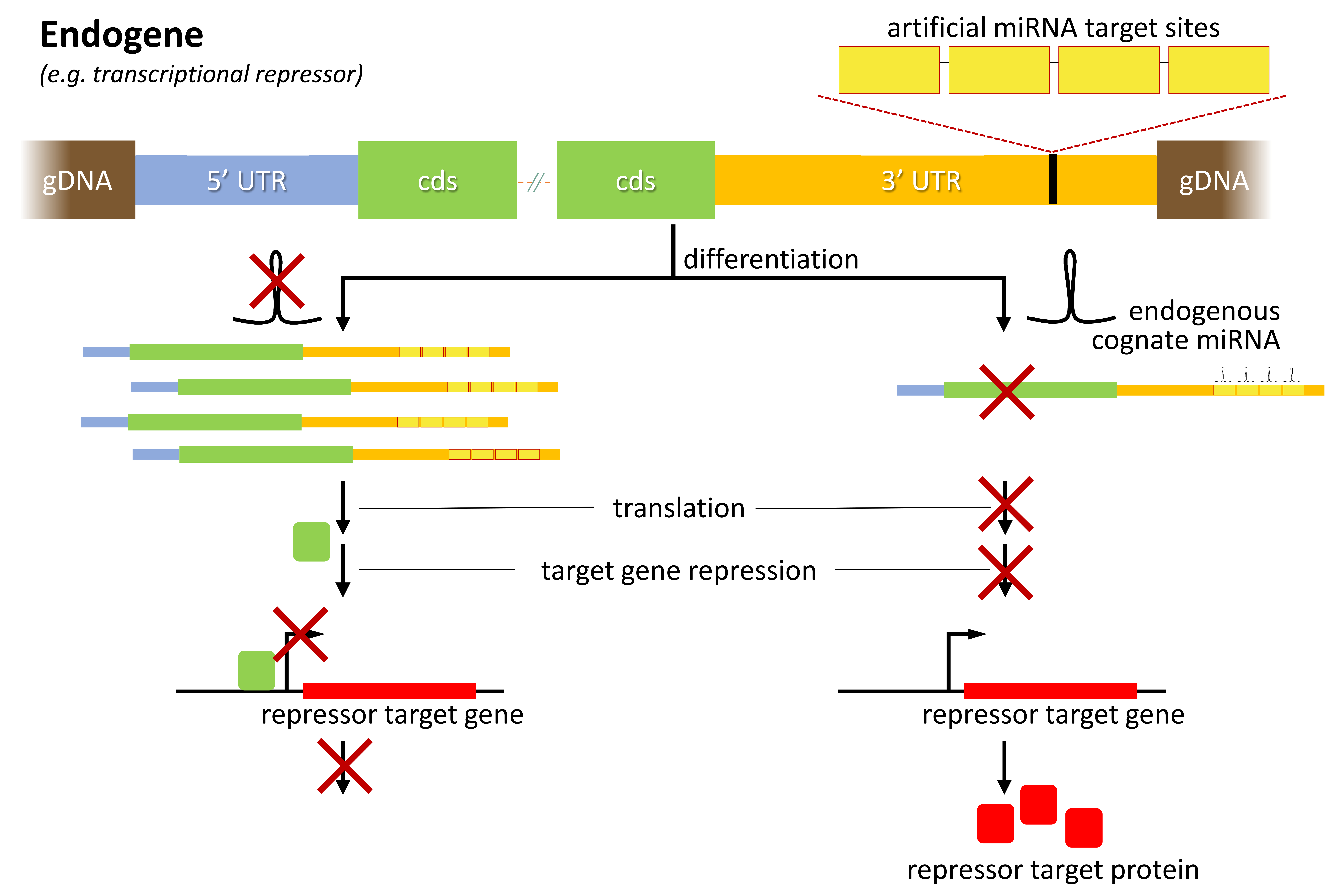{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Candidate ErythromiRs for TAMED in Adult Late-Erythroid Cells
2.2. Design Choices for TAMED of BCL11A
2.3. Validating Cleavage and Inertness of Shortlisted UTR Target Sites
2.4. NHEJ-Mediated Integration of MRSs of miR-451a as dsODNs
2.5. HDR-Mediated Integration of MRSs of miR-451a as ssODNs
2.6. Characterization of Monoclonal Cell Populations of HEK293T and HUDEP-2 Cells Bearing MRSs for miR-451a
3. Discussion
4. Materials and Methods
4.1. Culture of Human Primary Cells and Cell Lines
4.2. gRNA and ODN Design
4.3. Lentiviral Vector (LV) Construction and Production
4.4. Lentiviral Transduction of HUDEP-2 Cells
4.5. Nucleofection of Cells
4.6. Generation of Clonal Populations of HEK293T and HUDEP-2 Cells
4.7. DNA Analysis
4.8. T7 Endonuclease I (T7EI) Assay
4.9. Tracking of Indels by DEcomposition (TIDE)
4.10. Restriction Fragment Length Polymorphism Analysis of PCR-Amplified Fragments (PCR-RFLP)
4.11. Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC) Analysis of Globin Chains
4.12. Immunoblotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| bp | base pair |
| Cas | CRISPR-associated protein |
| Cpf1 | CRISPR from Prevotella and Francisella 1 |
| CRISPR | clustered regularly interspaced short palindromic repeat |
| DPBS | Dulbecco’s phosphate-buffered saline |
| ds | double-stranded |
| DSB | double-strand break |
| HDR | homology-directed repair |
| HEK293T | human embryonic kidney 293T cells |
| HPV16 | human papillomavirus 16 |
| hSCF | human Stem Cell Factor |
| HUDEP | human umbilical cord-blood-derived erythroid progenitor |
| gDNA | genomic DNA |
| gRNA | guide RNA |
| LV | lentiviral vector |
| miRNA | microRNA |
| MRS | miRNA recognition site |
| NHEJ | non-homologous end joining |
| nt | nucleotide |
| ODN | oligodeoxynucleotide |
| oligo | oligodeoxynucleotide |
| PCR-RFLP | restriction fragment length polymorphism analysis of PCR-amplified fragment |
| RP-HPLC | reversed-phase high-performance liquid chromatography |
| ss | single-stranded |
| T7EI | T7 endonuclease I |
| TAMED | tag-activated miRNA-mediated endogene deactivation |
| TIDE | Tracking of Indels by DEcomposition |
| UTR | untranslated region |
References
- Hinman, V.; Cary, G. The evolution of gene regulation: The gene regulation mechanisms necessary for the development of complex multicellular animals have been found in sponges. Elife 2017, 6. [Google Scholar] [CrossRef]
- Toscano, M.G.; Romero, Z.; Mũoz, P.; Cobo, M.; Benabdellah, K.; Martin, F. Physiological and tissue-specific vectors for treatment of inherited diseases. Gene Ther. 2011, 18, 117–127. [Google Scholar] [CrossRef]
- Chhatwal, J.P.; Hammack, S.E.; Jasnow, A.M.; Rainnie, D.G.; Ressler, K.J. Identification of cell-type-specific promoters within the brain using lentiviral vectors. Gene Ther. 2007, 14, 575–583. [Google Scholar] [CrossRef]
- Zheng, C.; Baum, B.J. Evaluation of promoters for use in tissue-specific gene delivery. Methods Mol. Biol. 2008, 434, 205–219. [Google Scholar] [CrossRef][Green Version]
- Sakamoto, K.; Rädler, P.D.; Wehde, B.L.; Triplett, A.A.; Shrestha, H.; Ferraiuolo, R.M.; Amari, F.; Coppola, V.; Klinakis, A.; Efstratiadis, A.; et al. Efficient tissue-type specific expression of target genes in a tetracycline-controlled manner from the ubiquitously active Eef1a1 locus. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Brown, B.D.; Venneri, M.A.; Zingale, A.; Sergi, L.S.; Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 2006, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Plank-Bazinet, J.; Krivega, I.; Dale, R.K.; Dean, A. Embryonic erythropoiesis and hemoglobin switching require transcriptional repressor ETO2 to modulate chromatin organization. Nucleic Acids Res. 2020, 48, 10226–10240. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Cantore, A.; Annoni, A.; Sergi, L.S.; Lombardo, A.; Della Valle, P.; D’Angelo, A.; Naldini, L. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood 2007, 110, 4144–4152. [Google Scholar] [CrossRef]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol 2007, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Gentner, B.; Visigalli, I.; Hiramatsu, H.; Lechman, E.; Ungari, S.; Giustacchini, A.; Schira, G.; Amendola, M.; Quattrini, A.; Martino, S.; et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. Transl. Med. 2010, 2, 58ra84. [Google Scholar] [CrossRef]
- Ungari, S.; Montepeloso, A.; Morena, F.; Cocchiarella, F.; Recchia, A.; Martino, S.; Gentner, B.; Naldini, L.; Biffi, A. Design of a regulated lentiviral vector for hematopoietic stem cell gene therapy of globoid cell leukodystrophy. Mol. Ther. - Methods Clin. Dev. 2015, 2, 15038. [Google Scholar] [CrossRef]
- Dhungel, B.; Ramlogan-Steel, C.A.; Layton, C.J.; Steel, J.C. miRNA122a regulation of gene therapy vectors targeting hepatocellular cancer stem cells. Oncotarget 2018, 9, 23577–23588. [Google Scholar] [CrossRef] [PubMed]
- Fay, E.J.; Langlois, R.A. MicroRNA-Attenuated Virus Vaccines. Non-coding RNA 2018, 4, 25. [Google Scholar] [CrossRef]
- De Ros, X.B.; Rovira-Rigau, M.; Fillat, C. Implications of MicroRNAs in oncolytic virotherapy. Front. Oncol. 2017, 7, 142. [Google Scholar] [CrossRef]
- Kelly, E.J.; Hadac, E.M.; Greiner, S.; Russell, S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008, 14, 1278–1283. [Google Scholar] [CrossRef]
- Wang, X.W.; Hu, L.F.; Hao, J.; Liao, L.Q.; Chiu, Y.T.; Shi, M.; Wang, Y. A microRNA-inducible CRISPR–Cas9 platform serves as a microRNA sensor and cell-type-specific genome regulation tool. Nat. Cell Biol. 2019, 21, 522–530. [Google Scholar] [CrossRef]
- Hirosawa, M.; Fujita, Y.; Parr, C.J.C.; Hayashi, K.; Kashida, S.; Hotta, A.; Woltjen, K.; Saito, H. Cell-type-specific genome editing with a microRNA-responsive CRISPR-Cas9 switch. Nucleic Acids Res. 2017, 45. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.D.; Aschenbrenner, S.; Grosse, S.; Rapti, K.; Domenger, C.; Fakhiri, J.; Mastel, M.; Börner, K.; Eils, R.; Grimm, D.; et al. Cell-specific CRISPR-Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef]
- Renaud, J.B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef]
- Fang, Q.; Shinkai, Y. A cost efficient protocol to introduce epitope tags by CRISPR-Cas9 mediated gene knock-in with asymmetric semi-double stranded template. MethodsX 2021, 8. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnolgy 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Dewari, P.S.; Southgate, B.; McCarten, K.; Monogarov, G.; O’duibhir, E.; Quinn, N.; Tyrer, A.; Leitner, M.C.; Plumb, C.; Kalantzaki, M.; et al. An efficient and scalable pipeline for epitope tagging in mammalian stem cells using Cas9 ribonucleoprotein. Elife 2018, 7. [Google Scholar] [CrossRef]
- Sawatsubashi, S.; Joko, Y.; Fukumoto, S.; Matsumoto, T.; Sugano, S.S. Development of versatile non-homologous end joining-based knock-in module for genome editing. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef]
- Michaels, Y.S.; Barnkob, M.B.; Barbosa, H.; Baeumler, T.A.; Thompson, M.K.; Andre, V.; Colin-York, H.; Fritzsche, M.; Gileadi, U.; Sheppard, H.M.; et al. Precise tuning of gene expression levels in mammalian cells. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Luc, S.; Huang, J.; McEldoon, J.L.; Somuncular, E.; Li, D.; Rhodes, C.; Mamoor, S.; Hou, S.; Xu, J.; Orkin, S.H. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016, 16. [Google Scholar] [CrossRef]
- Liu, P.; Keller, J.R.; Ortiz, M.; Tessarollo, L.; Rachel, R.A.; Nakamura, T.; Jenkins, N.A.; Copeland, N.G. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003, 4, 525–532. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Khaled, W.; Burke, S.; Li, P.; Chen, X.; Yang, W.; Jenkins, N.A.; Copeland, N.G.; Zhang, S.; et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med. 2012, 209, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Xu, J.; Ragoczy, T.; Ippolito, G.C.; Walkley, C.R.; Maika, S.D.; Fujiwara, Y.; Ito, M.; Groudine, M.; Bender, M.A.; et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature 2009, 460, 1093–1097. [Google Scholar] [CrossRef]
- Bauer, D.E.; Orkin, S.H. Hemoglobin switching’s surprise: The versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 2015, 33, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. - Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Smith, A.R.; Schiller, G.J.; Vercellotti, G.M.; Kwiatkowski, J.L.; Krishnamurti, L.; Lakshmanan Krishnamurti, E.B.; Williams, D.A.; Miller, W.P.; Woolfson, A.; Walters, M.C. Preliminary Results of a Phase 1/2 Clinical Study of Zinc Finger Nuclease-Mediated Editing of BCL11A in Autologous Hematopoietic Stem Cells for Transfusion-Dependent Beta Thalassemia. Blood 2019, 134, 3544. [Google Scholar]
- Frangoul, H.; Bobruff, Y.; Cappellini, M.D.; Corbacioglu, S.; Fernandez, C.M.; de la Fuente, J.; Grupp, S.A.; Handgretinger, R.; Ho, T.W.; Imren, S.; et al. Safety and Efficacy of CTX001 in Patients with Transfusion-Dependent β-Thalassemia and Sickle Cell Disease: Early Results from the Climb THAL-111 and Climb SCD-121 Studies of Autologous CRISPR-CAS9-Modified CD34+ Hematopoietic Stem and Progenitor Cells. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Papasavva, P.L.; Papaioannou, N.Y.; Patsali, P.; Kurita, R.; Nakamura, Y.; Sitarou, M.; Christou, S.; Kleanthous, M.; Lederer, C.W. Distinct miRNA Signatures and Networks Discern Fetal from Adult Erythroid Differentiation and Primary from Immortalized Erythroid Cells. Int. J. Mol. Sci. 2021, 22, 3626. [Google Scholar] [CrossRef]
- Doss, J.F.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.-T. A comprehensive joint analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genomics 2015, 16, 952. [Google Scholar] [CrossRef]
- Jin, H.L.; Kim, J.S.; Kim, Y.J.; Kim, S.J.; Broxmeyer, H.E.; Kim, K.S. Dynamic expression of specific miRNAs during erythroid differentiation of human embryonic stem cells. Mol. Cells 2012, 34, 177–183. [Google Scholar] [CrossRef]
- Choong, M.L.; Yang, H.H.; McNiece, I. MicroRNA expression profiling during human cord blood-derived CD34 cell erythropoiesis. Exp. Hematol. 2007, 35, 551–564. [Google Scholar] [CrossRef]
- Bissels, U.; Bosio, A.; Wagner, W. MicroRNAs are shaping the hematopoietic landscape. Haematologica 2012, 97, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Mohammdai-Asl, J.; Ramezani, A.; Norozi, F.; Malehi, A.S.; Asnafi, A.A.; Far, M.A.J.; Mousavi, S.H.; Saki, N. MicroRNAs in erythropoiesis and red blood cell disorders. Front. Biol. (Beijing). 2015, 10, 321–332. [Google Scholar] [CrossRef]
- Byon, J.C.H.; Papayannopoulou, T. MicroRNAs: Allies or Foes in erythropoiesis? J. Cell. Physiol. 2012, 227, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bissels, U.; Wild, S.; Tomiuk, S.; Holste, A.; Hafner, M.; Tuschl, T.; Bosio, A. Absolute quantification of microRNAs by using a universal reference. RNA 2009, 15, 2375–2384. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, K.; Li, Y.; Xia, N.; Nie, S.; Lv, B.; Zhang, M.; Tu, X.; Li, Q.; Tang, T.; et al. Down-regulation of microRNA-451a facilitates the activation and proliferation of CD4+ T cells by targeting Myc in patients with dilated cardiomyopathy. J. Biol. Chem. 2017, 292, 6004–6013. [Google Scholar] [CrossRef]
- Masaki, S.; Ohtsuka, R.; Abe, Y.; Muta, K.; Umemura, T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem. Biophys. Res. Commun. 2007, 364, 509–514. [Google Scholar] [CrossRef]
- Zhan, M.; Miller, C.P.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Song, C.Z. MicroRNA expression dynamics during murine and human erythroid differentiation. Exp. Hematol. 2007, 35, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, F.; Yu, D. miR-144/451 in hematopoiesis and beyond. ExRNA 2019, 1, 1–12. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef]
- Xu, J.; Sankaran, V.G.; Ni, M.; Menne, T.F.; Puram, R.V.; Kim, W.; Orkin, S.H. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010, 24, 783–789. [Google Scholar] [CrossRef]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef]
- Hoffman, Y.; Bublik, D.R.; Ugalde, A.P.; Elkon, R.; Biniashvili, T.; Agami, R.; Oren, M.; Pilpel, Y. 3’UTR Shortening Potentiates MicroRNA-Based Repression of Pro-differentiation Genes in Proliferating Human Cells. PLoS Genet. 2016, 12, 1–15. [Google Scholar] [CrossRef]
- Grimson, A.; Farh, K.K.H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioate Oligodeoxynucleotides: What Is Their Origin and What Is Unique About Them? Antisense Nucleic Acid Drug Dev. 2011, 10, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Horn, T.; Urdea, M.S. A Chemical 5′-Phosphorylation of Oligodeoxyribonucleotides. DNA 1986, 5, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Pattabhi, S.; Lotti, S.N.; Berger, M.P.; Singh, S.; Lux, C.T.; Jacoby, K.; Lee, C.; Negre, O.; Scharenberg, A.M.; Rawlings, D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther.—Nucleic Acids 2019, 17, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Malinin, N.L.; Lee, G.; Lazzarotto, C.R.; Li, Y.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Iafrate, A.J.; Le, L.P.; et al. Defining genome-wide CRISPR–Cas genome-editing nuclease activity with GUIDE-seq. Nat. Protoc. 2021. [Google Scholar] [CrossRef] [PubMed]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus–host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef]
- Boel, A.; De Saffel, H.; Steyaert, W.; Callewaert, B.; De Paepe, A.; Coucke, P.J.; Willaert, A. CRISPR/Cas9-mediated homology-directed repair by ssODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Okamoto, S.; Amaishi, Y.; Maki, I.; Enoki, T.; Mineno, J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Bollen, Y.; Post, J.; Koo, B.K.; Snippert, H.J.G. How to create state-of-the-art genetic model systems: Strategies for optimal CRISPR-mediated genome editing. Nucleic Acids Res. 2018, 46, 6435–6454. [Google Scholar] [CrossRef] [PubMed]
- Moir-Meyer, G.; Cheong, P.L.; Olijnik, A.A.; Brown, J.; Knight, S.; King, A.; Kurita, R.; Nakamura, Y.; Gibbons, R.J.; Higgs, D.R.; et al. Robust crispr/cas9 genome editing of the hudep-2 erythroid precursor line using plasmids and single-stranded oligonucleotide donors. Methods Protoc. 2018, 1, 28. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance crispr genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, Y.A.; Nguyen, D.T.; Chow, S.; Chung, R.S.; Guillemin, G.J.; Cole, N.J.; Hesselson, D. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos. Commun. Biol. 2019, 2, 1–9. [Google Scholar] [CrossRef]
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A.M.; et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Fazekas, A.J.; Steeves, R.; Newmaster, S.G. Improving sequencing quality from PCR products containing long mononucleotide repeats. Biotechniques 2010, 48, 277–283. [Google Scholar] [CrossRef]
- Suraweera, N.; Iacopetta, B.; Duval, A.; Compoint, A.; Tubacher, E.; Hamelin, R. Conservation of mononucleotide repeats within 3′ and 5′ untranslated regions and their instability in MSI-H colorectal cancer. Oncogene 2001, 20, 7472–7477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kieleczawa, J. Fundamentals of sequencing of difficult templates-An overview. J. Biomol. Tech. 2006, 17, 207–217. [Google Scholar]
- Merlin, S.; Follenzi, A. Transcriptional Targeting and MicroRNA Regulation of Lentiviral Vectors. Mol. Ther.-Methods Clin. Dev. 2019, 12, 223–232. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Sikorski, A.F. Editorial focus: Understanding off-target effects as the key to successful RNAi therapy. Cell. Mol. Biol. Lett. 2019, 24, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.D.; Senzer, N.; Cleary, M.A.; Nemunaitis, J. Comparative assessment of siRNA and shRNA off target effects: What is slowing clinical development. Cancer Gene Ther. 2009, 16, 807–809. [Google Scholar] [CrossRef]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by siRNA can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Orlando, S.J.; Santiago, Y.; DeKelver, R.C.; Freyvert, Y.; Boydston, E.A.; Moehle, E.A.; Choi, V.M.; Gopalan, S.M.; Lou, J.F.; Li, J.; et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res. 2010, 38, e152. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- IDT Alt-R HDR Donor Oligos. Available online: https://eu.idtdna.com/pages/products/crispr-genome-editing/alt-r-hdr-donor-oligos (accessed on 10 August 2018).
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Hunt, S.; Ninova, M.; Griffiths-Jones, S.; Ronshaugen, M. Target repression induced by endogenous microRNAs: Large differences, small effects. PLoS One 2014, 9, e104286. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.S.; Dohjima, T.; Bauer, G.; Li, H.; Li, M.J.; Ehsani, A.; Salvaterra, P.; Rossi, J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002, 20, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Vella, M.C.; Choi, E.Y.; Lin, S.Y.; Reinert, K.; Slack, F.J. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′UTR. Genes Dev 2004, 18, 132–137. [Google Scholar] [CrossRef]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Dusl, M.; Senderek, J.; Muller, J.S.; Vogel, J.G.; Pertl, A.; Stucka, R.; Lochmuller, H.; David, R.; Abicht, A. A 3’-UTR mutation creates a microRNA target site in the GFPT1 gene of patients with congenital myasthenic syndrome. Hum. Mol. Genet. 2015, 24, 3418–3426. [Google Scholar] [CrossRef]
- Wynendaele, J.; Böhnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3′ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef] [PubMed]
- Geisler, A.; Jungmann, A.; Kurreck, J.; Poller, W.; Katus, H.A.; Vetter, R.; Fechner, H.; Müller, O.J. MicroRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther. 2011, 18, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, M.A.; Leber, M.F.; Bossow, S.; Singh, M.; Engeland, C.E.; Albert, J.; Grossardt, C.; Jäger, D.; Von Kalle, C.; Ungerechts, G. MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Ther. 2014, 21, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xie, Q.; Zhang, H.; Ameres, S.L.; Hung, J.-H.; Su, Q.; He, R.; Mu, X.; Seher Ahmed, S.; Park, S.; et al. MicroRNA-regulated, systemically delivered rAAV9: A step closer to CNS-restricted transgene expression. Mol. Ther. 2011, 19, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lin, J.; Hong, M.; Choudhury, Y.; Balani, P.; Leung, D.; Dang, L.H.; Zhao, Y.; Zeng, J.; Wang, S. Combinatorial control of suicide gene expression by tissue-specific promoter and microRNA regulation for cancer therapy. Mol. Ther. 2009, 17, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P. a MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Ameres, S.L.; Martinez, J.; Schroeder, R. Molecular Basis for Target RNA Recognition and Cleavage by Human RISC. Cell 2007, 130, 101–112. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Breda, L.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Lampronti, I.; Borgatti, M.; Chiavilli, F.; Gamberini, M.R.; Satta, S.; et al. A validated cellular biobank for β-thalassemia. J. Transl. Med. 2016, 14, 255. [Google Scholar] [CrossRef]
- Zhang, F. CRISPR.MIT.EDU, Taken Offline in November 2018. Available online: https://zlab.bio/guide-design-resources (accessed on 1 September 2017).
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. MiRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, I.; Patsali, P.; Stephanou, C.; Antoniou, M.; Kleanthous, M.; Lederer, C.W. Measurement of lentiviral vector titre and copy number by cross-species duplex quantitative PCR. Gene Ther. 2016, 23, 113–118. [Google Scholar] [CrossRef]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica 2019, 104, e497–e501. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.-L.; Conklin, B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Bimboim, H.C.; Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Berg Rasmussen, H. Restriction Fragment Length Polymorphism Analysis of PCR-Amplified Fragments (PCR-RFLP) and Gel Electrophoresis - Valuable Tool for Genotyping and Genetic Fingerprinting. In Gel Electrophoresis—Principles and Basics; InTech Open: Rijeka, Croatia, 2012. [Google Scholar]
- Loucari, C.C.; Patsali, P.; van Dijk, T.B.; Stephanou, C.; Papasavva, P.; Zanti, M.; Kurita, R.; Nakamura, Y.; Christou, S.; Sitarou, M.; et al. Rapid and Sensitive Assessment of Globin Chains for Gene and Cell Therapy of Hemoglobinopathies. Hum. Gene Ther. Methods 2018, 29, 60–74. [Google Scholar] [CrossRef]
- Patsali, P.; Papasavva, P.; Stephanou, C.; Christou, S.; Sitarou, M.; Antoniou, M.N.; Lederer, C.W.; Kleanthous, M. Short-hairpin RNA against aberrant HBBIVSI-110(G>A) mRNA restores β-globin levels in a novel cell model and acts as mono- and combination therapy for β-thalassemia in primary hematopoietic stem cells. Haematologica 2018, 103, e419–e423. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papasavva, P.L.; Patsali, P.; Loucari, C.C.; Kurita, R.; Nakamura, Y.; Kleanthous, M.; Lederer, C.W. CRISPR Editing Enables Consequential Tag-Activated MicroRNA-Mediated Endogene Deactivation. Int. J. Mol. Sci. 2022, 23, 1082. https://doi.org/10.3390/ijms23031082
Papasavva PL, Patsali P, Loucari CC, Kurita R, Nakamura Y, Kleanthous M, Lederer CW. CRISPR Editing Enables Consequential Tag-Activated MicroRNA-Mediated Endogene Deactivation. International Journal of Molecular Sciences. 2022; 23(3):1082. https://doi.org/10.3390/ijms23031082
Chicago/Turabian StylePapasavva, Panayiota L., Petros Patsali, Constantinos C. Loucari, Ryo Kurita, Yukio Nakamura, Marina Kleanthous, and Carsten W. Lederer. 2022. "CRISPR Editing Enables Consequential Tag-Activated MicroRNA-Mediated Endogene Deactivation" International Journal of Molecular Sciences 23, no. 3: 1082. https://doi.org/10.3390/ijms23031082
APA StylePapasavva, P. L., Patsali, P., Loucari, C. C., Kurita, R., Nakamura, Y., Kleanthous, M., & Lederer, C. W. (2022). CRISPR Editing Enables Consequential Tag-Activated MicroRNA-Mediated Endogene Deactivation. International Journal of Molecular Sciences, 23(3), 1082. https://doi.org/10.3390/ijms23031082