
Discovering and Targeting Dynamic Drugging Pockets of Oncogenic Proteins: The Role of Magnesium in Conformational Changes of the G12D Mutated Kirsten Rat Sarcoma-Guanosine Diphosphate Complex



Abstract
1. Introduction
2. Results and Discussion
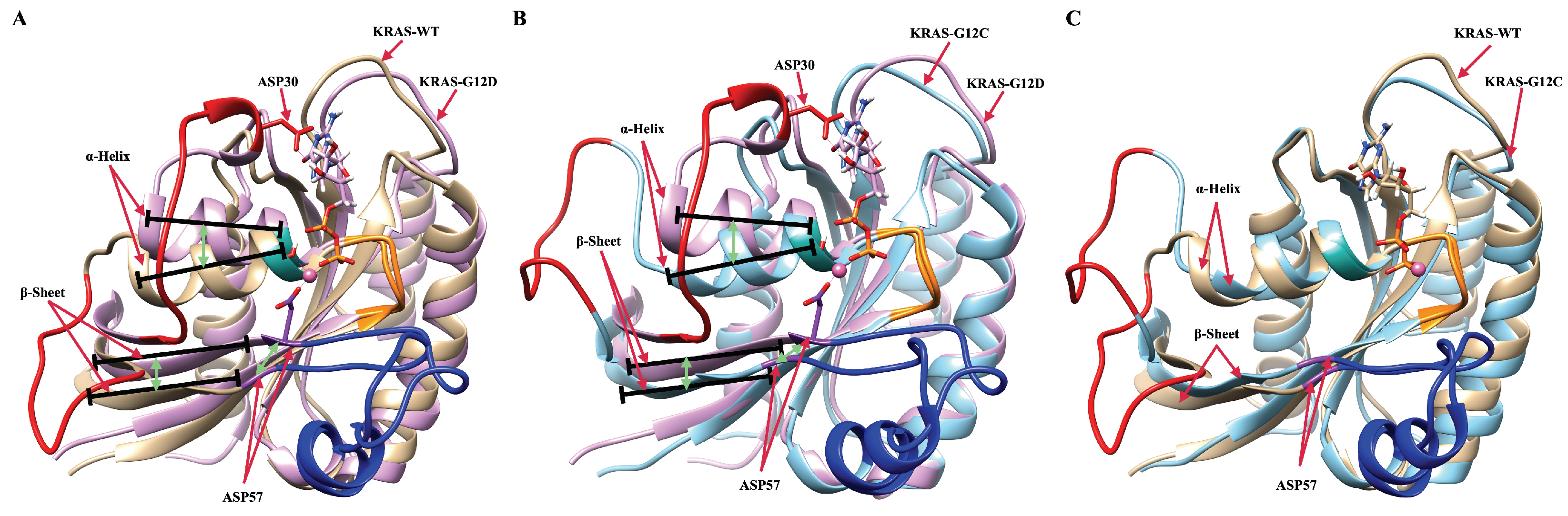
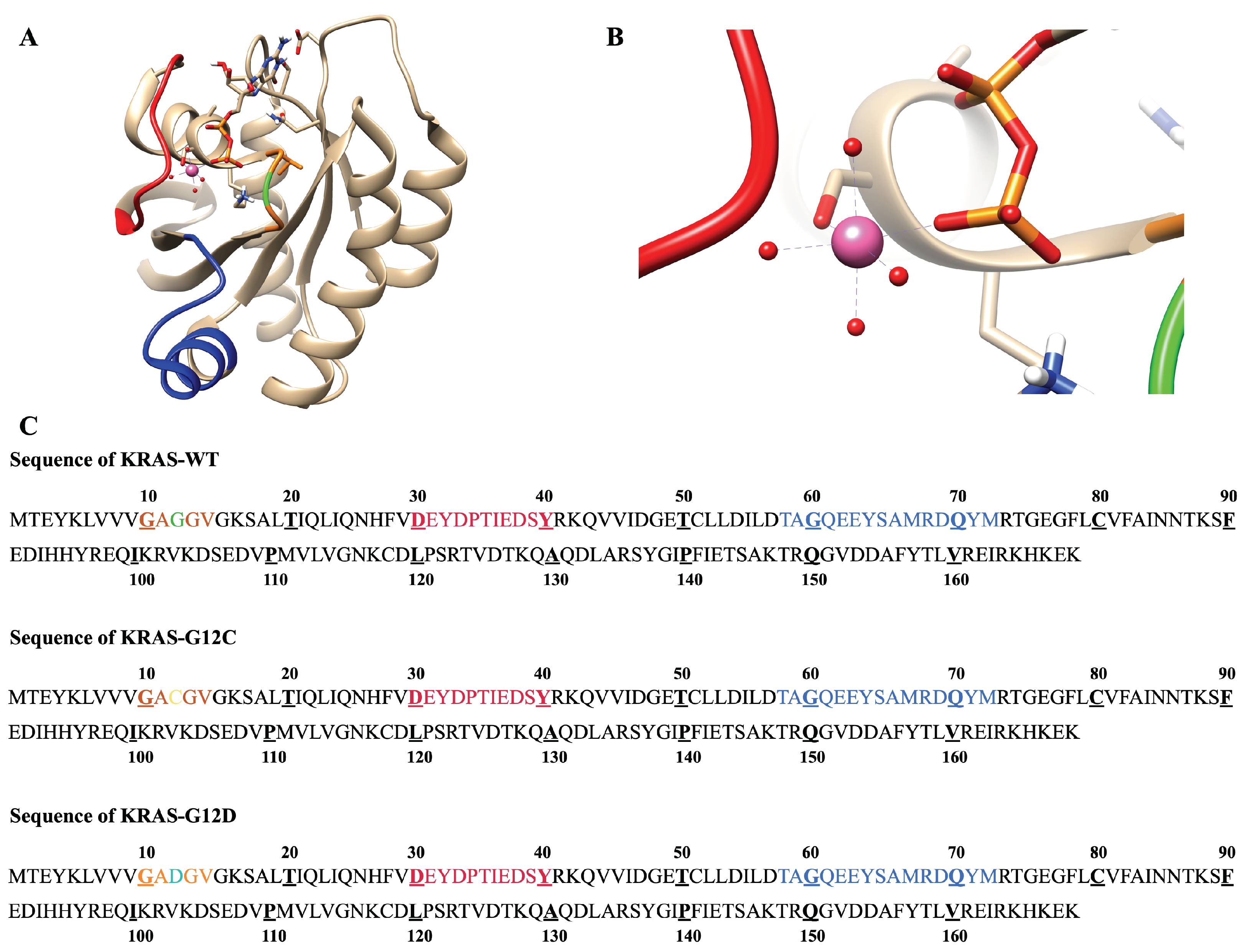
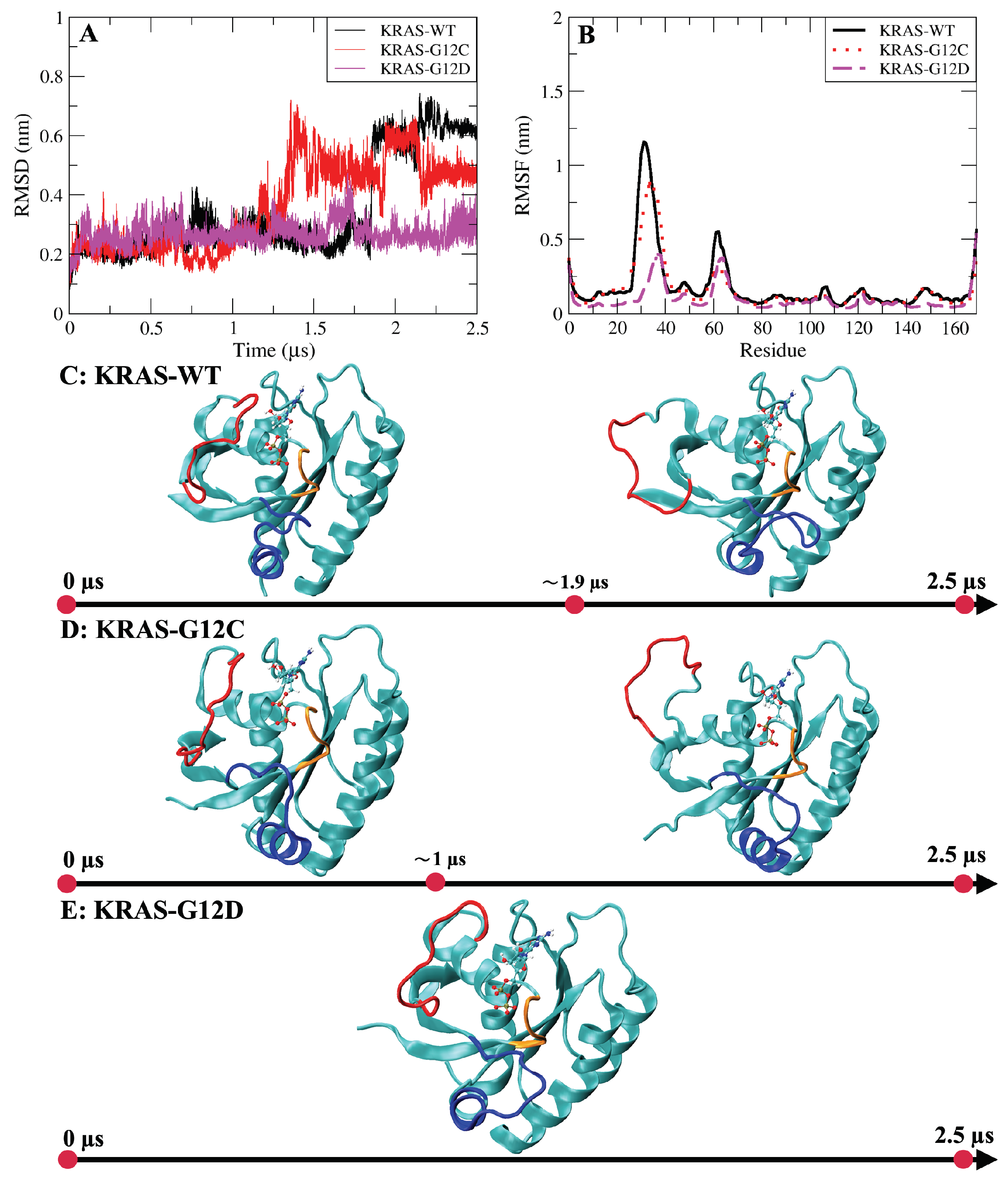
2.1. The Fluctuations and Stability of KRAS Protein Conformations with Different Mutation Isoforms
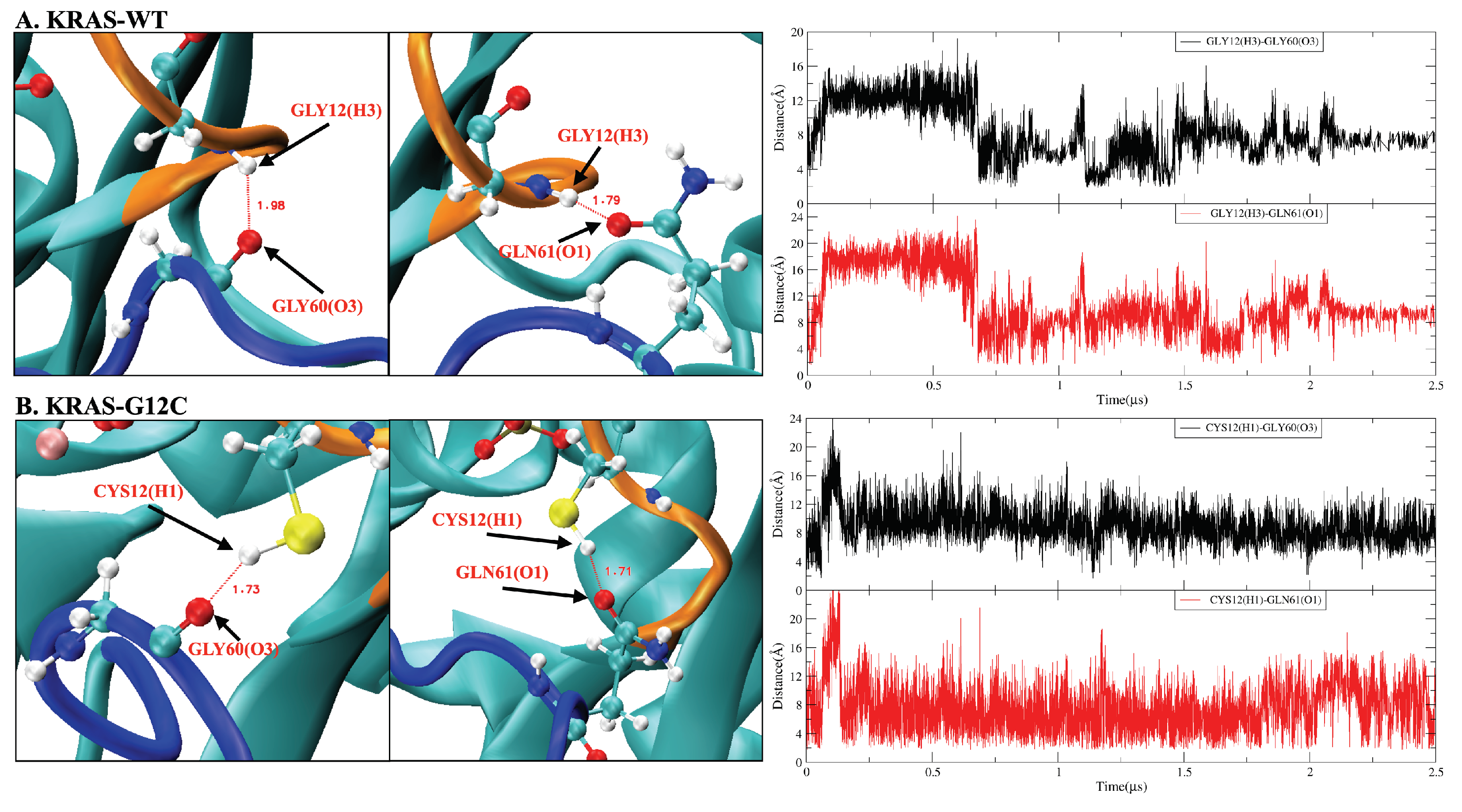
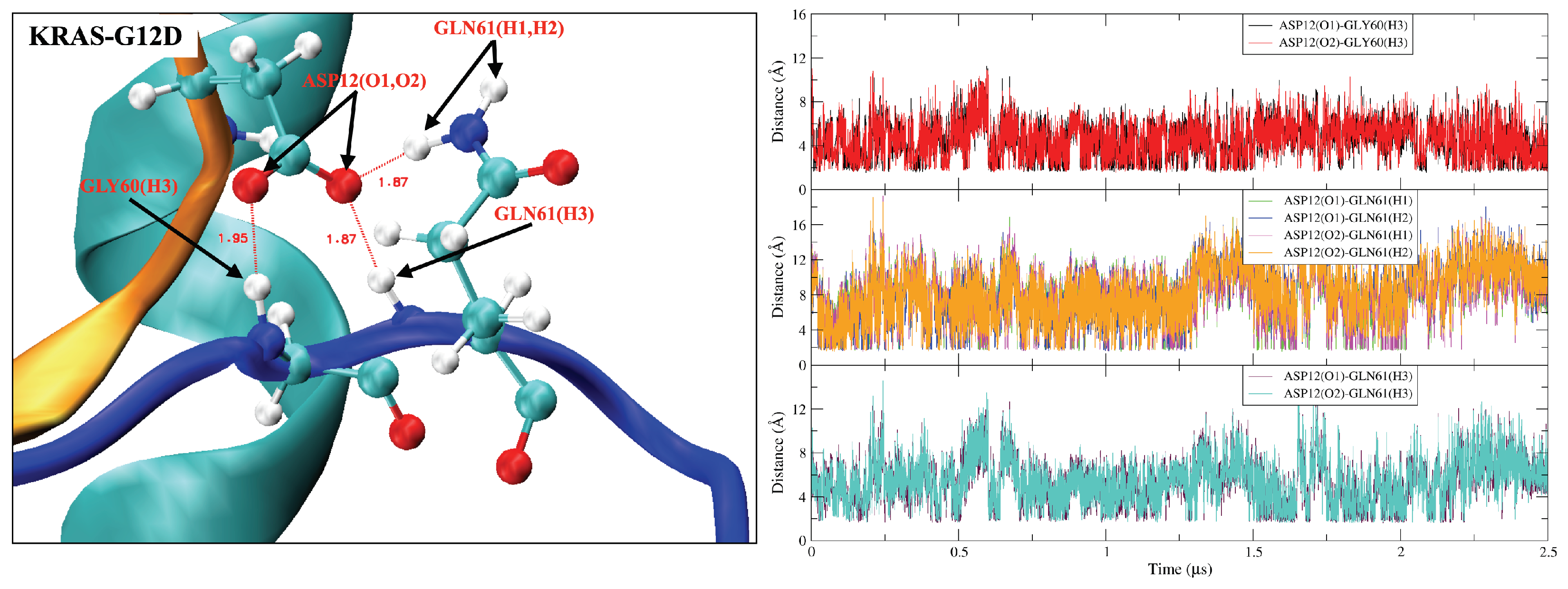
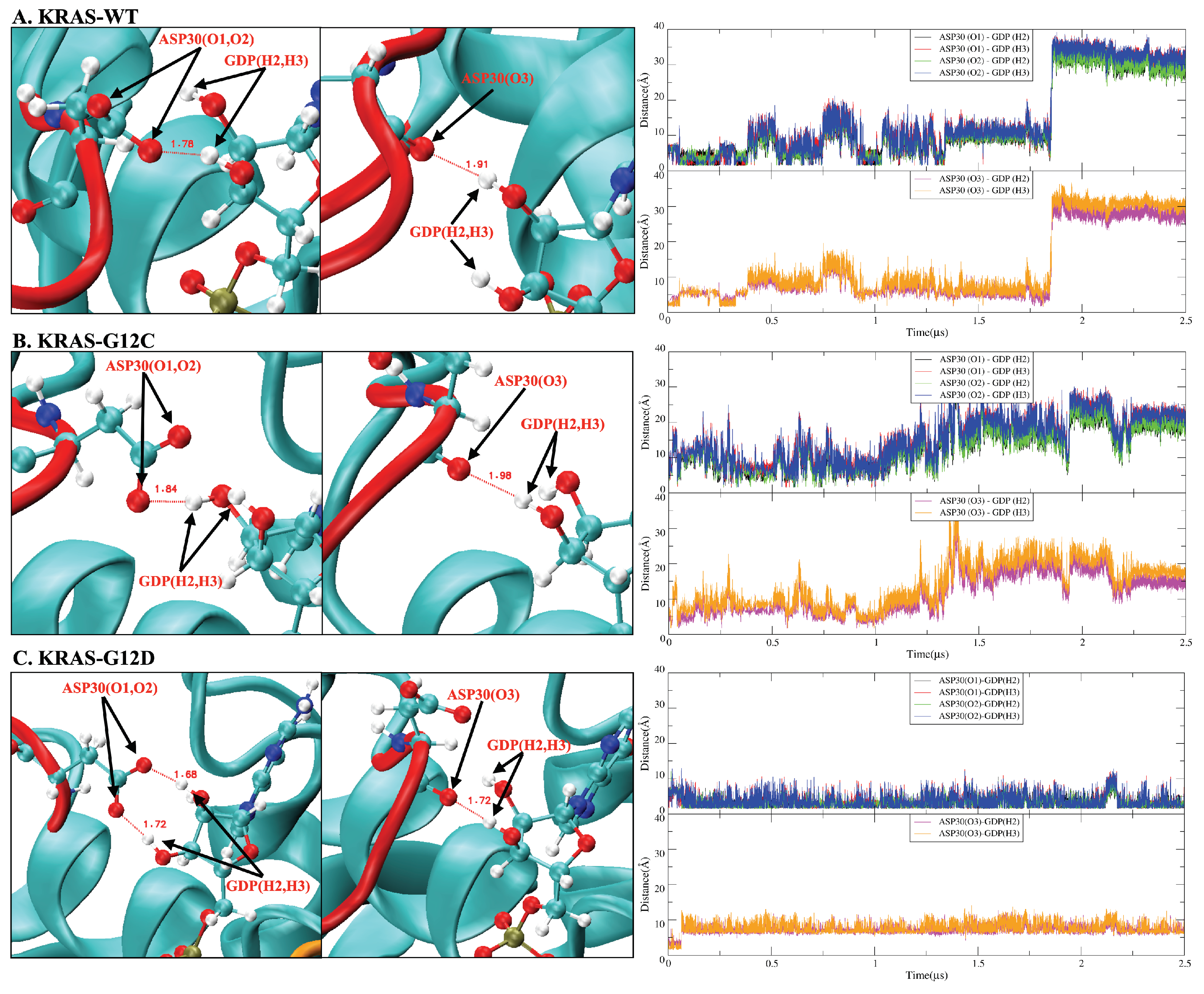
2.2. Mutation of GLY12 Enhances the Interaction between P-Loop and SW-II Domain
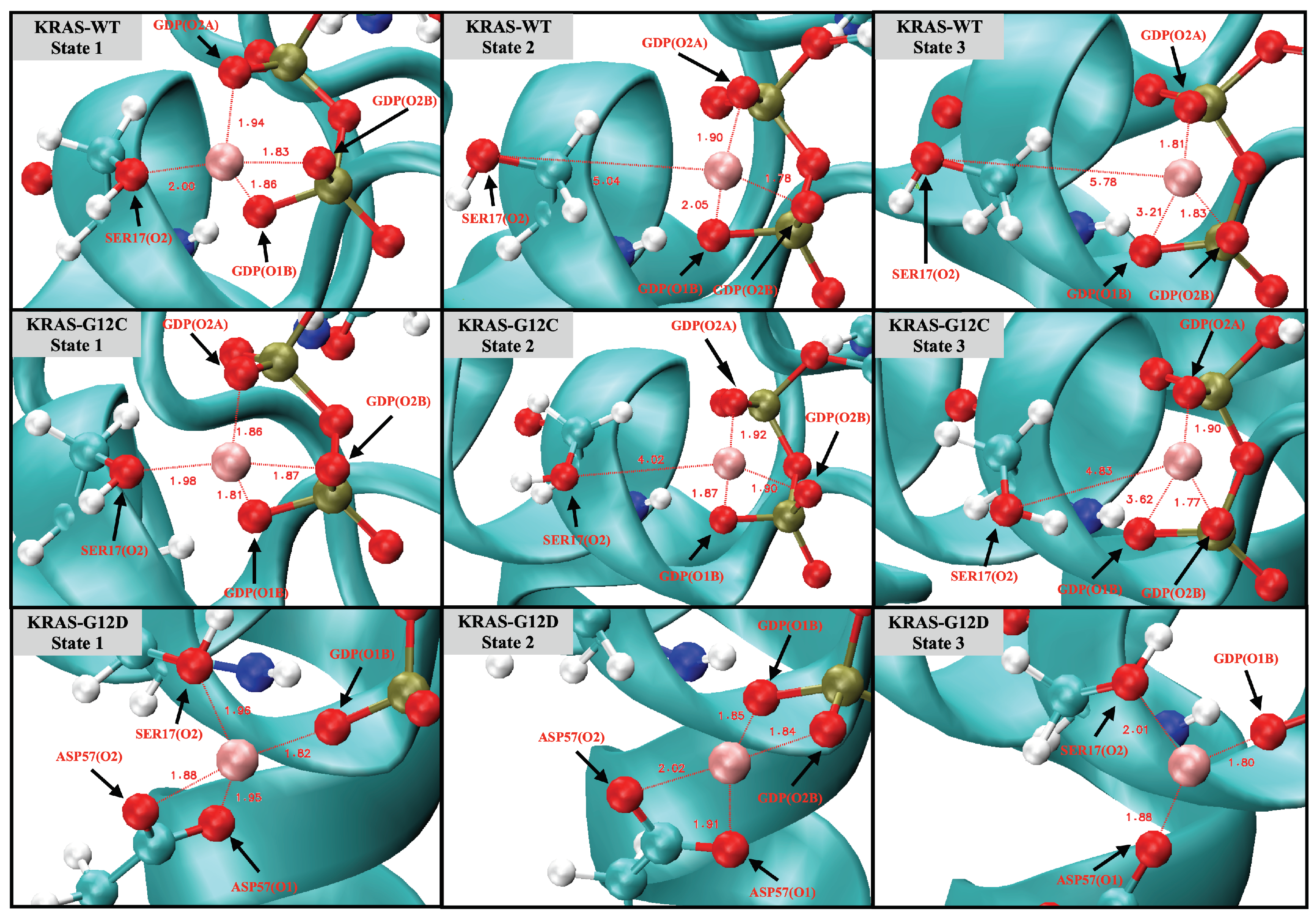
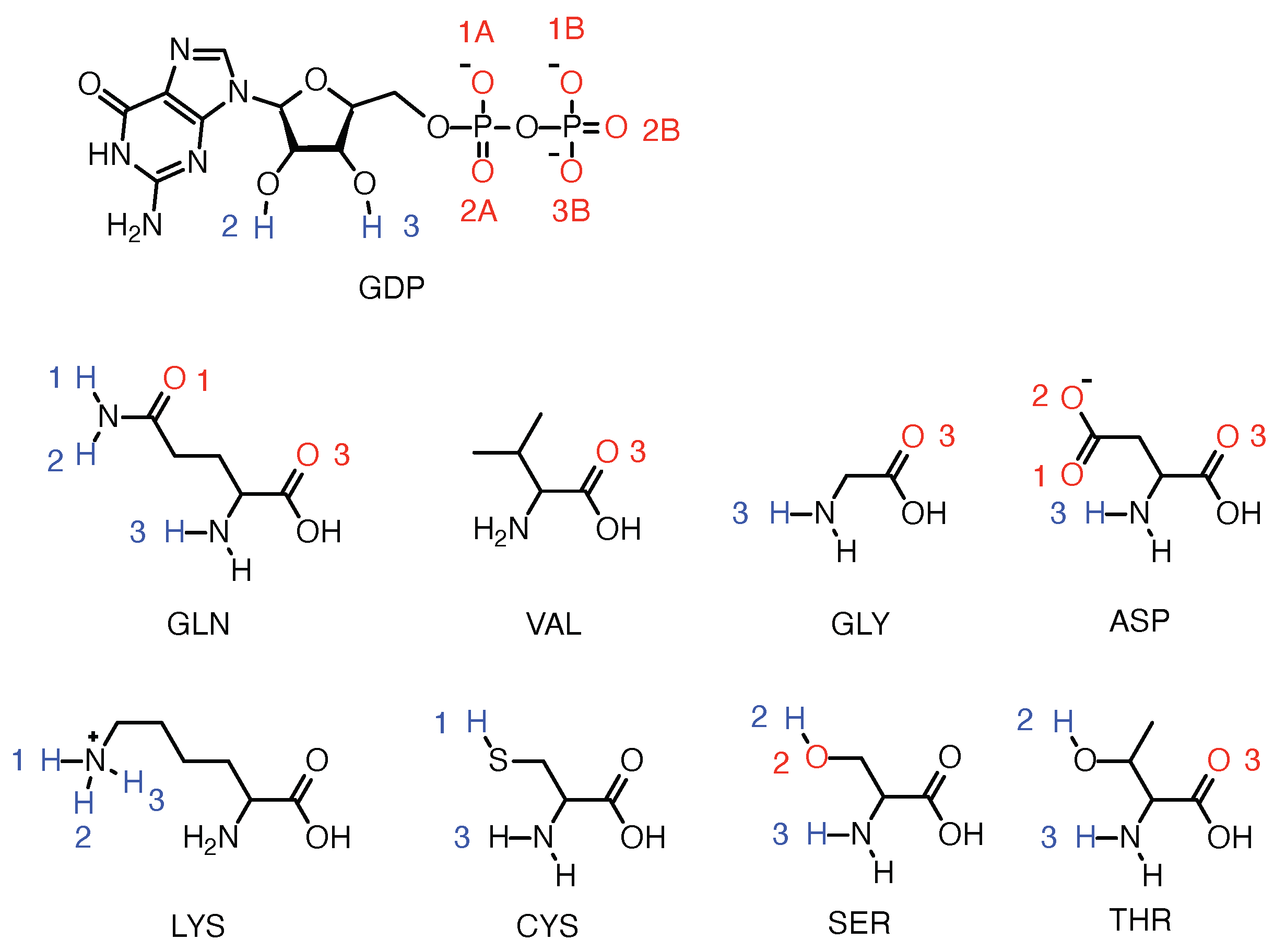
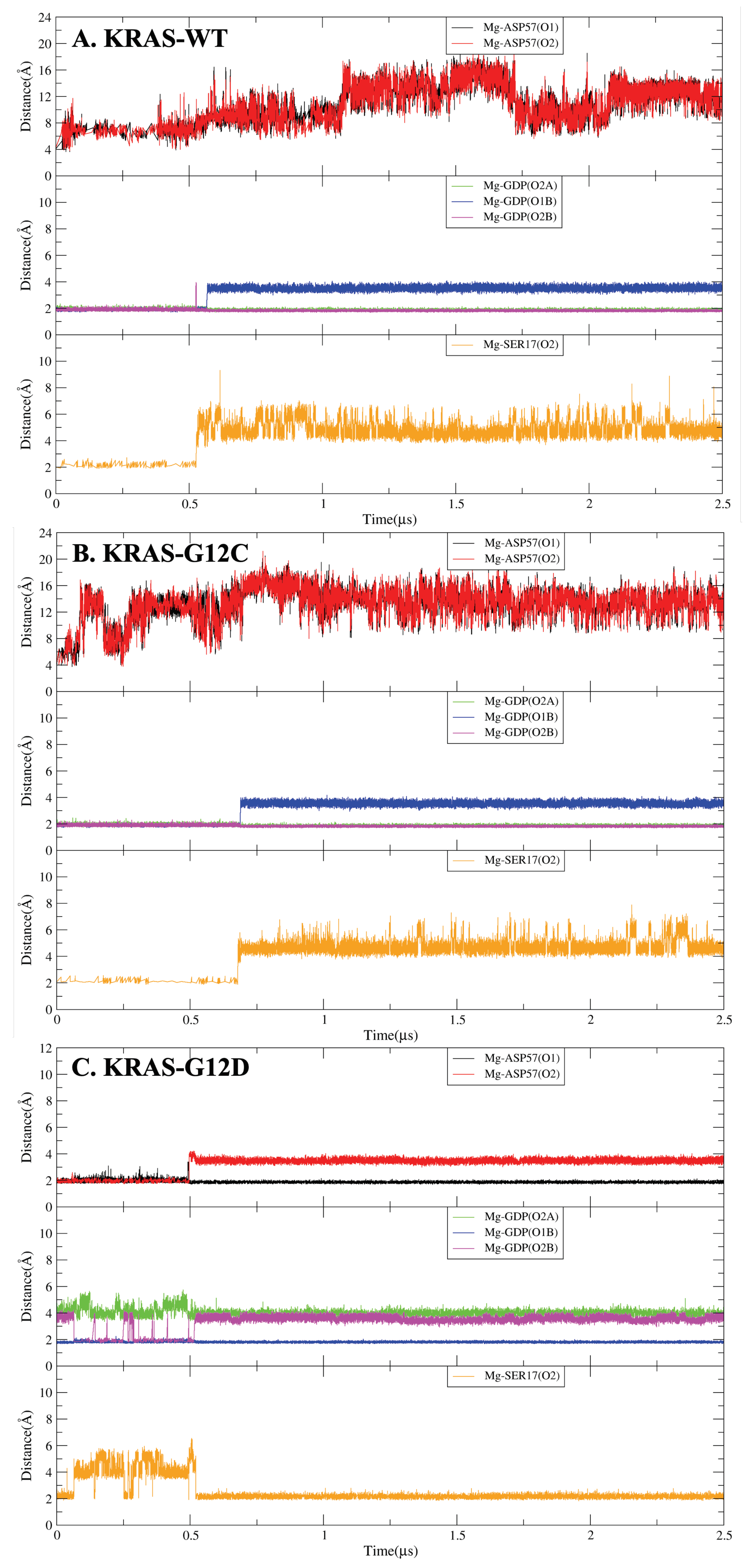
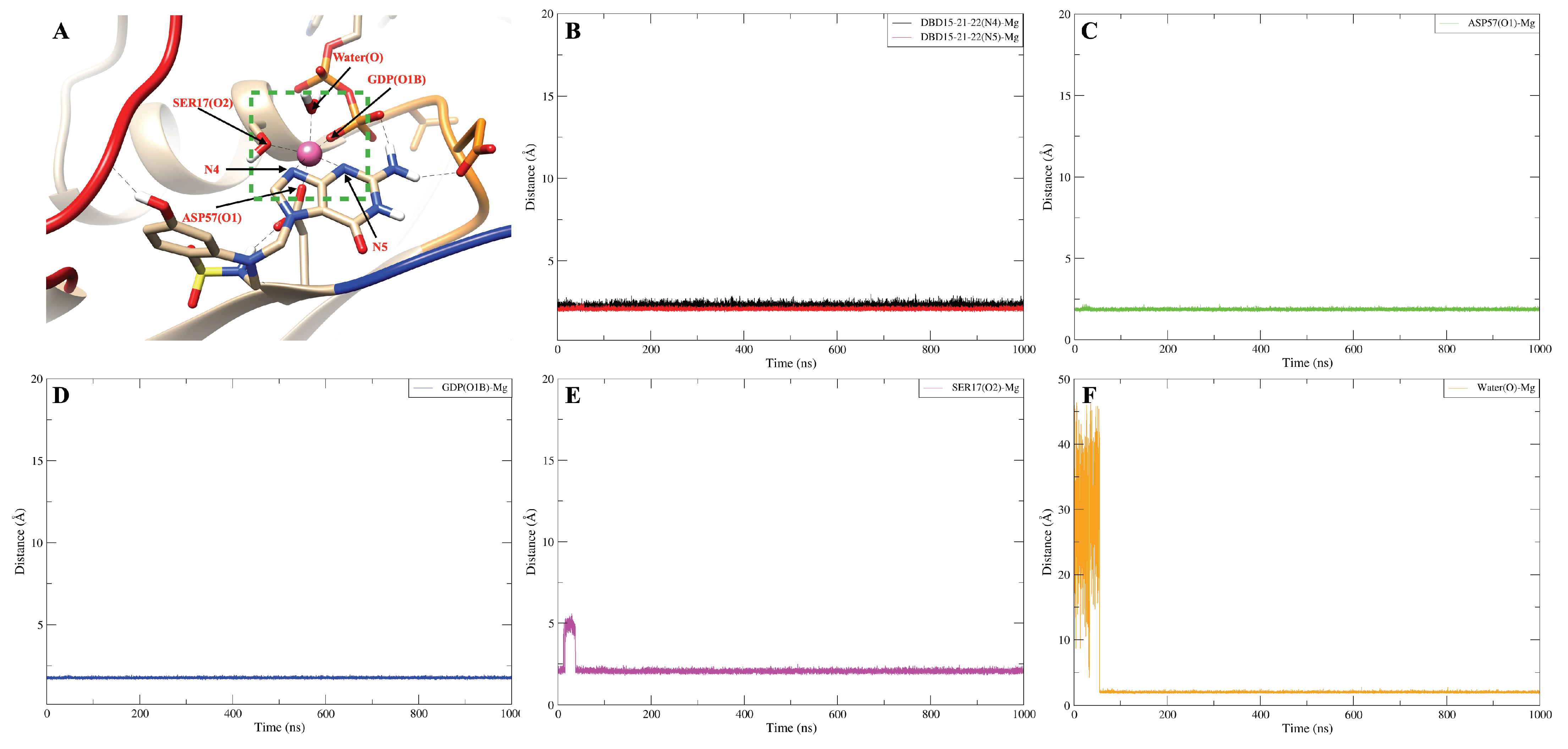
2.3. Strong Coordination Interactions between Mg and SER17, ASP57, GDP in KRAS-G12D
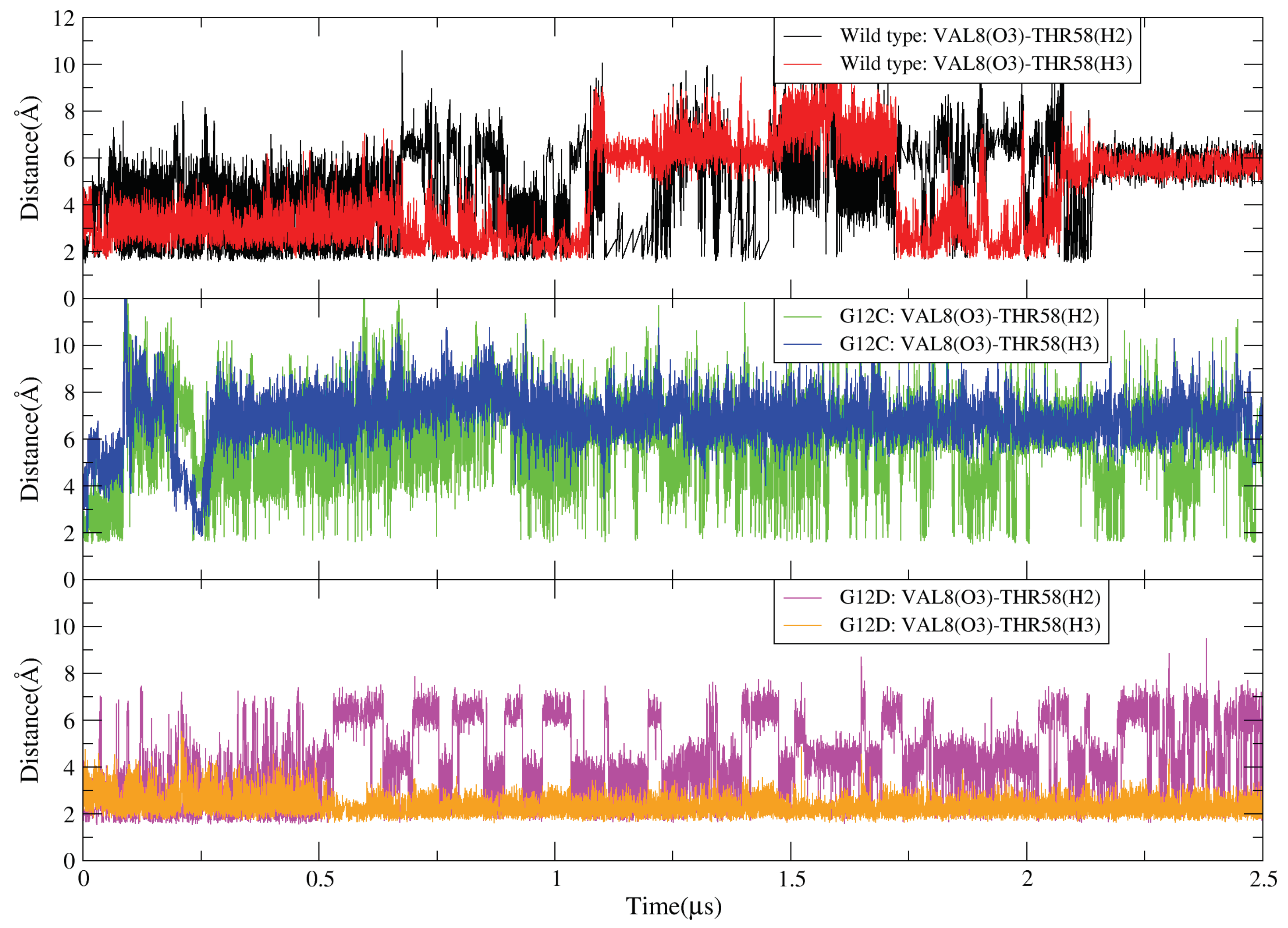
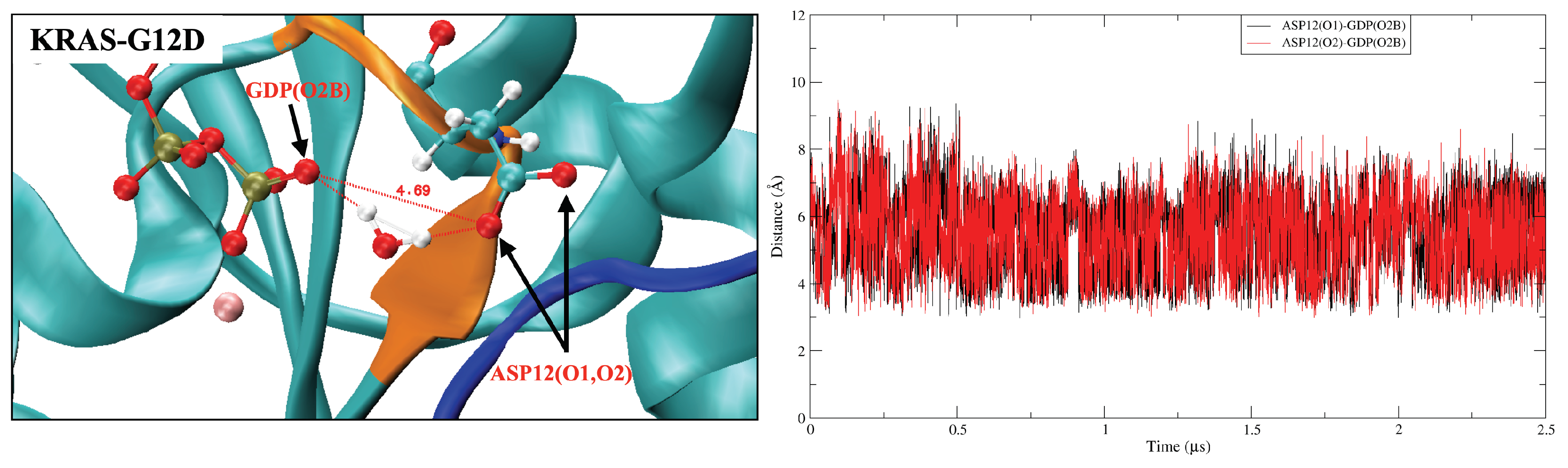
2.4. GDP Plays an Important Regulatory Role in the Conformational Change of SW-I
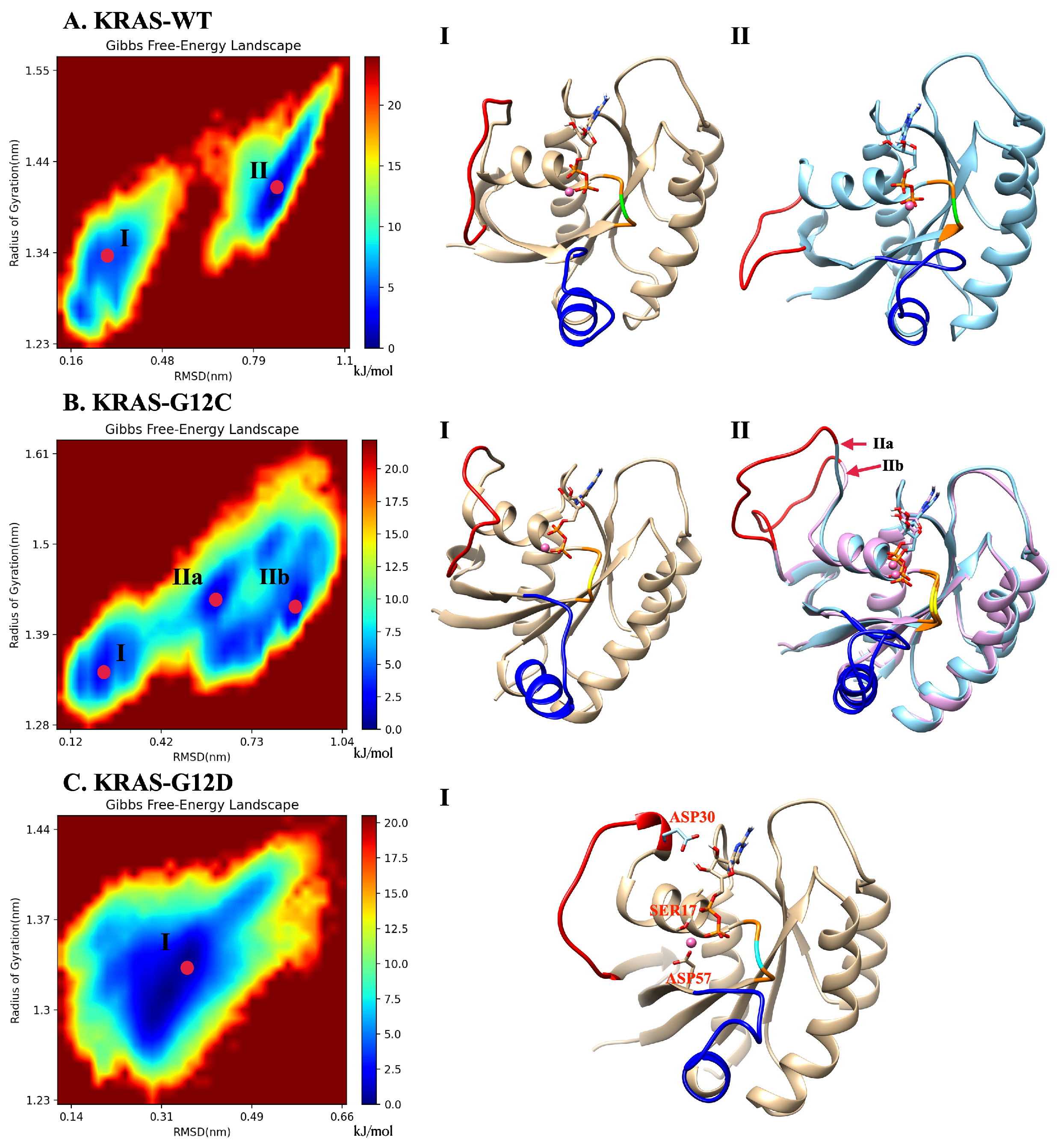
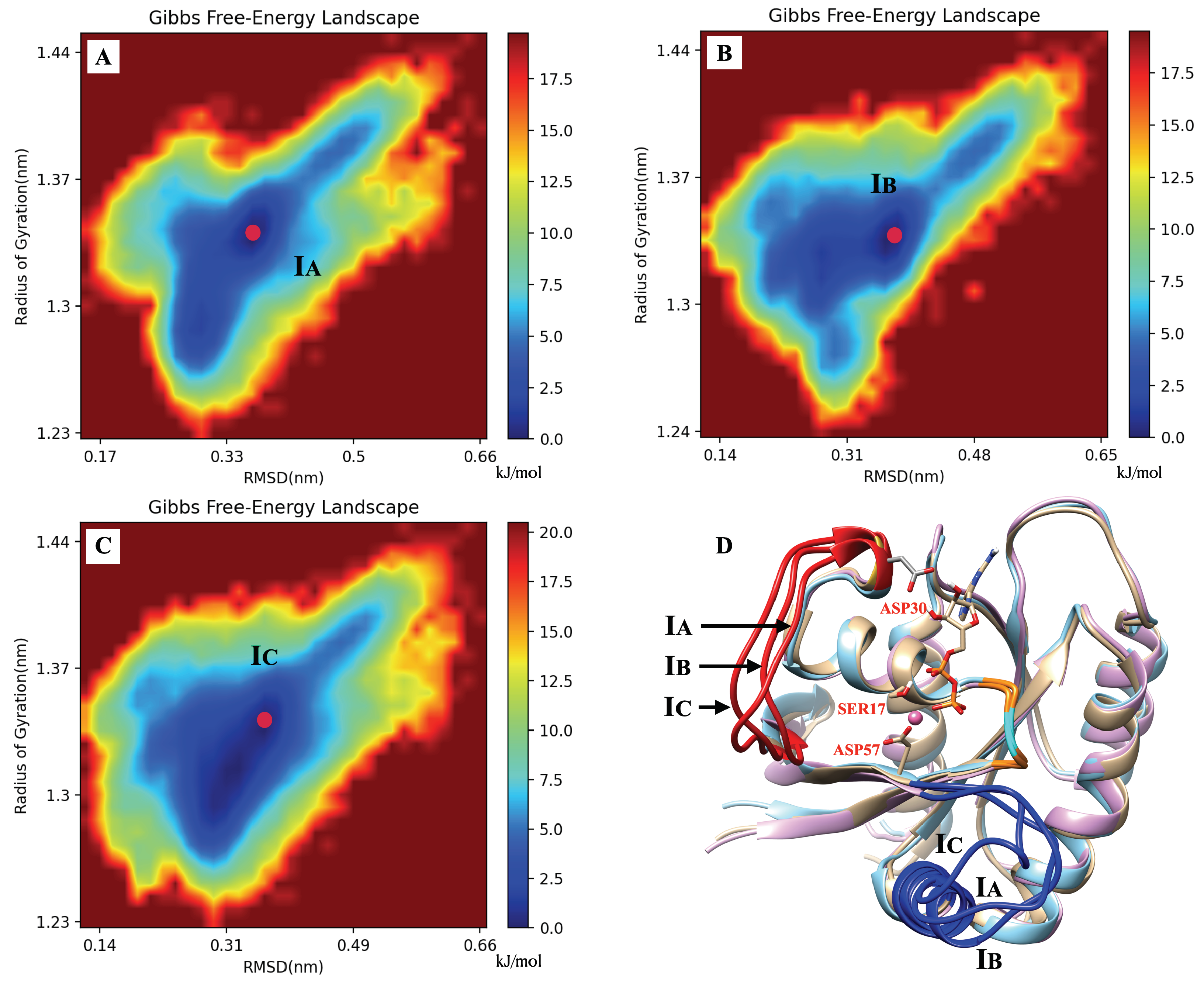
2.5. The Dominant Conformations of Wild-Type and Mutated KRAS
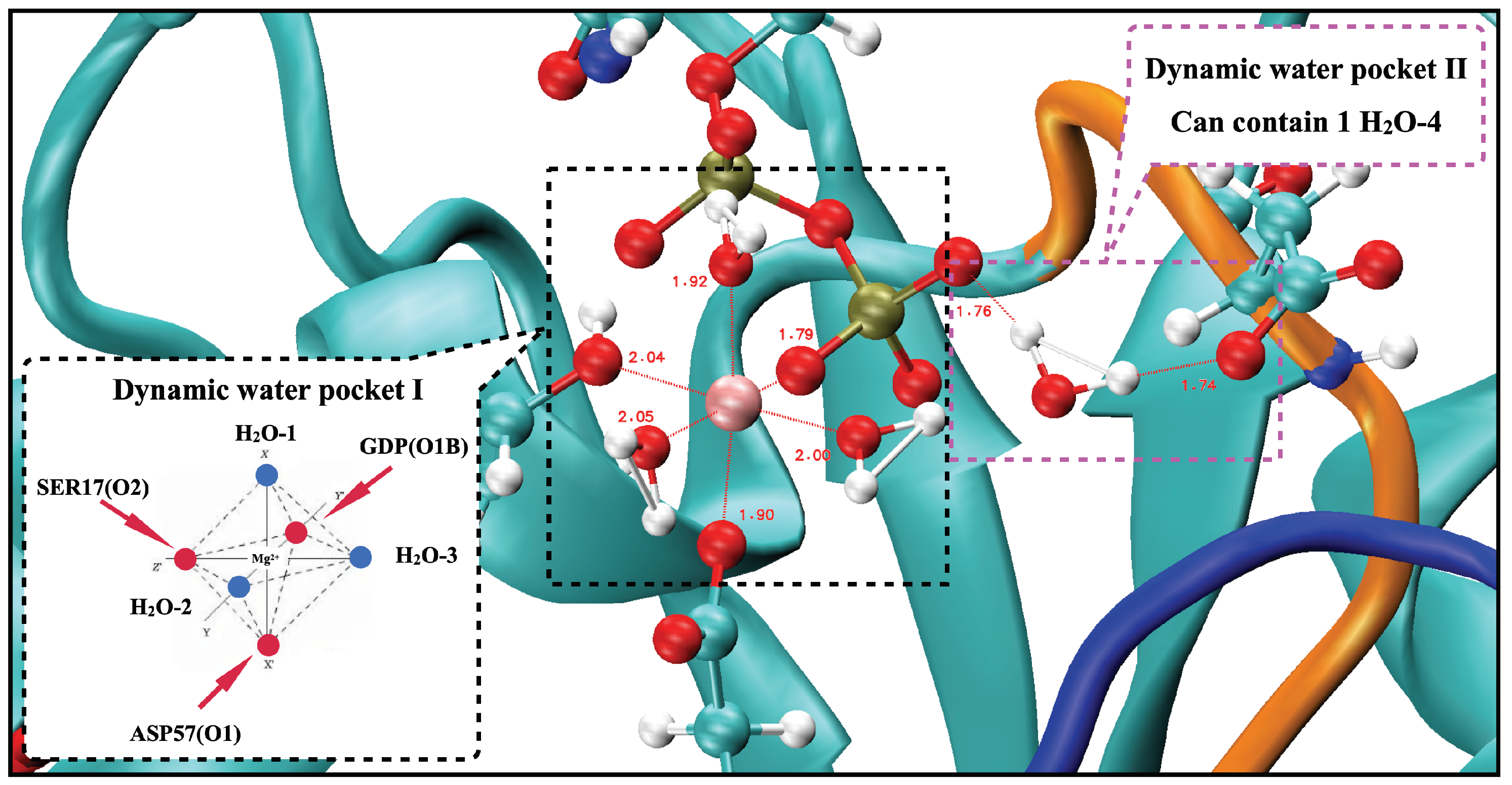
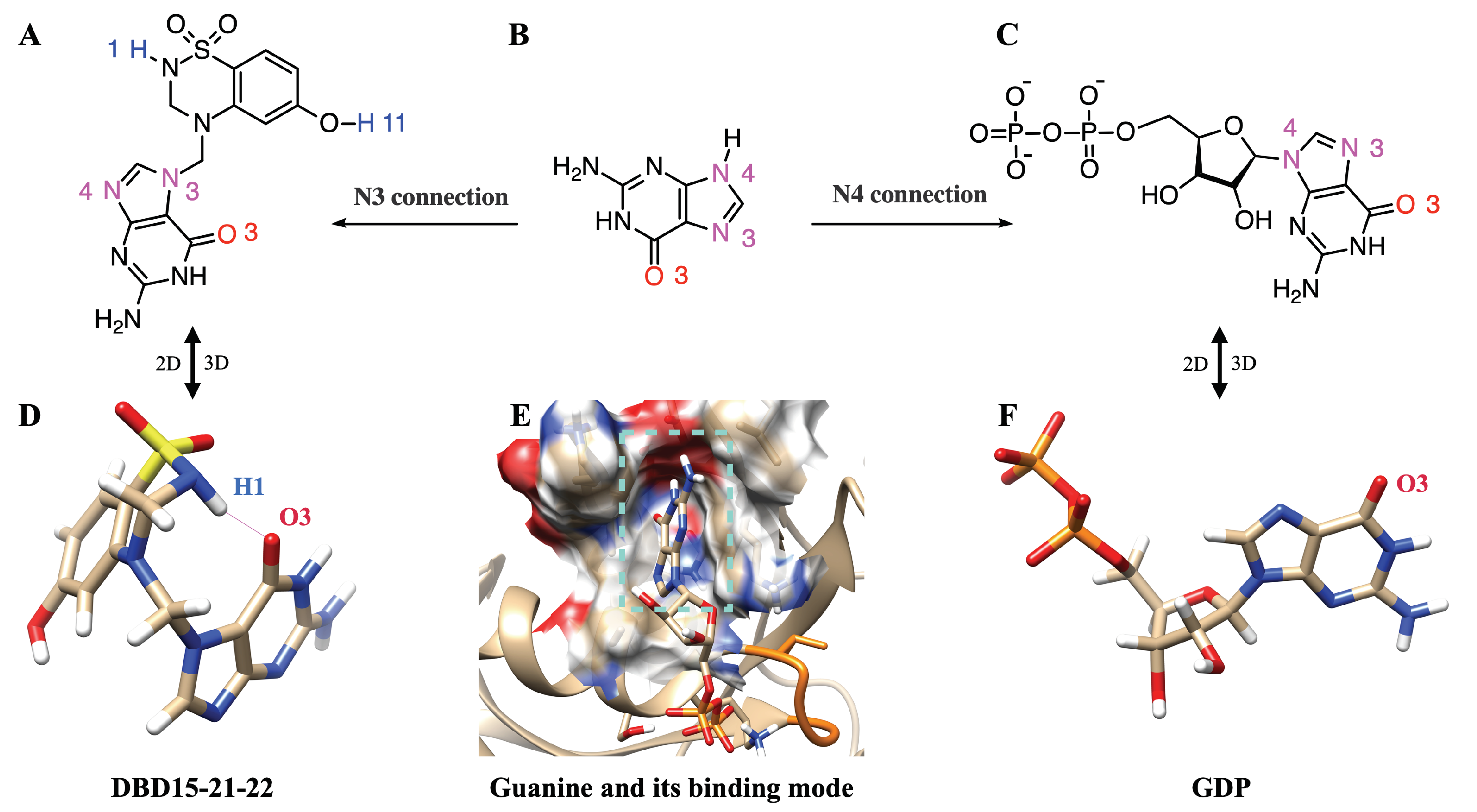
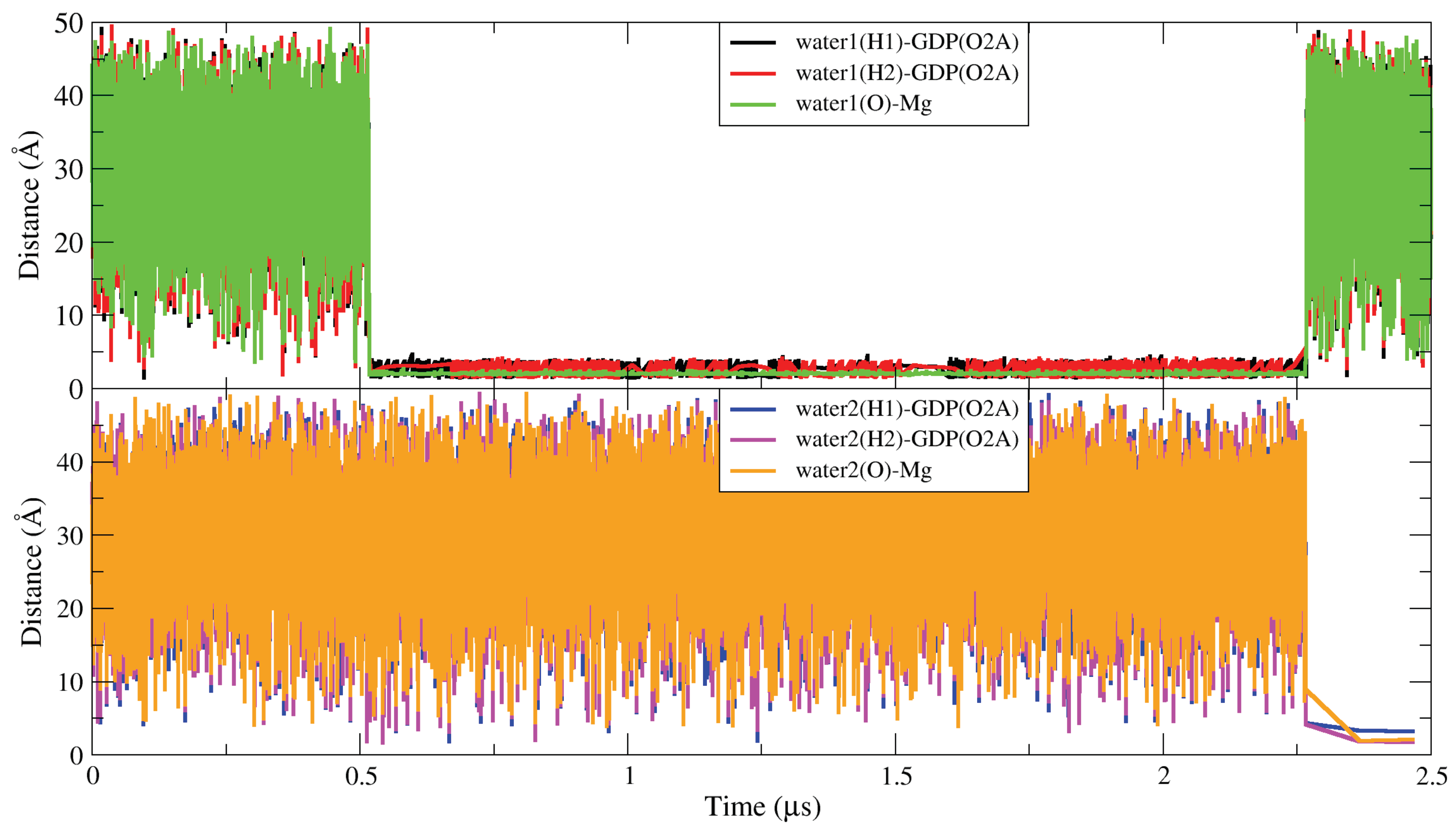
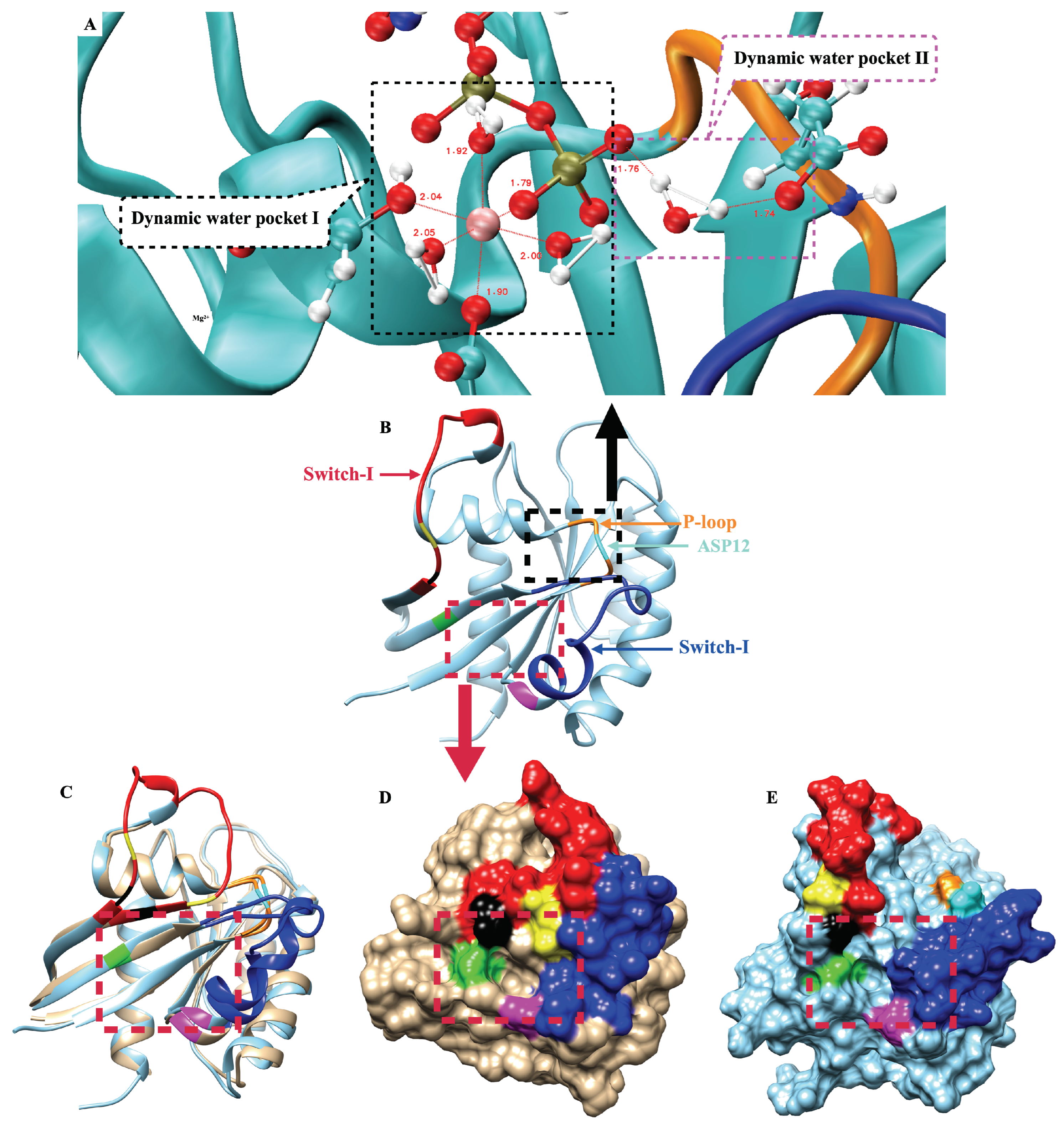
2.6. The Two Unique Druggable Dynamic Pockets on KRAS-G12D
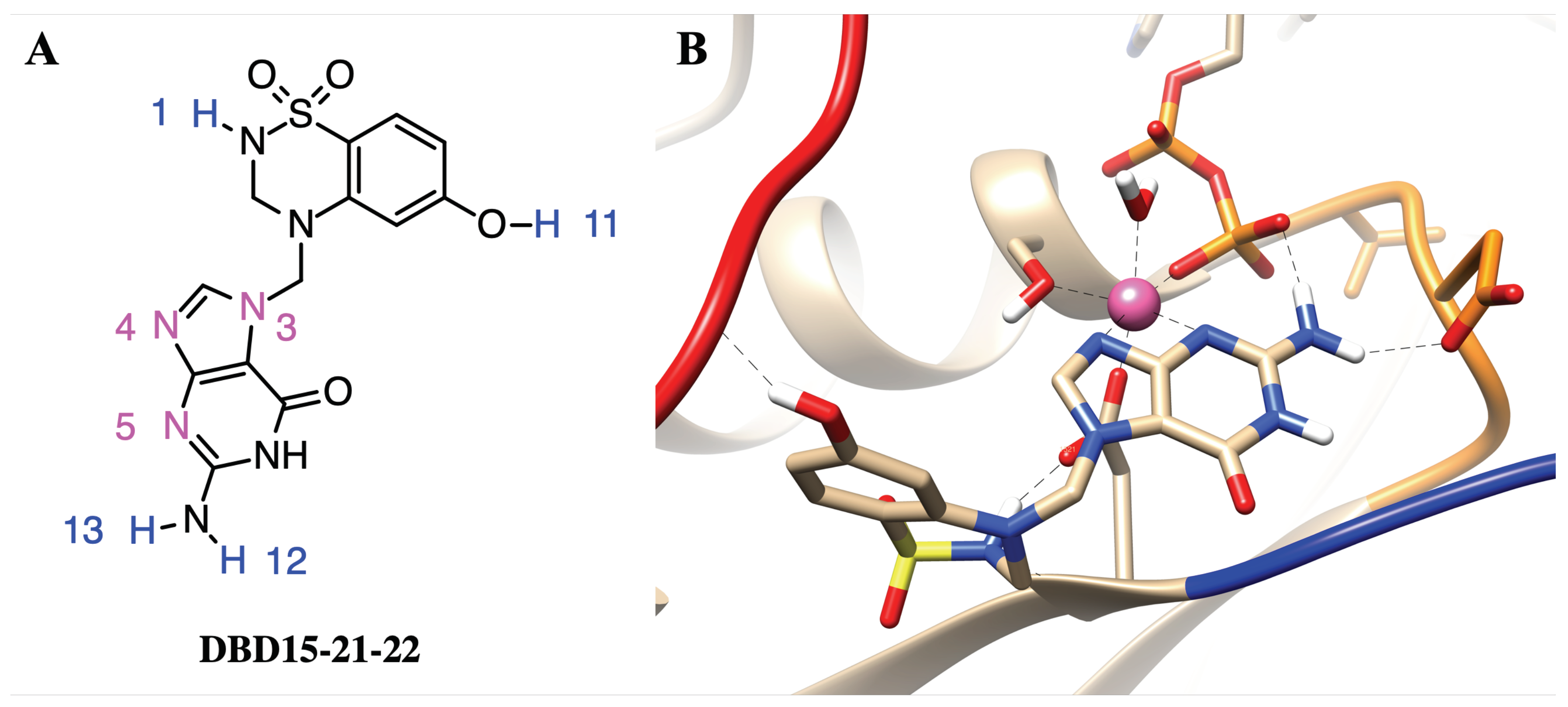
2.7. Structure-Based Drug Design and Targeting the Dynamic Water Pockets on KRAS-G12D
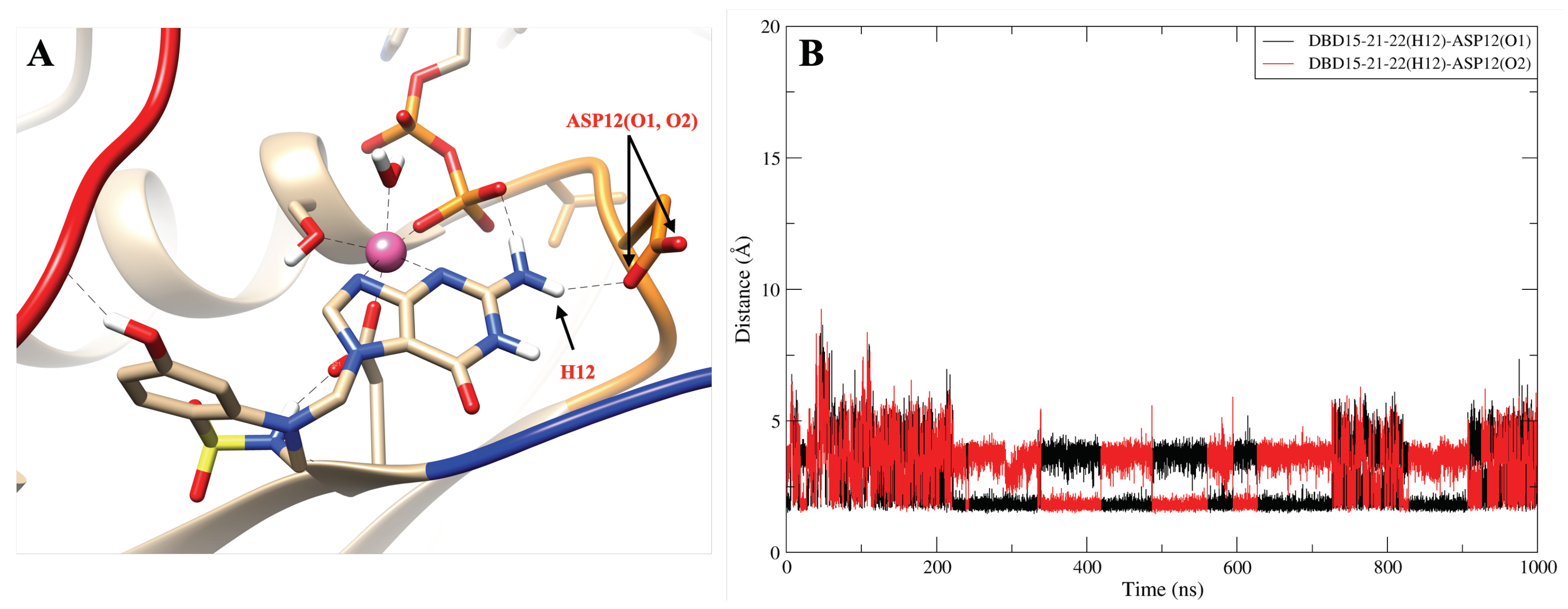
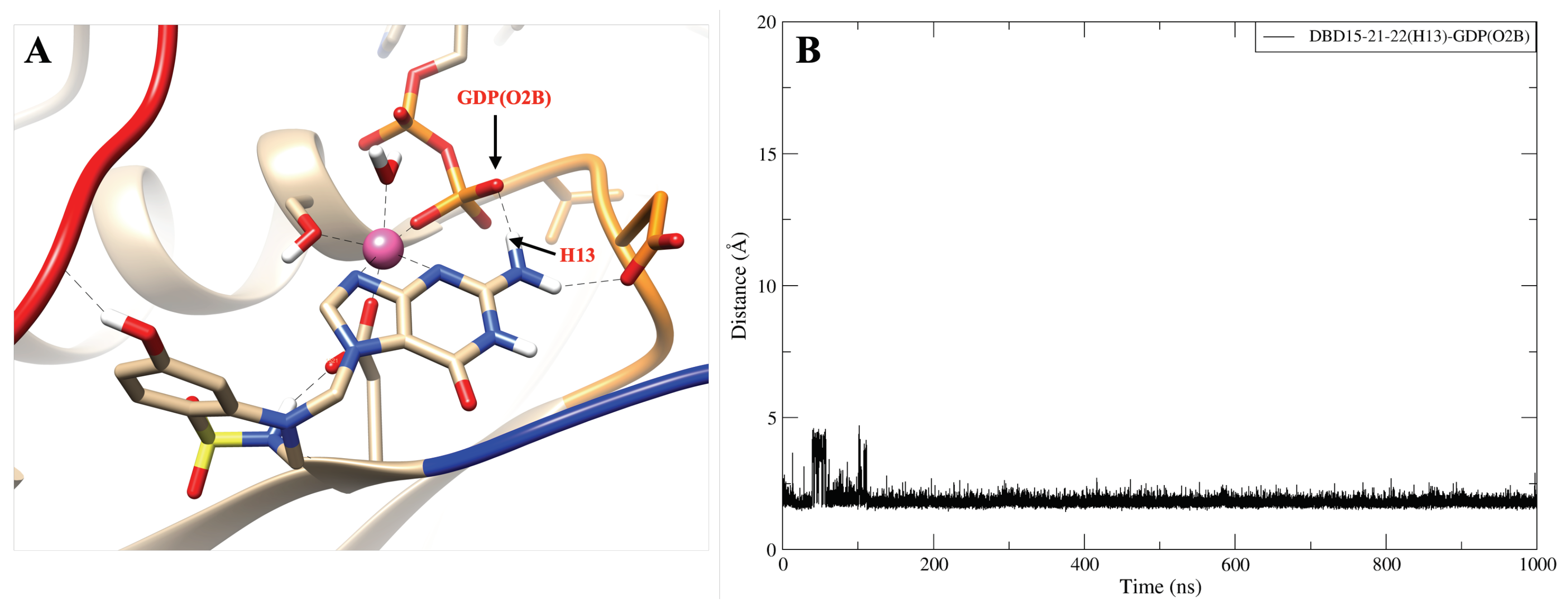
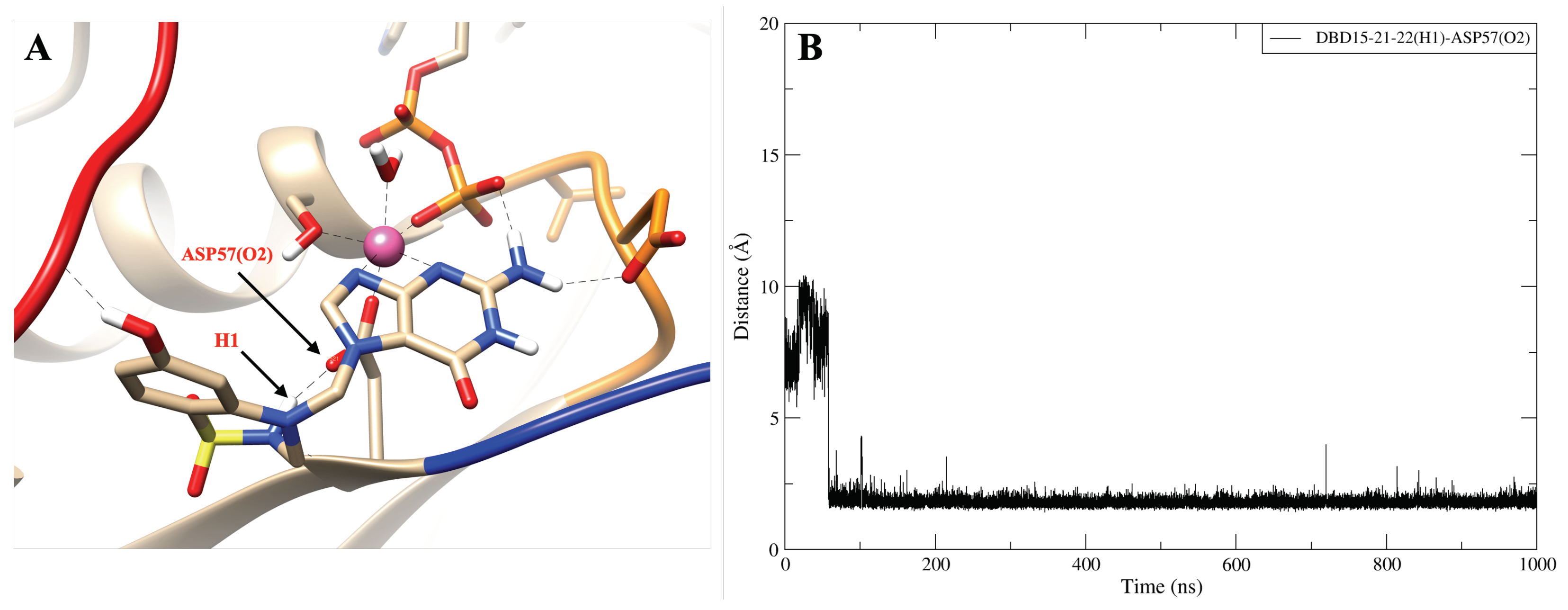
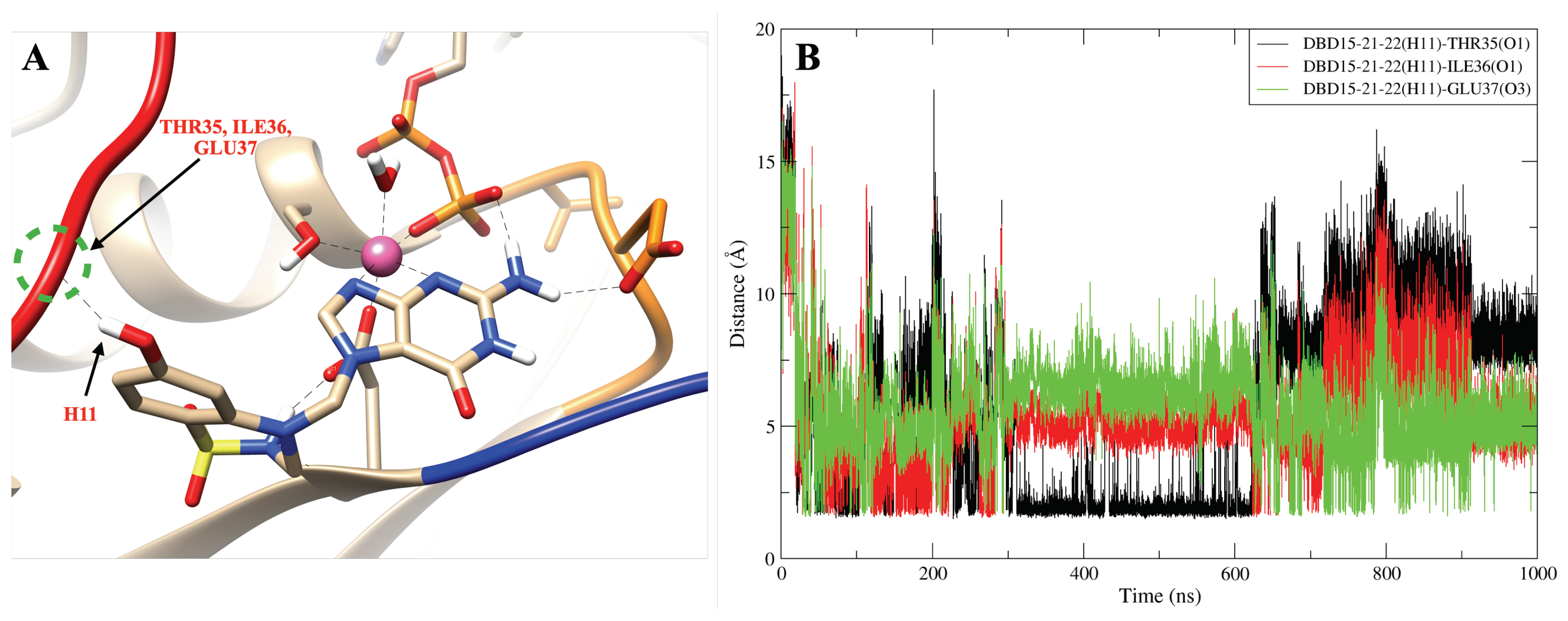
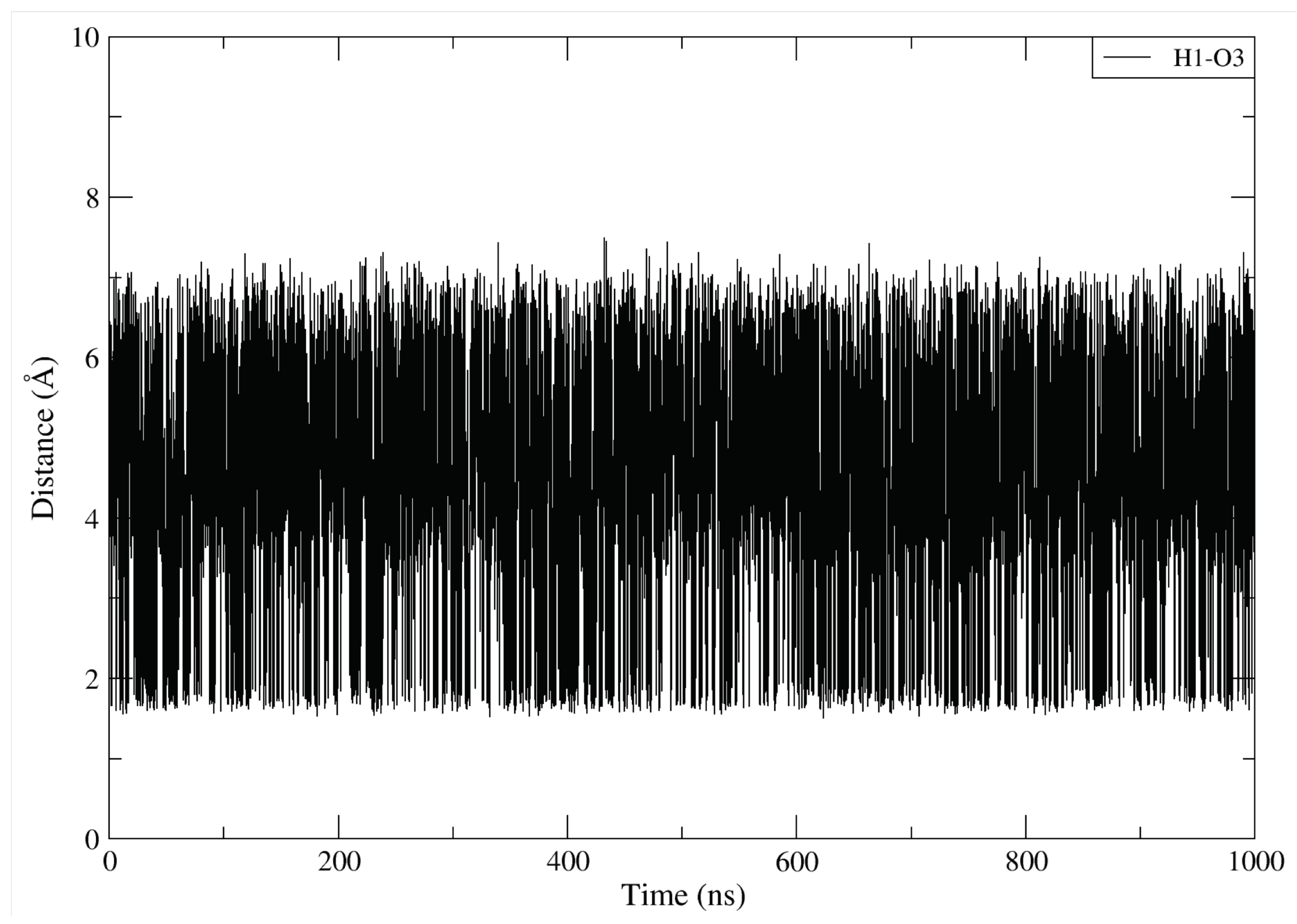
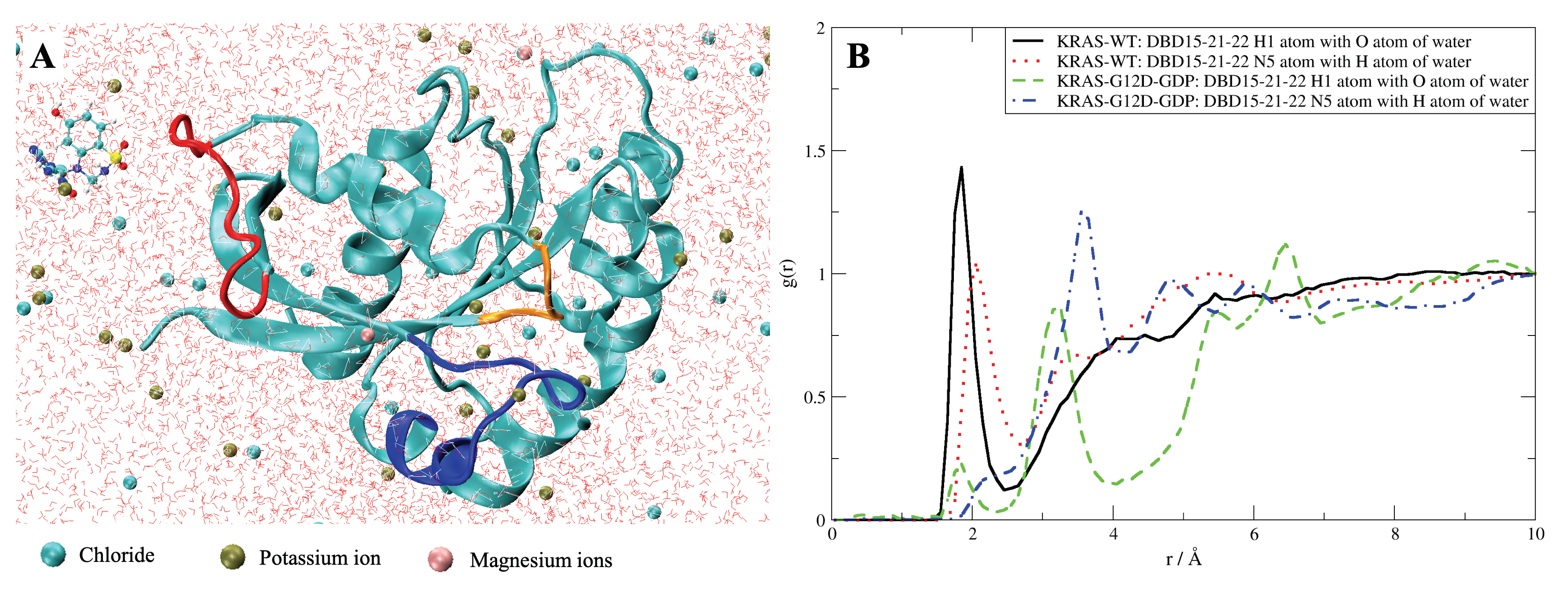
2.8. DBD15-21-22 Can Target KRAS-G12D and Bind Dynamic Water Pockets I and II
2.9. DBD15-21-22 Is Harmless to KRAS-WT
3. Methods and Materials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Initial Setups and Data for Molecular Dynamics Simulations



Appendix A.2. Atomic Distances and KRAS Conformations
Appendix A.3. Convergence of the Simulations

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full Name | Abbreviation |
|---|---|
| Aspartate | ASP |
| Cysteine | CYS |
| Glutamine | GLN |
| Glycine | GLY |
| Lysine | LYS |
| Serine | SER |
| Threonine | THR |
| Valine | VAL |
| Proline | PRO |
| Bond (Ion/Amino Acid (Atom)) | KRAS-WT | KRAS-G12C | KRAS-G12D |
|---|---|---|---|
| GLY12 (H3)-GLY60 (O3) | 1 | - | - |
| GLY12 (H3)-GLN61 (O1) | 7 | - | - |
| CYS12 (H1)-GLY60 (O3) | - | 1 | - |
| CYS12 (H1)-GLN61 (O1) | - | 8 | - |
| ASP12 (O1,O2)-GLY60 (H3) | - | - | 342 |
| ASP12 (O1,O2)-GLN61 (H1,H2) | - | - | 26 |
| ASP12 (O1,O2)-GLN61 (H3) | - | - | 113 |
| VAL8 (O3)-THR58 (H2) | 219 | 1050 | 422 |
| VAL8 (O3)-THR58 (H3) | 186 | 5 | 1403 |
| Mg-ASP57 (O1) | - | - | 2496 |
| Mg-ASP57 (O2) | - | - | 249 |
| Mg-SER17 (O2) | 252 | 329 | 2129 |
| Mg-GDP (O2A) | 2500 | 2500 | - |
| Mg-GDP (O1B) | 284 | 345 | 2500 |
| Mg-GDP (O2B) | 2500 | 2500 | 210 |
| ASP30 (O1,O2)-GDP (H2,H3) | 65 | 2 | 712 |
| ASP30 (O3)-GDP (H2,H3) | 30 | 2 | 48 |
| Bond (Drug/Ion/Amino Acid (Atom)) | KRAS-G12D |
|---|---|
| DBD15-21-22 (H12)-ASP12 (O1,O2) | 389 |
| DBD15-21-22 (H13)-GDP (O2B) | 970 |
| DBD15-21-22 (H1)-ASP57 (O2) | 932 |
| DBD15-21-22 (H11)-THR35 (O1) | 330 |
| DBD15-21-22 (H11)-ILE36 (O1) | 53 |
| DBD15-21-22 (H11)-GLU37 (O3) | 32 |
| DBD15-21-22 (N4)-Mg | 750 |
| DBD15-21-22 (N5)-Mg | 1000 |
| ASP57 (O1)-Mg | 1000 |
| SER17 (O2)-Mg | 964 |
| GDP (O1B)-Mg | 1000 |
| Water (O)-Mg | 931 |
References
- Cherfils, J.; Zeghouf, M. Regulation of small gtpases by gefs, gaps, and gdis. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.W.; Sarnecki, C.; Roberts, T.M.; Blenis, J. ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 1992, 68, 1041–1050. [Google Scholar] [CrossRef]
- Howe, L.R.; Leevers, S.J.; Gómez, N.; Nakielny, S.; Cohen, P.; Marshall, C.J. Activation of the MAP kinase pathway by the protein kinase raf. Cell 1992, 71, 335–342. [Google Scholar] [CrossRef]
- Sjölander, A.; Yamamoto, K.; Huber, B.E.; Lapetina, E.G. Association of p21ras with phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 7908–7912. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Hofer, F.; Fields, S.; Schneider, C.; Martin, G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef] [PubMed]
- Spaargaren, M.; Bischoff, J.R. Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc. Natl. Acad. Sci. USA 1994, 91, 12609–12613. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yao, H.; Wang, H.; Cheng, P.; Long, D. Microsecond timescale dynamics of GDP-bound Ras underlies the formation of novel inhibitor-binding pockets. Angew. Chem. 2016, 128, 15858–15861. [Google Scholar] [CrossRef]
- Mott, H.R.; Owen, D. Allostery and dynamics in small G proteins. Biochem. Soc. Trans. 2018, 46, 1333–1343. [Google Scholar] [CrossRef]
- Pálfy, G.; Menyhárd, D.K.; Perczel, A. Dynamically encoded reactivity of Ras enzymes: Opening new frontiers for drug discovery. Cancer Metastasis Rev. 2020, 39, 1075–1089. [Google Scholar] [CrossRef]
- Chen, J.; Wang, L.; Wang, W.; Sun, H.; Pang, L.; Bao, H. Conformational transformation of switch domains in GDP/K-Ras induced by G13 mutants: An investigation through Gaussian accelerated molecular dynamics simulations and principal component analysis. Comput. Biol. Med. 2021, 135, 104639. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Wang, W.; Pang, L.; Zhang, Q.; Liu, X. Mutation-induced impacts on the switch transformations of the GDP-and GTP-bound K-ras: Insights from multiple replica Gaussian accelerated molecular dynamics and free energy analysis. J. Chem. Inf. Model. 2021, 61, 1954–1969. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and PatientsTherapeutic Insight from the KRASG12C Inhibitor MRTX849. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Sheridan, C. Oncologists greet Lumakras: The world’s first KRAS inhibitor. Nat. Biotechnol. 2021, 39, 1032–1034. [Google Scholar] [CrossRef]
- Mann, J.E. Sotorasib (Lumakras™). Oncol. Times 2021, 43, 12–17. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef]
- Kessler, D.; Bergner, A.; Böttcher, J.; Fischer, G.; Döbel, S.; Hinkel, M.; Müllauer, B.; Weiss-Puxbaum, A.; McConnell, D.B. Drugging all RAS isoforms with one pocket. Future Med. Chem. 2020, 12, 1911–1923. [Google Scholar] [CrossRef]
- Mao, Z.; Xiao, H.; Shen, P.; Yang, Y.; Xue, J.; Yang, Y.; Shang, Y.; Zhang, L.; Li, X.; Zhang, Y.; et al. KRAS (G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022, 8, 5. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Y.; Bos, P.H.; Chambers, J.M.; Dupont, M.M.; Stockwell, B.R. K-RasG12D has a potential allosteric small molecule binding site. Biochemistry 2019, 58, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent small-molecule pan-RAS inhibitors. Cell 2017, 168, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, R.; Hu, Q.; Peacock, H.; Peacock, D.M.; Dai, S.; Shokat, K.M.; Suga, H. GTP-state-selective cyclic peptide ligands of K-Ras (G12D) block its interaction with Raf. ACS Cent. Sci. 2020, 6, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 2021, 65, 3123–3133. [Google Scholar] [CrossRef]
- Zhang, Z.; Guiley, K.Z.; Shokat, K.M. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras (G12S). Nat. Chem. Biol. 2022, 18, 1177–1183. [Google Scholar] [CrossRef]
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef]
- Zhang, Z.; Morstein, J.; Ecker, A.K.; Guiley, K.Z.; Shokat, K.M. Chemoselective covalent modification of K-Ras (G12R) with a small molecule electrophile. J. Am. Chem. Soc. 2022, 144, 15916–15921. [Google Scholar] [CrossRef]
- Zheng, Q.; Peacock, D.M.; Shokat, K.M. Drugging the next undruggable KRAS allele-Gly12Asp. J. Med. Chem. 2022, 65, 3119–3122. [Google Scholar] [CrossRef]
- Grudzien, P.; Jang, H.; Leschinsky, N.; Nussinov, R.; Gaponenko, V. Conformational Dynamics Allows Sampling of an “Active-like” State by Oncogenic K-Ras-GDP. J. Mol. Biol. 2022, 434, 167695. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mao, Y.; Gu, X.; Zhou, Y.; Long, D. Unveiling the “invisible” druggable conformations of GDP-bound inactive Ras. Proc. Natl. Acad. Sci. USA 2021, 118, e2024725118. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The structural basis of oncogenic mutations G12, G13 and Q61 in small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef]
- Prakash, P.; Hancock, J.F.; Gorfe, A.A. Binding hotspots on K-ras: Consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins Struct. Funct. Bioinform. 2015, 83, 898–909. [Google Scholar] [CrossRef]
- Chen, C.C.; Er, T.K.; Liu, Y.Y.; Hwang, J.K.; Barrio, M.J.; Rodrigo, M.; Garcia-Toro, E.; Herreros-Villanueva, M. Computational analysis of KRAS mutations: Implications for different effects on the KRAS p. G12D and p. G13D mutations. PLoS ONE 2013, 8, e55793. [Google Scholar] [CrossRef]
- Lu, H.; Marti, J. Long-lasting Salt Bridges Provide the Anchoring Mechanism of Oncogenic Kirsten Rat Sarcoma Proteins at Cell Membranes. J. Phys. Chem. Lett. 2020, 11, 9938–9945. [Google Scholar] [CrossRef]
- Lu, H.; Marti, J. Predicting the conformational variability of oncogenic GTP-bound G12D mutated KRas-4B proteins at zwitterionic model cell membranes. Nanoscale 2022, 14, 3148–3158. [Google Scholar] [CrossRef]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- John, J.; Rensland, H.; Schlichting, I.; Vetter, I.; Borasio, G.D.; Goody, R.S.; Wittinghofer, A. Kinetic and structural analysis of the Mg (2+)-binding site of the guanine nucleotide-binding protein p21H-ras. J. Biol. Chem. 1993, 268, 923–929. [Google Scholar] [CrossRef]
- Bock, C.W.; Kaufman, A.; Glusker, J.P. Coordination of water to magnesium cations. Inorg. Chem. 1994, 33, 419–427. [Google Scholar] [CrossRef]
- Pálfy, G.; Menyhárd, D.K.; Ákontz-Kiss, H.; Vida, I.; Batta, G.; Tőke, O.; Perczel, A. The Importance of Mg2+-free State in Nucleotide Exchange of Oncogenic K-Ras Mutants. Chem. A Eur. J. 2022, 28, e202201449. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.; Sczakiel, G.; Feuerstein, J.; John, J.; Goody, R.S.; Wittinghofer, A. Expression of p21 proteins in Escherichia coli and stereochemistry of the nucleotide-binding site. EMBO J. 1986, 5, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Self, A.J. The effect of Mg2+ on the guanine nucleotide exchange rate of p21N-ras. J. Biol. Chem. 1986, 261, 10963–10965. [Google Scholar] [CrossRef]
- Feuerstein, J.; Goody, R.S.; Wittinghofer, A. Preparation and characterization of nucleotide-free and metal ion-free p21 “apoprotein”. J. Biol. Chem. 1987, 262, 8455–8458. [Google Scholar] [CrossRef]
- Novello, F.C.; Sprague, J.M. Benzothiadiazine dioxides as novel diuretics. J. Am. Chem. Soc. 1957, 79, 2028–2029. [Google Scholar] [CrossRef]
- Hu, Z.; Martí, J.; Lu, H. Structure of benzothiadiazine at zwitterionic phospholipid cell membranes. J. Chem. Phys. 2021, 155, 154303. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Jang, H. Oncogenic Ras isoforms signaling specificity at the membrane. Cancer Res. 2018, 78, 593–602. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef]
- Stein, S.A.M.; Loccisano, A.E.; Firestine, S.M.; Evanseck, J.D. Principal components analysis: A review of its application on molecular dynamics data. Annu. Rep. Comput. Chem. 2006, 2, 233–261. [Google Scholar]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed]
- Marti, J. Dynamic properties of hydrogen-bonded networks in supercritical water. Phys. Rev. E 2000, 61, 449. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Walia, P.; Labbe, C.; Jao, K.; Leighl, N.B. Targeting the KRAS pathway in non-small cell lung cancer. Oncologist 2016, 21, 1450–1460. [Google Scholar] [CrossRef]
- Lietman, C.D.; Johnson, M.L.; McCormick, F.; Lindsay, C.R. More to the RAS Story: KRASG12C Inhibition, Resistance Mechanisms, and Moving Beyond KRASG12C. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–13. [Google Scholar] [CrossRef]
- Walter, E.R.; Hogg, C.; Parker, D.; Williams, J.G. Designing magnesium-selective ligands using coordination chemistry principles. Coord. Chem. Rev. 2021, 428, 213622. [Google Scholar] [CrossRef]
- Hu, Z.; Marti, J. In silico drug design of benzothiadiazine derivatives interacting with phospholipid cell membranes. Membranes 2022, 12, 331. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Elsevier: Amsterdam, The Netherlands, 2001; Volume 1. [Google Scholar]
- Nagy, G.; Gordillo, M.; Guàrdia, E.; Marti, J. Liquid water confined in carbon nanochannels at high temperatures. J. Phys. Chem. B 2007, 111, 12524–12530. [Google Scholar] [CrossRef]
- Marrink, S.J.; Corradi, V.; Souza, P.C.; Ingolfsson, H.I.; Tieleman, D.P.; Sansom, M.S. Computational modeling of realistic cell membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef]
- Padro, J.; Marti, J.; Guardia, E. Molecular dynamics simulation of liquid water at 523 K. J. Phys. Condens. Matter 1994, 6, 2283. [Google Scholar] [CrossRef]
- Padro, J.; Marti, J. Response to “Comment on ‘An interpretation of the low-frequency spectrum of liquid water’”[J. Chem. Phys. 118, 452 (2003)]. J. Chem. Phys. 2004, 120, 1659–1660. [Google Scholar] [CrossRef]
- Calero, C.; Marti, J.; Guàrdia, E. 1H nuclear spin relaxation of liquid water from molecular dynamics simulations. J. Phys. Chem. B 2015, 119, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Martí, J.; Padró, J. Rotation-vibration coupling in liquid water investigated by molecular dynamics simulation. Mol. Simul. 1999, 23, 55–62. [Google Scholar] [CrossRef]
- Marti, J.; Padró, J.; Guardia, E. Computer simulation of molecular motions in liquids: Infrared spectra of water and heavy water. Mol. Simul. 1993, 11, 321–336. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks III, C.L.; Mackerell Jr, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell Jr, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34, D302–D305. [Google Scholar] [CrossRef]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Marti, J.; Csajka, F. The aqueous solvation of sodium chloride: A Monte Carlo transition path sampling study. J. Chem. Phys. 2000, 113, 1154–1161. [Google Scholar] [CrossRef]
- Dellago, C.; Bolhuis, P.G.; Geissler, P.L. Transition path sampling. Adv. Chem. Phys. 2002, 123, 1–78. [Google Scholar]
- Marti, J.; Csajka, F. Transition path sampling study of flip-flop transitions in model lipid bilayer membranes. Phys. Rev. E 2004, 69, 061918. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A. Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2020, 2, 200–212. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Marti, J. Discovering and Targeting Dynamic Drugging Pockets of Oncogenic Proteins: The Role of Magnesium in Conformational Changes of the G12D Mutated Kirsten Rat Sarcoma-Guanosine Diphosphate Complex. Int. J. Mol. Sci. 2022, 23, 13865. https://doi.org/10.3390/ijms232213865
Hu Z, Marti J. Discovering and Targeting Dynamic Drugging Pockets of Oncogenic Proteins: The Role of Magnesium in Conformational Changes of the G12D Mutated Kirsten Rat Sarcoma-Guanosine Diphosphate Complex. International Journal of Molecular Sciences. 2022; 23(22):13865. https://doi.org/10.3390/ijms232213865
Chicago/Turabian StyleHu, Zheyao, and Jordi Marti. 2022. "Discovering and Targeting Dynamic Drugging Pockets of Oncogenic Proteins: The Role of Magnesium in Conformational Changes of the G12D Mutated Kirsten Rat Sarcoma-Guanosine Diphosphate Complex" International Journal of Molecular Sciences 23, no. 22: 13865. https://doi.org/10.3390/ijms232213865
APA StyleHu, Z., & Marti, J. (2022). Discovering and Targeting Dynamic Drugging Pockets of Oncogenic Proteins: The Role of Magnesium in Conformational Changes of the G12D Mutated Kirsten Rat Sarcoma-Guanosine Diphosphate Complex. International Journal of Molecular Sciences, 23(22), 13865. https://doi.org/10.3390/ijms232213865