
Identification of Competing Endogenous RNAs (ceRNAs) Network Associated with Drought Tolerance in Medicago truncatula with Rhizobium Symbiosis

 ,
, 
Abstract
1. Introduction
2. Result
2.1. Morphology of M. truncatula with Nodules under Drought Treatment
2.2. Transcriptome Sequencing Feature Analysis
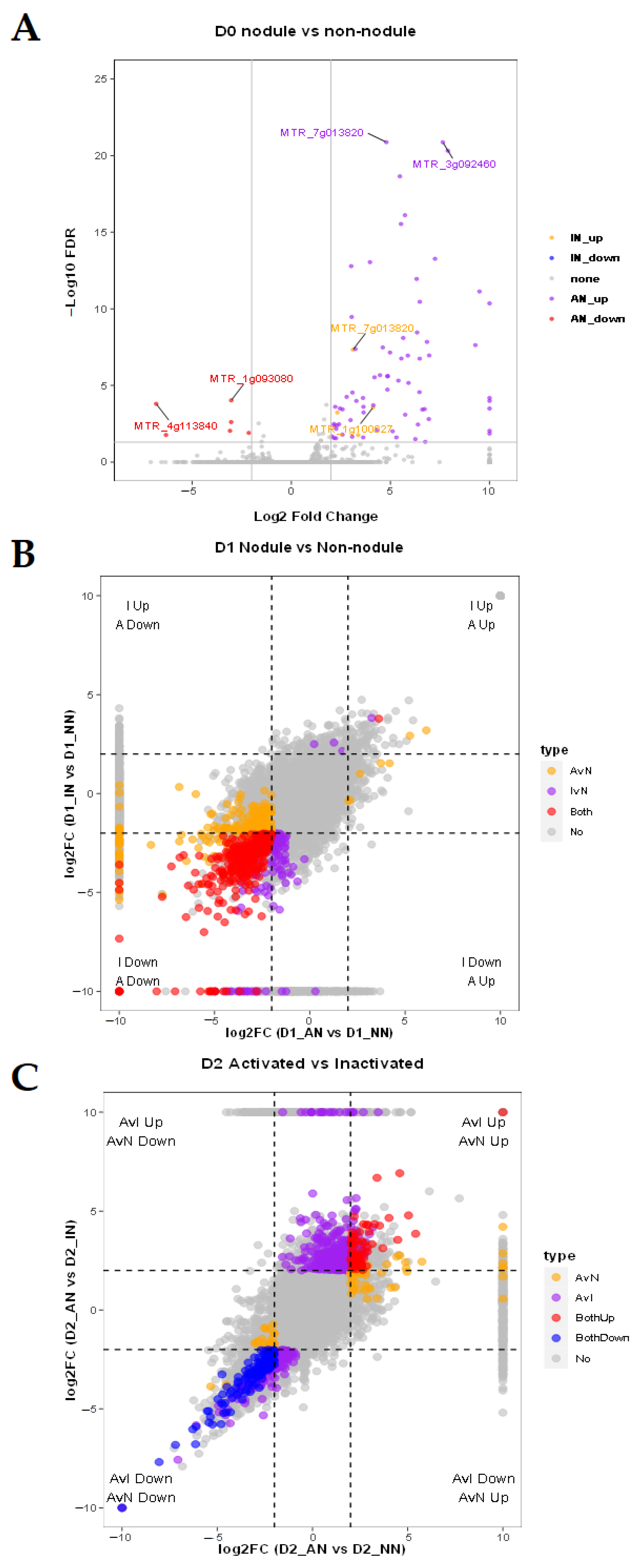
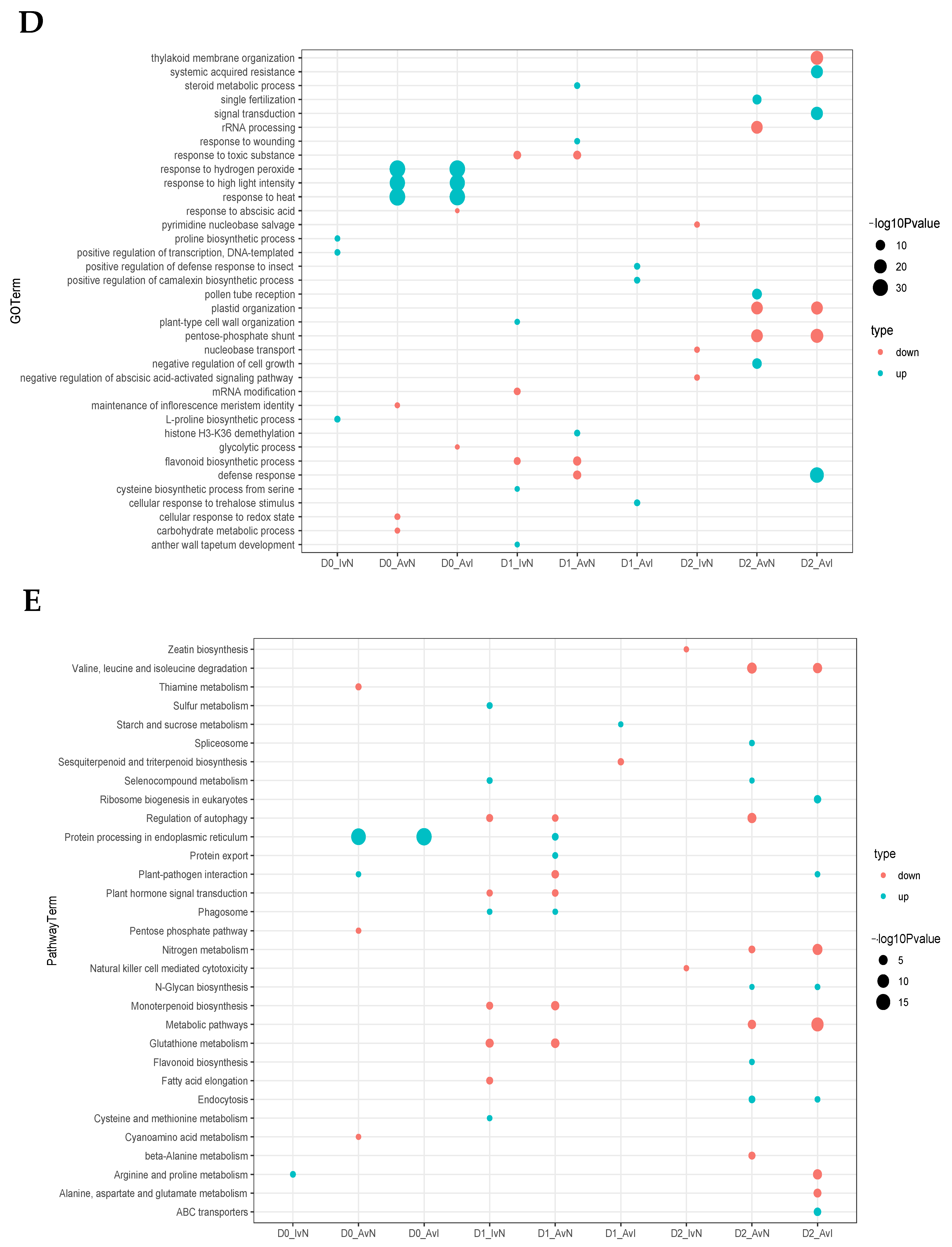
2.3. Different Expression Patterns Analysis of mRNA
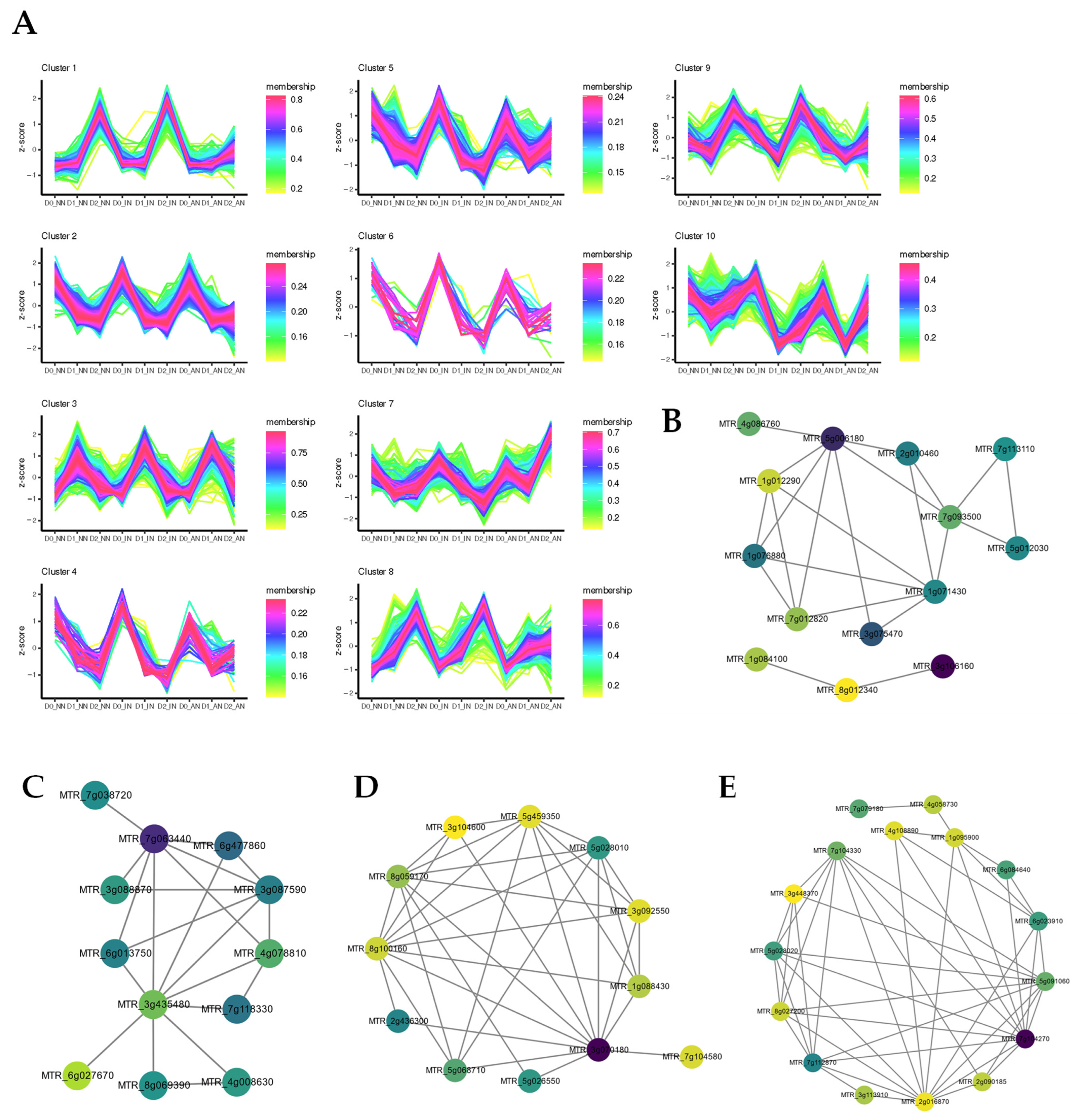
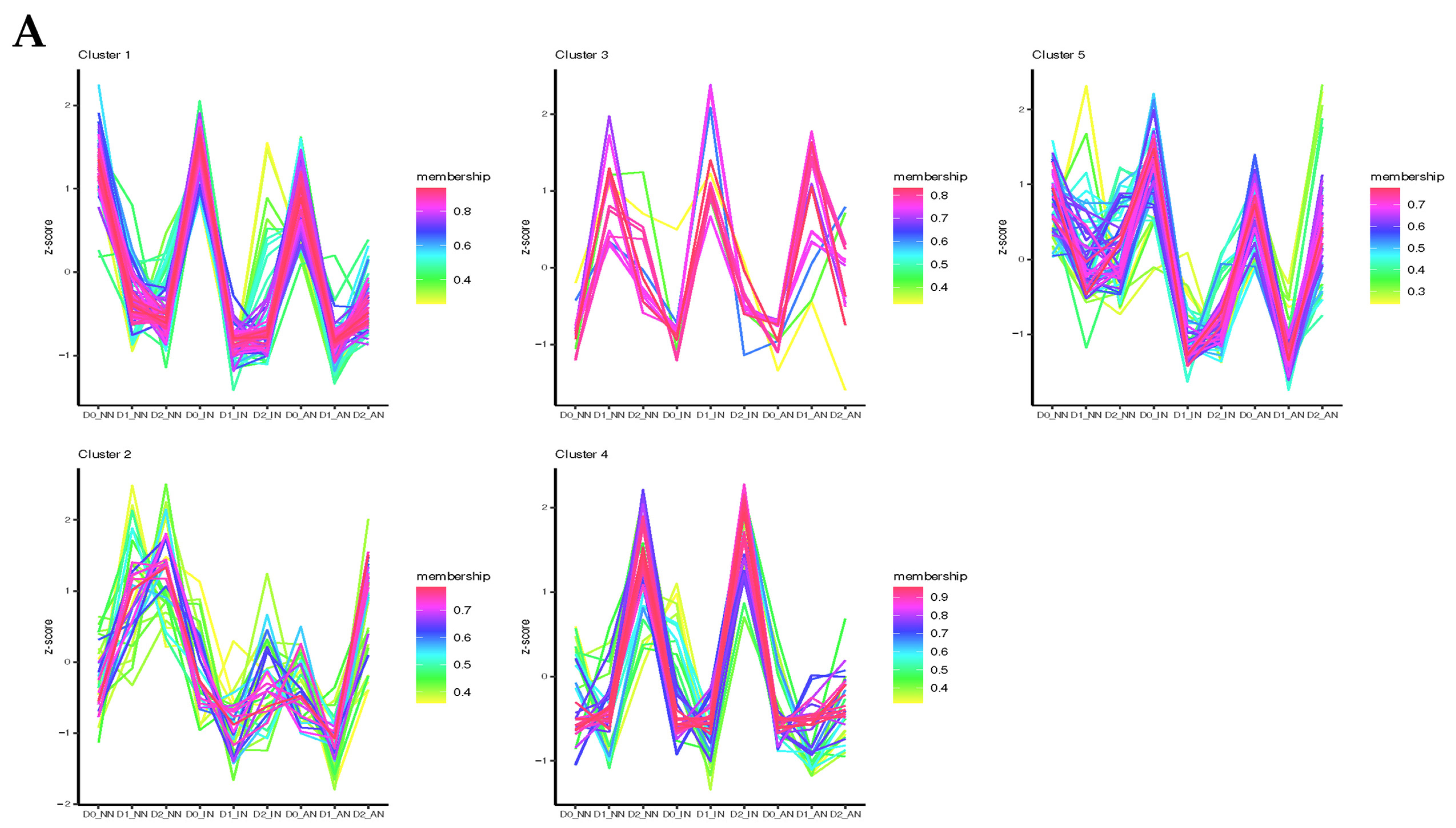
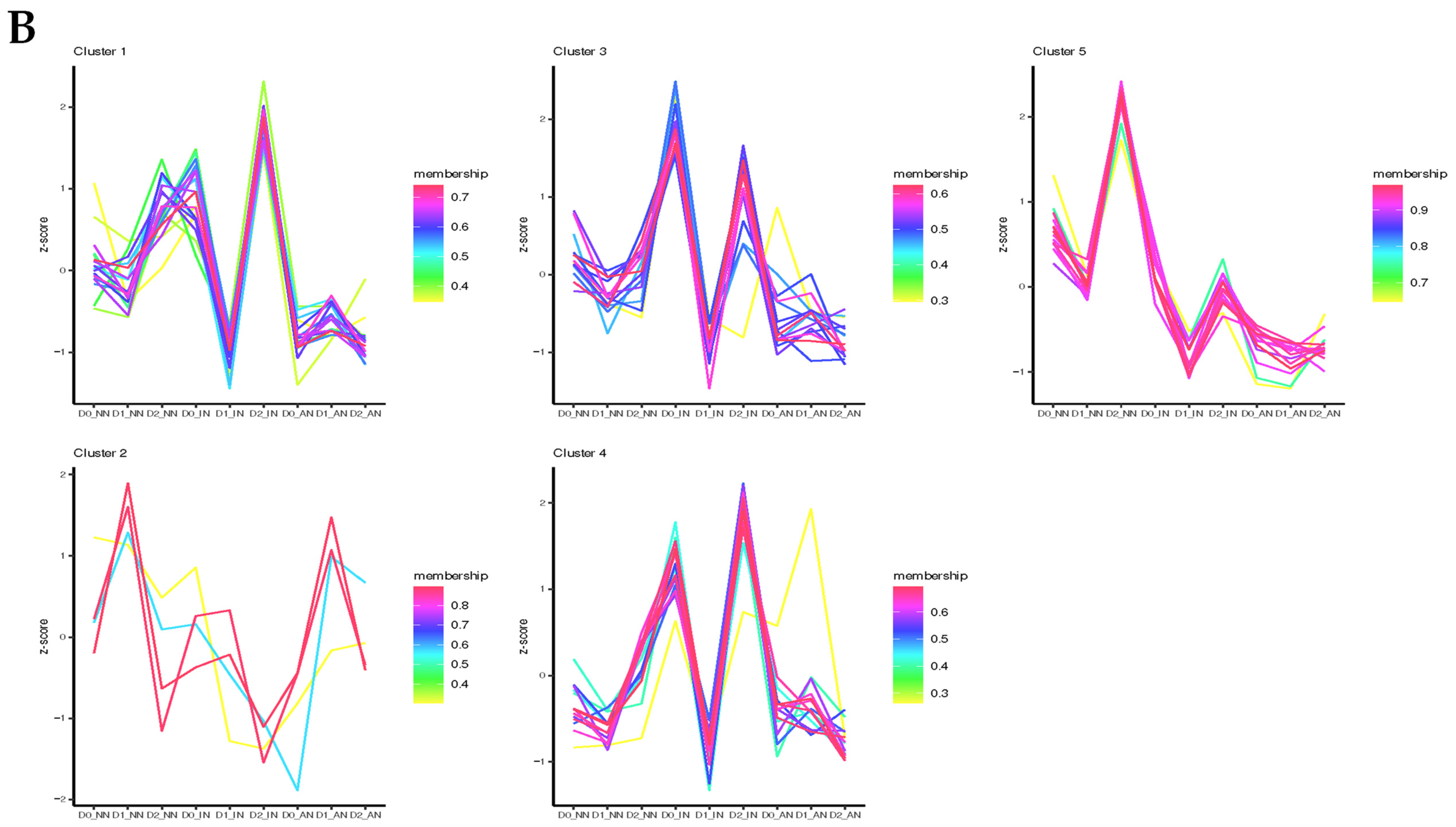
2.4. Feature Analysis and Expression Patterns of ncRNAs
2.5. Analysis of miRNA-Target Genes
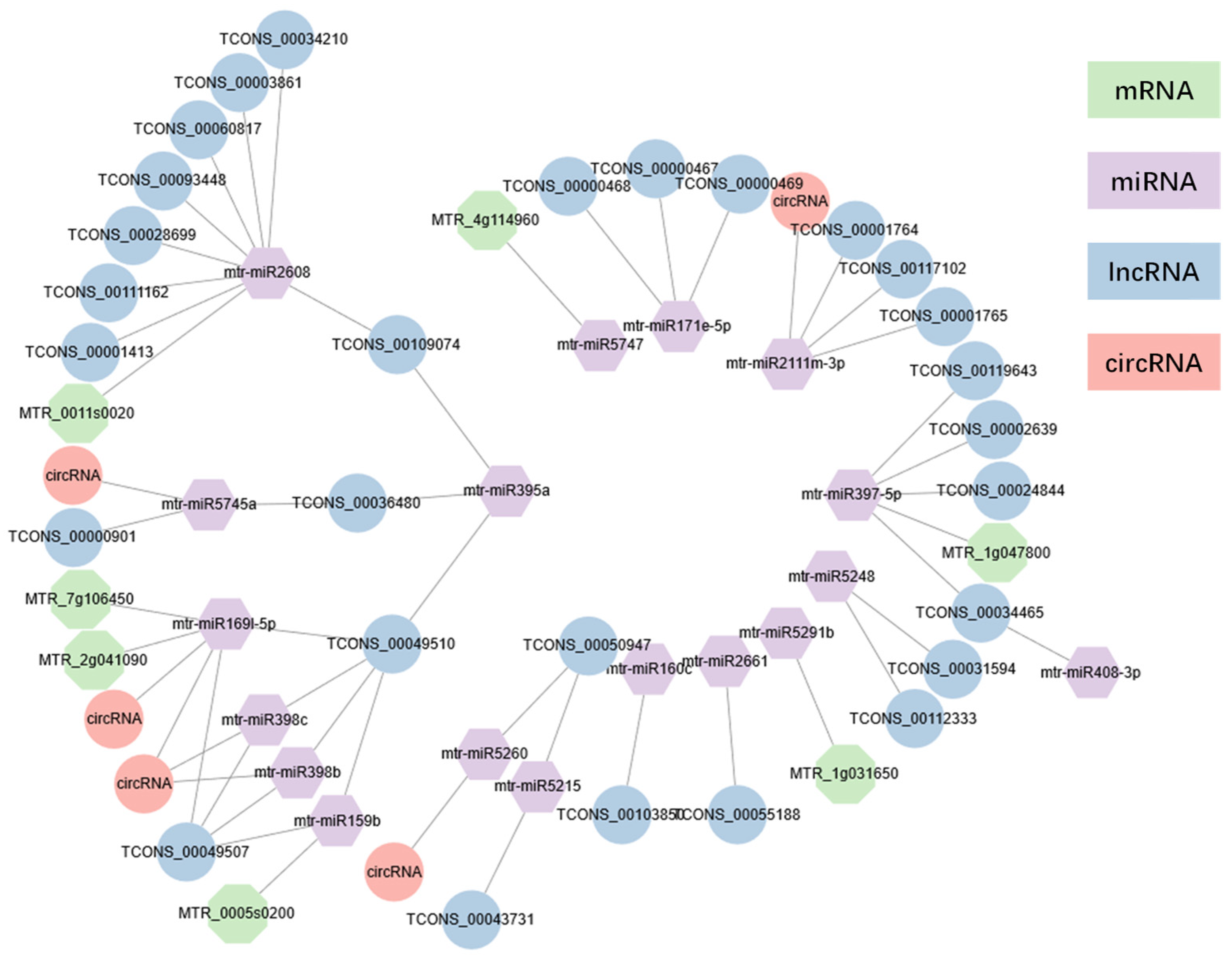
2.6. Construction of ceRNA-miRNA-Target Genes Regulatory Networks
3. Discussion
3.1. SNF-Triggered Pathways Enhanced Drought Tolerance in M. truncatula
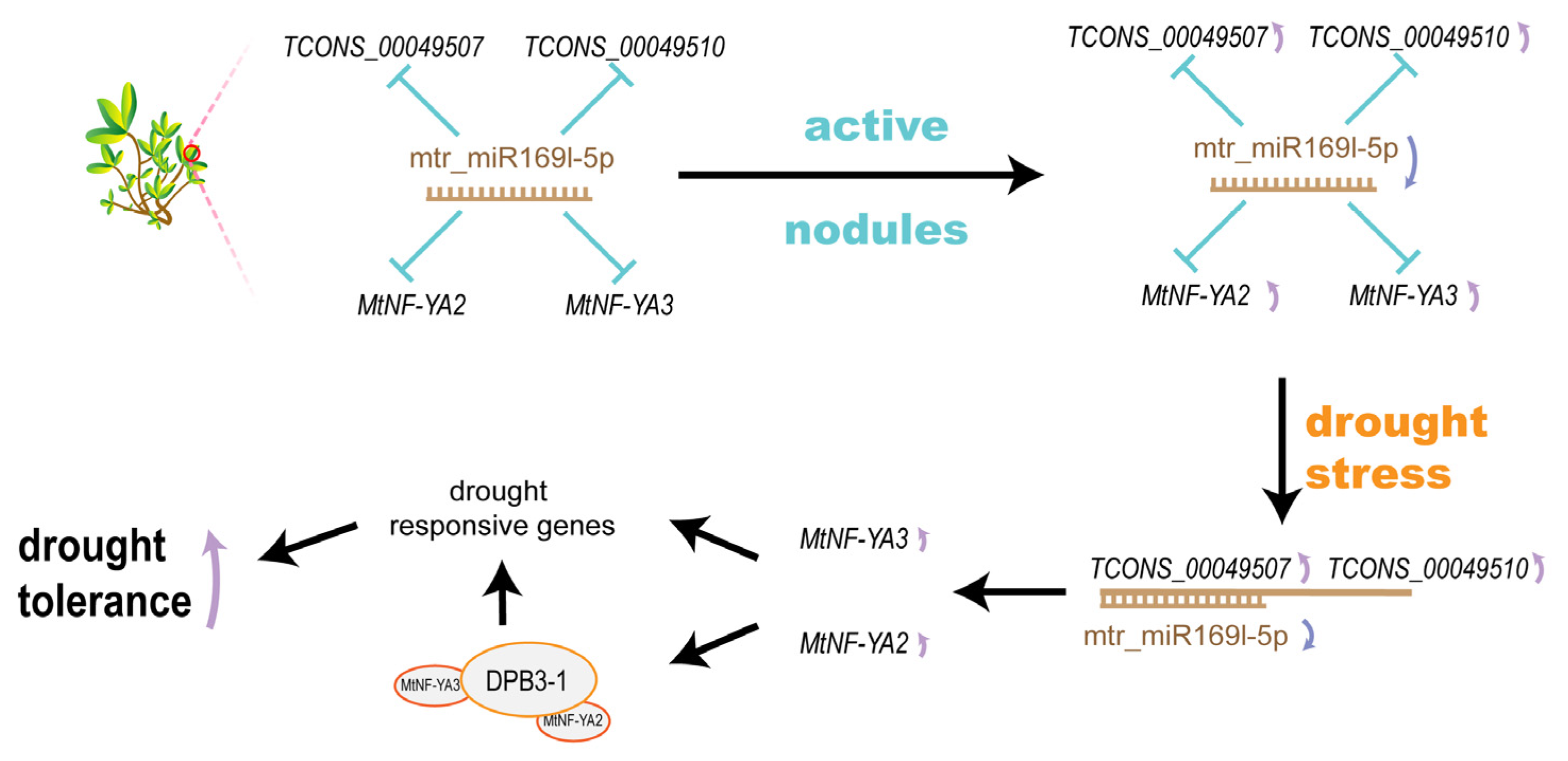
3.2. SNF-Related miRNA Contributed to Drought Tolerance
3.3. SNF-Induced ceRNA Network Impact on Drought Tolerance
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. Transcriptome Sequencing
4.3. Different Expression Pattern of Transcriptome
4.4. Feature Analysis of Transcriptome
4.5. Identification of ceRNA Network
4.6. qRT-PCR Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NN | no nodule treatment |
| IN | inactive nodule treatment |
| AN | active nodule treatment |
| D0 | no drought stress treatment |
| D1 | mild drought stress treatment |
| D2 | severe drought stress treatment |
| ceRNAs | competing endogenous RNAs |
| SNF | symbiotic nitrogen fixation |
| circRNAs | circular RNAs |
| lncRNAs | long noncoding RNAs |
| sRNAs | small noncoding RNAs |
| miRNAs | microRNAs |
| siRNAs | small-interfering RNAs |
| DEGs | differentially expressed genes |
| DElncRNAs | differentially expressed lncRNAs |
| DEcircRNAs | differentially expressed circRNAs |
| DEmiRNAs | differentially expressed miRNAs |
| pri-miRNA | primary-miRNA |
| Pol II | RNA polymerase II |
| pre-miRNA | precursor-miRNA |
| DCL1 | Dicer-like protein 1 |
| RISC | RNA-induced silencing complex |
| AGO | Argonaute |
| MREs | miRNA response elements |
| NF-Y | NUCLEAR FACTOR-Y |
| NF-YA | nuclear transcription factor Y subunit A |
| GO | gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| WGCNA | weighted gene co-expression network analysis |
| LLO | lipid-linked oligosaccharide |
| Dol | dolichol |
| FDR | false discovery rate |
References
- National Centers for Environmental Information. DROUGHT: Monitoring Economic, Environmental, and Social Impacts; National Centers for Environmental Information: Hancock County, MS, USA, 2018. [Google Scholar]
- Kunert, K.J.; Vorster, B.J.; Fenta, B.A.; Kibido, T.; Dionisio, G.; Foyer, C.H. Drought Stress Responses in Soybean Roots and Nodules. Front. Plant Sci. 2016, 7, 1015. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kumar Patel, M.; Kumar, N.; Bajpai, A.B.; Siddique, K.H.M. Metabolomics and Molecular Approaches Reveal Drought Stress Tolerance in Plants. Int. J. Mol. Sci. 2021, 22, 9108. [Google Scholar] [CrossRef] [PubMed]
- Gelaw, T.A.; Sanan-Mishra, N. Non-Coding RNAs in Response to Drought Stress. Int. J. Mol. Sci. 2021, 22, 12519. [Google Scholar] [CrossRef]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017, 258, 122–136. [Google Scholar] [CrossRef] [PubMed]
- López-Galiano, M.J.; García-Robles, I.; González-Hernández, A.I.; Camañes, G.; Vicedo, B.; Real, M.D.; Rausell, C. Expression of miR159 Is Altered in Tomato Plants Undergoing Drought Stress. Plants 2019, 8, 201. [Google Scholar] [CrossRef]
- Lindstrom, K.; Mousavi, S.A. Effectiveness of nitrogen fixation in rhizobia. Microb. Biotechnol. 2020, 13, 1314–1335. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Liu, W.; Nandety, R.S.; Crook, A.; Mysore, K.S.; Pislariu, C.I.; Frugoli, J.; Dickstein, R.; Udvardi, M.K. Celebrating 20 Years of Genetic Discoveries in Legume Nodulation and Symbiotic Nitrogen Fixation. Plant Cell 2020, 32, 15–41. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, P.; Li, B.; Hu, T. Effect of nodules on dehydration response in alfalfa (Medicago sativa L.). Environ. Exp. Bot. 2013, 86, 29–34. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Zhang, P.; Cao, Y.; Hu, T.; Yang, P. Rhizobium symbiosis contribution to short-term salt stress tolerance in alfalfa (Medicago sativa L.). Plant Soil 2016, 402, 247–261. [Google Scholar] [CrossRef]
- Sulieman, S.; Van Ha, C.; Nasr Esfahani, M.; Watanabe, Y.; Nishiyama, R.; Pham, C.T.; Van Nguyen, D.; Tran, L.S. DT2008: A promising new genetic resource for improved drought tolerance in soybean when solely dependent on symbiotic N2 fixation. BioMed Res. Int. 2015, 2015, 687213. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Liao, J.-Y.; Li, Z.-Y.; Yu, Y.; Zhang, J.-P.; Li, Q.-F.; Qu, L.-H.; Shu, W.-S.; Chen, Y.-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J. 2017, 89, 577–589. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, M.; Zhang, X.; Liu, M.; Yang, C.; Chen, Y.; Chen, R.; Wen, J.; Mysore, K.S.; Zhang, W.-H. Novel phosphate deficiency-responsive long non-coding RNAs in the legume model plant Medicago truncatula. J. Exp. Bot. 2017, 68, 5937–5948. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cui, J.; Meng, J.; Luan, Y. Interactions and links among the noncoding RNAs in plants under stresses. Theor. Appl. Genet. 2020, 133, 3235–3248. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Chen, Y.; Wu, Z.-K.; Foster, F.S.; Yang, Z.; Li, X.; Yang, B.B. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur. Heart J. 2017, 38, 1402–1412. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef]
- Borges, F.; Martienssen, R.A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 2015, 16, 727–741. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Yang, T.; Ma, H.; Zhang, J.; Wu, T.; Yao, Y. Systematic identification of long noncoding RNAs expressed during light-induced anthocyanin accumulation in apple fruit. Plant J. 2019, 100, 572–590. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Cui, J.; Hou, X.; Yang, G.; Xiao, Y.; Han, L.; Meng, J.; Luan, Y. Sl-lncRNA15492 interacts with Sl-miR482a and affects Solanum lycopersicum immunity against Phytophthora infestans. Plant J. 2020, 103, 1561–1574. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, Y.; Qi, X. LncRNA TCONS_00021861 is functionally associated with drought tolerance in rice (Oryza sativa L.) via competing endogenous RNA regulation. BMC Plant Biol. 2021, 21, 410. [Google Scholar] [CrossRef] [PubMed]
- Okuma, N.; Soyano, T.; Suzaki, T.; Kawaguchi, M. MIR2111-5 locus and shoot-accumulated mature miR2111 systemically enhance nodulation depending on HAR1 in Lotus japonicus. Nat. Commun. 2020, 11, 5192. [Google Scholar] [CrossRef]
- Tsikou, D.; Yan, Z.; Holt, D.B.; Abel, N.B.; Reid, D.E.; Madsen, L.H.; Bhasin, H.; Sexauer, M.; Stougaard, J.; Markmann, K. Systemic control of legume susceptibility to rhizobial infection by a mobile microRNA. Science 2018, 362, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Cui, X.; Yan, Q.; Gan, S.; Xue, D.; Dou, D.; Guo, N.; Xing, H. Overexpression of gma-miR1510a/b suppresses the expression of a NB-LRR domain gene and reduces resistance to Phytophthora sojae. Gene 2017, 621, 32–39. [Google Scholar] [CrossRef]
- Reynoso, M.A.; Blanco, F.A.; Bailey-Serres, J.; Crespi, M.; Zanetti, M.E. Selective recruitment of mRNAs and miRNAs to polyribosomes in response to rhizobia infection in Medicago truncatula. Plant J. 2013, 73, 289–301. [Google Scholar] [CrossRef]
- Sorin, C.; Declerck, M.; Christ, A.; Blein, T.; Ma, L.; Lelandais-Brière, C.; Njo, M.F.; Beeckman, T.; Crespi, M.; Hartmann, C. A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol. 2014, 202, 1197–1211. [Google Scholar] [CrossRef]
- Yu, Y.; Ni, Z.; Wang, Y.; Wan, H.; Hu, H.; Jiang, Q.; Sun, X.; Zhang, H. Overexpression of soybean miR169c confers increased drought stress sensitivity in transgenic Arabidopsis thaliana. Plant Sci. 2019, 285, 68–78. [Google Scholar] [CrossRef]
- Xing, L.; Zhu, M.; Luan, M.; Zhang, M.; Jin, L.; Liu, Y.; Zou, J.; Wang, L.; Xu, M. miR169q and NUCLEAR FACTOR YA8 enhance salt tolerance by activating PEROXIDASE1 expression in response to ROS. Plant Physiol. 2021, 188, 608–623. [Google Scholar] [CrossRef] [PubMed]
- Laloum, T.; De Mita, S.; Gamas, P.; Baudin, M.; Niebel, A. CCAAT-box binding transcription factors in plants: Y so many? Trends Plant Sci. 2013, 18, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Ma, H.; Chen, S.; Gu, T.; Gong, J.; Fu, Y.; Ma, H.; Chen, S.; Gu, T.; Gong, J. Control of proline accumulation under drought via a novel pathway comprising the histone methylase CAU1 and the transcription factor ANAC055. J. Exp. Bot. 2018, 69, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Ma, K.; Lu, Z.; Lu, J.; Cui, J.; Wang, L.; Jin, B. Physiological, Transcriptomic, and Metabolic Responses of Ginkgo biloba L. to Drought, Salt, and Heat Stresses. Biomolecules 2020, 10, 1635. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Wang, F.; Ma, L.; Qi, Z.; Liu, S.; Chen, C.; Wu, J.; Wang, P.; Yang, C.; Wu, Y.; et al. Physiological and Biochemical Mechanisms and Cytology of Cold Tolerance in Brassica napus. Front. Plant Sci. 2020, 11, 1241. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Jalmi, S.K.; Bhagat, P.K.; Verma, N.; Sinha, A.K. A bHLH transcription factor, MYC2, imparts salt intolerance by regulating proline biosynthesis in Arabidopsis. FEBS J. 2020, 287, 2560–2576. [Google Scholar] [CrossRef]
- Fang, H.; Wright, T.; Jinn, J.-R.; Guo, W.; Zhang, N.; Wang, X.; Wang, Y.-J.; Xu, J. Engineering hydroxyproline-O-glycosylated biopolymers to reconstruct the plant cell wall for improved biomass processability. Biotechnol. Bioeng. 2020, 117, 945–958. [Google Scholar] [CrossRef]
- Zhang, G.; Hou, X.; Wang, L.; Xu, J.; Chen, J.; Fu, X.; Shen, N.; Nian, J.; Jiang, Z.; Hu, J.; et al. PHOTO-SENSITIVE LEAF ROLLING 1 encodes a polygalacturonase that modifies cell wall structure and drought tolerance in rice. New Phytol. 2021, 229, 890–901. [Google Scholar] [CrossRef]
- Sós-Hegedűs, A.; Domonkos, Á.; Tóth, T.; Gyula, P.; Kaló, P.; Szittya, G. Suppression of NB-LRR genes by miRNAs promotes nitrogen-fixing nodule development in Medicago truncatula. Plant Cell Environ. 2020, 43, 1117–1129. [Google Scholar] [CrossRef]
- Udvardi, M.; Poole, P.S. Transport and Metabolism in Legume-Rhizobia Symbioses. Annu. Rev. Plant Biol. 2013, 64, 781–805. [Google Scholar] [CrossRef]
- He, M.; Dijkstra, F.A. Drought effect on plant nitrogen and phosphorus: A meta-analysis. New Phytol. 2014, 204, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Weaver Natalie, D.; Kesarwani, M.; Dong, X. Induction of Protein Secretory Pathway Is Required for Systemic Acquired Resistance. Science 2005, 308, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Pattison, R.J.; Amtmann, A. N-glycan production in the endoplasmic reticulum of plants. Trends Plant Sci. 2009, 14, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Swiezewska, E.; Danikiewicz, W. Polyisoprenoids: Structure, biosynthesis and function. Prog. Lipid Res. 2005, 44, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ohyama, K.; Boudet, J.; Chen, Z.; Yang, J.; Zhang, M.; Muranaka, T.; Maurel, C.; Zhu, J.-K.; Gong, Z. Dolichol biosynthesis and its effects on the unfolded protein response and abiotic stress resistance in Arabidopsis. Plant Cell 2008, 20, 1879–1898. [Google Scholar] [CrossRef]
- Hou, S.; Liu, D.; He, P. Phytocytokines function as immunological modulators of plant immunity. Stress Biol. 2021, 1, 8. [Google Scholar] [CrossRef]
- Wan, J.; He, M.; Hou, Q.; Zou, L.; Yang, Y.; Wei, Y.; Chen, X. Cell wall associated immunity in plants. Stress Biol. 2021, 1, 3. [Google Scholar] [CrossRef]
- Nguyen, D.Q.; Brown, C.W.; Pegler, J.L.; Eamens, A.L.; Grof, C.P.L. Molecular Manipulation of MicroRNA397 Abundance Influences the Development and Salt Stress Response of Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 7879. [Google Scholar] [CrossRef]
- Li, L.; Yang, K.; Wang, S.; Lou, Y.; Zhu, C.; Gao, Z. Genome-wide analysis of laccase genes in moso bamboo highlights PeLAC10 involved in lignin biosynthesis and in response to abiotic stresses. Plant Cell Rep. 2020, 39, 751–763. [Google Scholar] [CrossRef]
- Huang, J.-H.; Qi, Y.-P.; Wen, S.-X.; Guo, P.; Chen, X.-M.; Chen, L.-S. Illumina microRNA profiles reveal the involvement of miR397a in Citrus adaptation to long-term boron toxicity via modulating secondary cell-wall biosynthesis. Sci. Rep. 2016, 6, 22900. [Google Scholar] [CrossRef]
- Huang, J.-H.; Zhang, L.-Y.; Lin, X.-J.; Gao, Y.; Zhang, J.; Huang, W.-L.; Zhao, D.; Ferrarezi, R.S.; Fan, G.-C.; Chen, L.-S. CsiLAC4 modulates boron flow in Arabidopsis and Citrus via high-boron-dependent lignification of cell walls. New Phytol. 2022, 233, 1257–1273. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Tang, Y.; Jia, P.; Zeng, Y.; Wang, B.; Wu, P.; Quan, Y.; Chen, A.; Li, Y.; Wu, J. A Cotton Lignin Biosynthesis Gene, GhLAC4, Fine-Tuned by ghr-miR397 Modulates Plant Resistance Against Verticillium dahliae. Front. Plant Sci. 2021, 12, 743795. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, L.; Chang, L.; Cao, Y.; Zhang, T.; Wang, Y.; Liu, Y.; Zhang, P.; Sun, X.; Wu, Y.; et al. Effect of rhizobia symbiosis on lignin levels and forage quality in alfalfa (Medicago sativa L.). Agric. Ecosyst. Environ. 2016, 233, 55–59. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, G.; Singh, J.; Khan, A.; Fazio, G.; Saltzgiver, M.; Xia, R. Laccase Directed Lignification Is One of the Major Processes Associated With the Defense Response Against Pythium ultimum Infection in Apple Roots. Front. Plant Sci. 2021, 12, 629776. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, Q.; Zhu, Z.; Wan, H.; Qin, Y.; Zhang, H. Integrated analyses of the transcriptome and small RNA of the hemiparasitic plant Monochasma savatieri before and after establishment of parasite-host association. BMC Plant Biol. 2021, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, J.; Hu, Y.; Fang, C.; Yu, Y.; Yang, J.; Zhu, B.; Ruan, Y.-L.; Zhu, Z. Overexpression of sly-miR398b increased salt sensitivity likely via regulating antioxidant system and photosynthesis in tomato. Environ. Exp. Bot. 2021, 181, 104273. [Google Scholar] [CrossRef]
- Lin, K.-Y.; Wu, S.-Y.; Hsu, Y.-H.; Lin, N.-S. MiR398-regulated antioxidants contribute to Bamboo mosaic virus accumulation and symptom manifestation. Plant Physiol. 2022, 188, 593–607. [Google Scholar] [CrossRef]
- De la Rosa, C.; Covarrubias, A.A.; Reyes, J.L. A dicistronic precursor encoding miR398 and the legume-specific miR2119 coregulates CSD1 and ADH1 mRNAs in response to water deficit. Plant Cell Environ. 2019, 42, 133–144. [Google Scholar] [CrossRef]
- Leyva-González, M.A.; Ibarra-Laclette, E.; Cruz-Ramírez, A.; Herrera-Estrella, L. Functional and transcriptome analysis reveals an acclimatization strategy for abiotic stress tolerance mediated by Arabidopsis NF-YA family members. PLoS ONE 2012, 7, e48138. [Google Scholar] [CrossRef]
- Deng, Z.; Wu, H.; Li, D.; Li, L.; Wang, Z.; Yuan, W.; Xing, Y.; Li, C.; Liang, D. Root-to-Shoot Long-Distance Mobile miRNAs Identified from Nicotiana Rootstocks. Int. J. Mol. Sci. 2021, 22, 12821. [Google Scholar] [CrossRef]
- Li, J.; Duan, Y.; Sun, N.; Wang, L.; Feng, S.; Fang, Y.; Wang, Y. The miR169n-NF-YA8 regulation module involved in drought resistance in Brassica napus L. Plant Sci. 2021, 313, 111062. [Google Scholar] [CrossRef]
- Singroha, G.; Sharma, P.; Sunkur, R. Current status of microRNA-mediated regulation of drought stress responses in cereals. Physiol. Plant. 2021, 172, 1808–1821. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-X.; Oono, Y.; Zhu, J.; He, X.-J.; Wu, J.-M.; Iida, K.; Lu, X.-Y.; Cui, X.; Jin, H.; Zhu, J.-K. The Arabidopsis NFYA5 Transcription Factor Is Regulated Transcriptionally and Posttranscriptionally to Promote Drought Resistance. Plant Cell 2008, 20, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-J.; Yu, T.-F.; Li, X.-H.; Cao, X.-Y.; Ma, J.; Chen, J.; Zhou, Y.-B.; Chen, M.; Ma, Y.-Z.; Zhang, J.-H.; et al. Overexpression of GmNFYA5 confers drought tolerance to transgenic Arabidopsis and soybean plants. BMC Plant Biol. 2020, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-J.; Fu, J.-D.; Tang, Y.-M.; Yu, T.-F.; Yin, Z.-G.; Chen, J.; Zhou, Y.-B.; Chen, M.; Xu, Z.-S.; Ma, Y.-Z. GmNFYA13 Improves Salt and Drought Tolerance in Transgenic Soybean Plants. Front. Plant Sci. 2020, 11, 587244. [Google Scholar] [CrossRef]
- Sato, H.; Mizoi, J.; Tanaka, H.; Maruyama, K.; Qin, F.; Osakabe, Y.; Morimoto, K.; Ohori, T.; Kusakabe, K.; Nagata, M.; et al. Arabidopsis DPB3-1, a DREB2A interactor, specifically enhances heat stress-induced gene expression by forming a heat stress-specific transcriptional complex with NF-Y subunits. Plant Cell 2014, 26, 4954–4973. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.I. The water culture method for growing plants without soil. Calif. Agric. Exp. Stn. 1950, 347, 32. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, D.; Bertin, N.; Hidalgo, C.A.; Venkatesan, K.; Tu, D.; Lee, D.; Rosenberg, J.; Svrzikapa, N.; Blanc, A.; Carnec, A.; et al. Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nat. Biotechnol. 2007, 25, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.I.; Ireland, A.; Lomax, J.; Ashburner, M.; Collins, R.; Eilbeck, K.; Lewis, S.; Mungall, C.; Richter, J.; et al. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006, 34, D322–D326. [Google Scholar] [CrossRef]
- Ramoni, M.F.; Sebastiani, P.; Kohane, I.S. Cluster analysis of gene expression dynamics. Proc. Natl. Acad. Sci. USA 2002, 99, 9121–9126. [Google Scholar] [CrossRef]
- Miller, L.D.; Long, P.M.; Wong, L.; Mukherjee, S.; McShane, L.M.; Liu, E.T. Optimal gene expression analysis by microarrays. Cancer Cell 2002, 2, 353–361. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Cui, J.; Yang, G.; He, X.; Meng, J.; Luan, Y. Comparative transcriptome analysis shows the defense response networks regulated by miR482b. Plant Cell Rep. 2019, 38, 1–13. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Without Drought (D0) | Mild Drought (D1) | Severe Drought (D2) |
|---|---|---|---|
| No Nodule (NN) | D0_NN | D1_NN | D2_NN |
| Inactive Nodule (IN) | D0_IN | D1_IN | D2_IN |
| Active Nodule (AN) | D0_AN | D1_AN | D2_AN |
| Treatment | miRNA | mRNA | lncRNA | CircRNA |
|---|---|---|---|---|
| D0 | mtr-miR2661 | TCONS_00055188 | ||
| mtr-miR397-5p | TCONS_00119643 | |||
| TCONS_00034465 | ||||
| TCONS_00002639 | ||||
| TCONS_00024844 | ||||
| mtr-miR408-3p | TCONS_00034465 | |||
| mtr-miR5215 | TCONS_00043731 | |||
| mtr-miR5248 | TCONS_00031594 | |||
| TCONS_00112333 | ||||
| D1 | mtr-miR2608 | MTR_0011s0020 | TCONS_00060817 | |
| TCONS_00003861 | ||||
| TCONS_00001413 | ||||
| TCONS_00109074 | ||||
| TCONS_00034210 | ||||
| TCONS_00093448 | ||||
| TCONS_00111162 | ||||
| TCONS_00028699 | ||||
| D2 | mtr-miR159b | MTR_0005s0200 | TCONS_00049507 | |
| TCONS_00049510 | ||||
| mtr-miR169l-5p | MTR_7g106450 | TCONS_00049507 | mtr_circ_0000090 | |
| MTR_2g041090 | TCONS_00049510 | mtr_circ_0000202 | ||
| mtr-miR397-5p | MTR_1g047800 | |||
| mtr-miR5291b | MTR_1g031650 | |||
| mtr-miR5747 | MTR_4g114960 | |||
| mtr-miR160c | TCONS_00103850 | |||
| mtr-miR171e-5p | TCONS_00000469 | |||
| TCONS_00000468 | ||||
| TCONS_00000467 | ||||
| mtr-miR2111m-3p | TCONS_00001765 | mtr_circ_0000167 | ||
| TCONS_00001764 | ||||
| TCONS_00117102 | ||||
| mtr-miR395a | TCONS_00036480 | |||
| TCONS_00109074 | ||||
| TCONS_00049510 | ||||
| mtr-miR398b | TCONS_00049507 | mtr_circ_0000202 | ||
| mtr-miR398c | TCONS_00049510 | |||
| mtr-miR5215 | TCONS_00050947 | |||
| mtr-miR5260 | TCONS_00050947 | mtr_circ_0000037 | ||
| mtr-miR5745a | TCONS_00000901 | mtr_circ_0000112 | ||
| TCONS_00036480 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, J.; Yang, P.; Wang, Y.; Qu, Q.; An, J.; Fu, B.; Hu, X.; Zhou, Y.; Hu, T.; Cao, Y. Identification of Competing Endogenous RNAs (ceRNAs) Network Associated with Drought Tolerance in Medicago truncatula with Rhizobium Symbiosis. Int. J. Mol. Sci. 2022, 23, 14237. https://doi.org/10.3390/ijms232214237
Jing J, Yang P, Wang Y, Qu Q, An J, Fu B, Hu X, Zhou Y, Hu T, Cao Y. Identification of Competing Endogenous RNAs (ceRNAs) Network Associated with Drought Tolerance in Medicago truncatula with Rhizobium Symbiosis. International Journal of Molecular Sciences. 2022; 23(22):14237. https://doi.org/10.3390/ijms232214237
Chicago/Turabian StyleJing, Jiaxian, Peizhi Yang, Yue Wang, Qihao Qu, Jie An, Bingzhe Fu, Xiaoning Hu, Yi Zhou, Tianming Hu, and Yuman Cao. 2022. "Identification of Competing Endogenous RNAs (ceRNAs) Network Associated with Drought Tolerance in Medicago truncatula with Rhizobium Symbiosis" International Journal of Molecular Sciences 23, no. 22: 14237. https://doi.org/10.3390/ijms232214237
APA StyleJing, J., Yang, P., Wang, Y., Qu, Q., An, J., Fu, B., Hu, X., Zhou, Y., Hu, T., & Cao, Y. (2022). Identification of Competing Endogenous RNAs (ceRNAs) Network Associated with Drought Tolerance in Medicago truncatula with Rhizobium Symbiosis. International Journal of Molecular Sciences, 23(22), 14237. https://doi.org/10.3390/ijms232214237