
Genotype Complements the Phenotype: Identification of the Pathogenicity of an LMNA Splice Variant by Nanopore Long-Read Sequencing in a Large DCM Family

 ,
, 
 , ,
, ,  , ,
, , 
Abstract
1. Introduction
2. Results
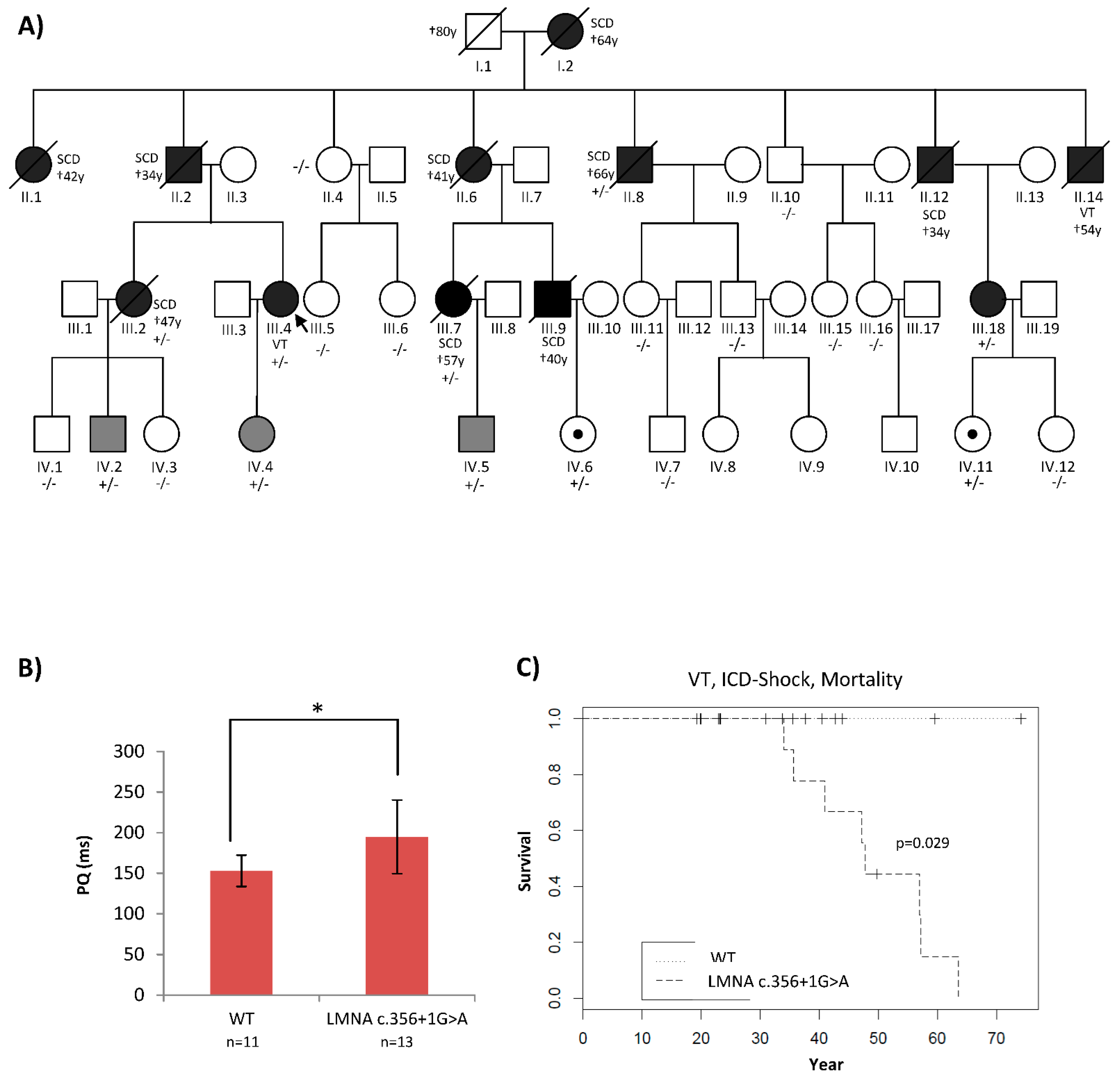
2.1. Patients’ Phenotypes Were Suggestive of a Laminopathy
2.2. Traditional Diagnostic Methods Revealed Two LMNA Variants with Unclear Pathogenicity
2.3. High-Coverage NGS (SureSelect) Detected Four LMNA Variants
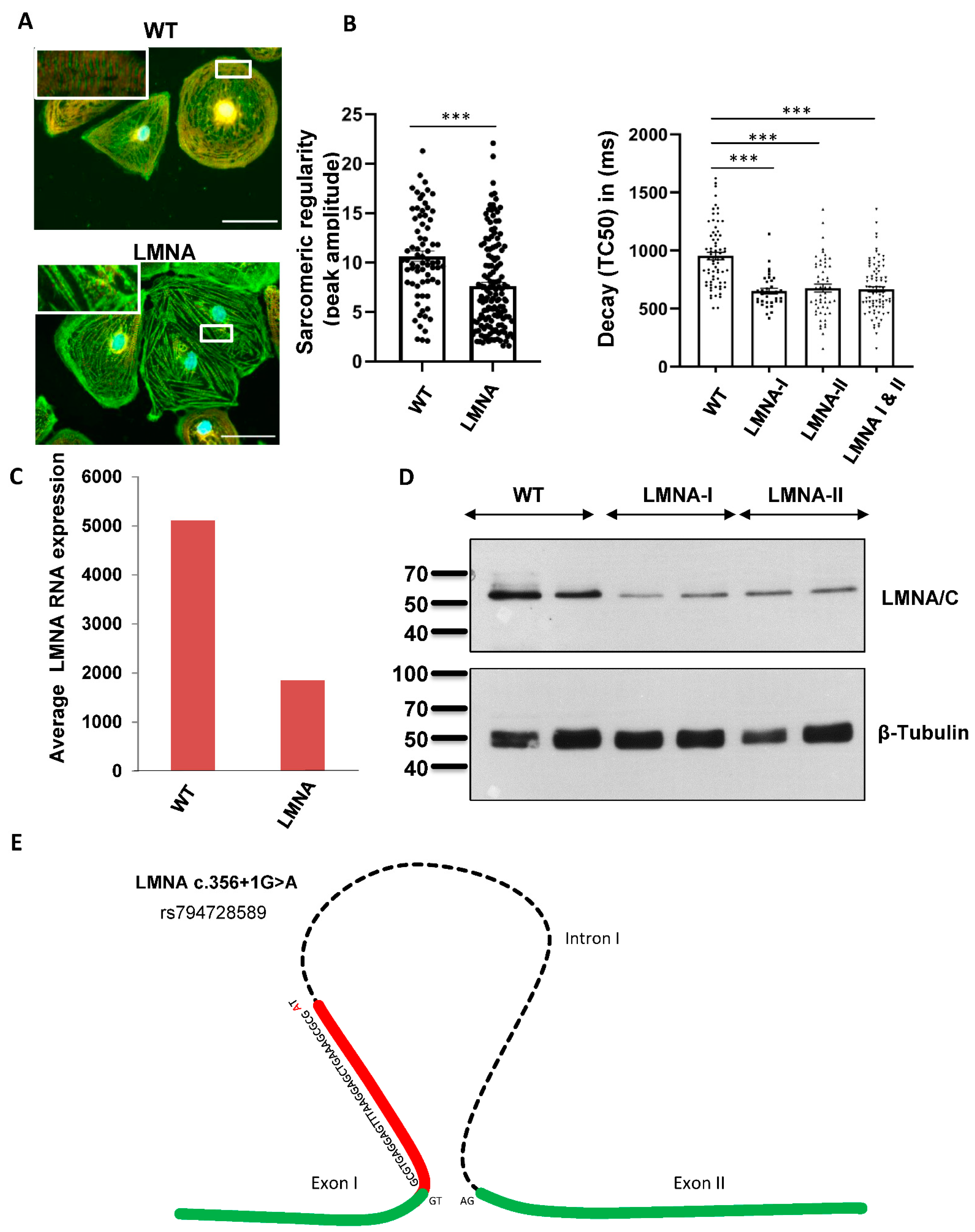
2.4. Functional Analyses Revealed the Pathogenicity of rs794728589
3. Discussion
4. Material and Methods
4.1. DNA and RNA Sequencing
4.2. Long-Read RNA Sequencing of cDNA Libraries
4.3. Generation of Patient-Specific Induced Pluripotent Stem Cells (ps-iPSCs)
4.4. Cell Culture and Cardiac Differentiation of iPSCs
4.5. Assessment of Sarcomeric Regularity of iPSC-CMs
4.6. Calcium Imaging: Fluo-4
4.7. Protein Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Katus, H.A.; Meder, B. Precision medicine for cardiovascular disease: Learning lessons from cardiomyopathies. Herz 2018, 43, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Rebs, S.; El-Battrawy, I.; Chasan, S.; Krause, T.; Haas, J.; Zhong, R.; Liao, Z.; Xu, Q.; Zhou, X.; et al. Identification of SCN5a p.C335R Variant in a Large Family with Dilated Cardiomyopathy and Conduction Disease. Int. J. Mol. Sci. 2021, 22, 12990. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef]
- Sammani, A.; Kayvanpour, E.; Bosman, L.P.; Sedaghat-Hamedani, F.; Proctor, T.; Gi, W.T.; Broezel, A.; Jensen, K.; Katus, H.A.; Te Riele, A.; et al. Predicting sustained ventricular arrhythmias in dilated cardiomyopathy: A meta-analysis and systematic review. ESC Heart Fail. 2020, 7, 1430–1441. [Google Scholar] [CrossRef]
- Moretti, M.; Merlo, M.; Barbati, G.; Di Lenarda, A.; Brun, F.; Pinamonti, B.; Gregori, D.; Mestroni, L.; Sinagra, G. Prognostic impact of familial screening in dilated cardiomyopathy. Eur. J. Heart Fail. 2010, 12, 922–927. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Rusconi, P.; Wilkinson, J.D.; Sleeper, L.A.; Lu, M.; Cox, G.F.; Towbin, J.A.; Colan, S.D.; Webber, S.A.; Canter, C.E.; Ware, S.M.; et al. Differences in Presentation and Outcomes Between Children With Familial Dilated Cardiomyopathy and Children With Idiopathic Dilated Cardiomyopathy: A Report From the Pediatric Cardiomyopathy Registry Study Group. Circ. Heart Fail. 2017, 10, 2637. [Google Scholar] [CrossRef]
- Shestak, A.G.; Bukaeva, A.A.; Saber, S.; Zaklyazminskaya, E.V. Allelic Dropout Is a Common Phenomenon That Reduces the Diagnostic Yield of PCR-Based Sequencing of Targeted Gene Panels. Front. Genet. 2021, 12, 620337. [Google Scholar] [CrossRef] [PubMed]
- Robyns, T.; Kuiperi, C.; Willems, R.; Corveleyn, A.; Nuyens, D. Targeted capture sequencing in a large LQTS family reveals a new pathogenic mutation c.2038delG in KCNH2 initially missed due to allelic dropout. Acta Cardiol. 2015, 70, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Blessing, F.; Fimpler, L.; Wenzel, F. Nanopore Sequencing in a Clinical Routine Laboratory: Challenges and Opportunities. Clin. Lab. 2020, 66, 191114. [Google Scholar] [CrossRef] [PubMed]
- Bowden, R.; Davies, R.W.; Heger, A.; Pagnamenta, A.T.; de Cesare, M.; Oikkonen, L.E.; Parkes, D.; Freeman, C.; Dhalla, F.; Patel, S.Y.; et al. Sequencing of human genomes with nanopore technology. Nat. Commun. 2019, 10, 1869. [Google Scholar] [CrossRef]
- Leija-Salazar, M.; Sedlazeck, F.J.; Toffoli, M.; Mullin, S.; Mokretar, K.; Athanasopoulou, M.; Donald, A.; Sharma, R.; Hughes, D.; Schapira, A.H.V.; et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genom. Med. 2019, 7, e564. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wu, J.; Sun, H.; Briganti, F.; Meder, B.; Wei, W.; Steinmetz, L.M. Single-molecule, full-length transcript isoform sequencing reveals disease-associated RNA isoforms in cardiomyocytes. Nat. Commun. 2021, 12, 4203. [Google Scholar] [CrossRef]
- Muchir, A.; Wu, W.; Choi, J.C.; Iwata, S.; Morrow, J.; Homma, S.; Worman, H.J. Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2012, 21, 4325–4333. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Borchert, T.; Hubscher, D.; Guessoum, C.I.; Lam, T.D.; Ghadri, J.R.; Schellinger, I.N.; Tiburcy, M.; Liaw, N.Y.; Li, Y.; Haas, J.; et al. Catecholamine-Dependent beta-Adrenergic Signaling in a Pluripotent Stem Cell Model of Takotsubo Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 975–991. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ID | Age | Gender | Affected | Splice Mutation | R644C Variant | Age at Onset | DCM | NYHA | EF | Rhythm | Conduction Disease | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I.2 | 64 | F | Yes | (+/−) | NA | 57 | NA | NA | NA | AF | AVB-III° | PM 60y, SCD 64y |
| II.1 | 42 | F | Yes | NA | NA | 37 | NA | NA | NA | AF | - | PM 40y, SCD 42y |
| II.3 | 34 | M | Yes | (+/−) | NA | 32 | NA | NA | NA | AF | - | SCD 34y |
| II.4 | 72 | F | No | −/− | +/− | - | No | I | 60 | AF | - | - |
| II.6 | 41 | F | Yes | (+/−) | NA | 37 | NA | NA | NA | AF | AVB-III° | PM 37y, SCD41y |
| II.8 | 66 | M | Yes | +/− | +/− | 55 | Yes | III | 35 | AF | AVB-I°, LAH | PM 59y, SCD 66y |
| II.10 | 77 | M | No | −/− | +/− | - | No | II | 58 | SR | - | - |
| II.12 | 66 | M | Yes | (+/−) | NA | 28 | NA | NA | NA | SR | AVB-II° | PM 30y, SCD 34y |
| II.14 | 54 | M | Yes | NA | NA | 36 | NA | NA | NA | NA | AVB-III° | PM 51y, VT, ICD 52y, Death 54y |
| III.2 | 45 | F | Yes | +/− | - | 44 | Yes | I | 40 | SR | AVB-I°, LAH | nsVT, ICD 45y, SCD 47y |
| III.4 | 52 | F | Yes | +/− | - | 40 | Yes | III | 50 | AF | AVB-I°, LBBB | Prophylactic ICD 40y, VT and ICD therapy 47y, CRT-D 52y |
| III.5 | 38 | F | No | −/− | −/− | - | No | I | 65 | SR | - | |
| III.6 | 36 | F | No | −/− | −/− | - | No | I | 60 | SR | - | |
| III.7 | 57 | F | Yes | +/− | −/− | 46 | NA | I | NA | NA | NA | SCD 57y |
| III.9 | 40 | M | Yes | NA | NA | NA | NA | NA | NA | NA | NA | SCD 40y |
| III.11 | 42 | F | No | −/− | +/− | - | No | I | 60 | SR | - | |
| III.13 | 40 | M | No | −/− | +/− | - | No | I | 60 | SR | NA | |
| III.15 | 44 | F | No | −/− | +/− | - | No | I | 60 | SR | - | |
| III.16 | 39 | F | No | −/− | +/− | - | No | I | 63 | SR | - | |
| III.18 | 53 | F | Yes | +/− | −/− | 38 | Yes | III | 33 | SR | AVB-III°, LBBB | Prophylactic ICD 42y, CRT-D 52y |
| IV.1 | 22 | M | No | −/− | −/− | - | No | I | 60 | SR | - | |
| IV.2 | 35 | M | Yes | +/− | −/− | 26 | early DCM | I | 50 | SR | LPH | |
| IV.3 | 20 | F | No | −/− | −/− | - | No | I | 60 | SR | - | |
| IV.4 | 19 | F | No | +/− | −/− | - | early DCM | I | 50 | SR | - | |
| IV. 5 | 23 | M | Yes | +/− | −/− | 23 | No | I | 60 | SR | AVB-I° | |
| IV. 6 | 23 | F | No | +/− | −/− | - | No | I | 55 | SR | - | |
| IV.7 | 19 | M | No | −/− | −/− | - | No | I | 64 | SR | - | |
| IV.11 | 27 | F | No | +/− | −/− | - | No | I | 55 | SR | - | |
| IV.12 | 20 | F | No | −/− | −/− | - | No | I | 55 | SR | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedaghat-Hamedani, F.; Rebs, S.; Kayvanpour, E.; Zhu, C.; Amr, A.; Müller, M.; Haas, J.; Wu, J.; Steinmetz, L.M.; Ehlermann, P.; et al. Genotype Complements the Phenotype: Identification of the Pathogenicity of an LMNA Splice Variant by Nanopore Long-Read Sequencing in a Large DCM Family. Int. J. Mol. Sci. 2022, 23, 12230. https://doi.org/10.3390/ijms232012230
Sedaghat-Hamedani F, Rebs S, Kayvanpour E, Zhu C, Amr A, Müller M, Haas J, Wu J, Steinmetz LM, Ehlermann P, et al. Genotype Complements the Phenotype: Identification of the Pathogenicity of an LMNA Splice Variant by Nanopore Long-Read Sequencing in a Large DCM Family. International Journal of Molecular Sciences. 2022; 23(20):12230. https://doi.org/10.3390/ijms232012230
Chicago/Turabian StyleSedaghat-Hamedani, Farbod, Sabine Rebs, Elham Kayvanpour, Chenchen Zhu, Ali Amr, Marion Müller, Jan Haas, Jingyan Wu, Lars M. Steinmetz, Philipp Ehlermann, and et al. 2022. "Genotype Complements the Phenotype: Identification of the Pathogenicity of an LMNA Splice Variant by Nanopore Long-Read Sequencing in a Large DCM Family" International Journal of Molecular Sciences 23, no. 20: 12230. https://doi.org/10.3390/ijms232012230
APA StyleSedaghat-Hamedani, F., Rebs, S., Kayvanpour, E., Zhu, C., Amr, A., Müller, M., Haas, J., Wu, J., Steinmetz, L. M., Ehlermann, P., Streckfuss-Bömeke, K., Frey, N., & Meder, B. (2022). Genotype Complements the Phenotype: Identification of the Pathogenicity of an LMNA Splice Variant by Nanopore Long-Read Sequencing in a Large DCM Family. International Journal of Molecular Sciences, 23(20), 12230. https://doi.org/10.3390/ijms232012230