
Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately?

 , , ,
, , ,
Abstract
1. Introduction
2. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma Development and Progression
2.1. Fatty Acids
2.2. CD36
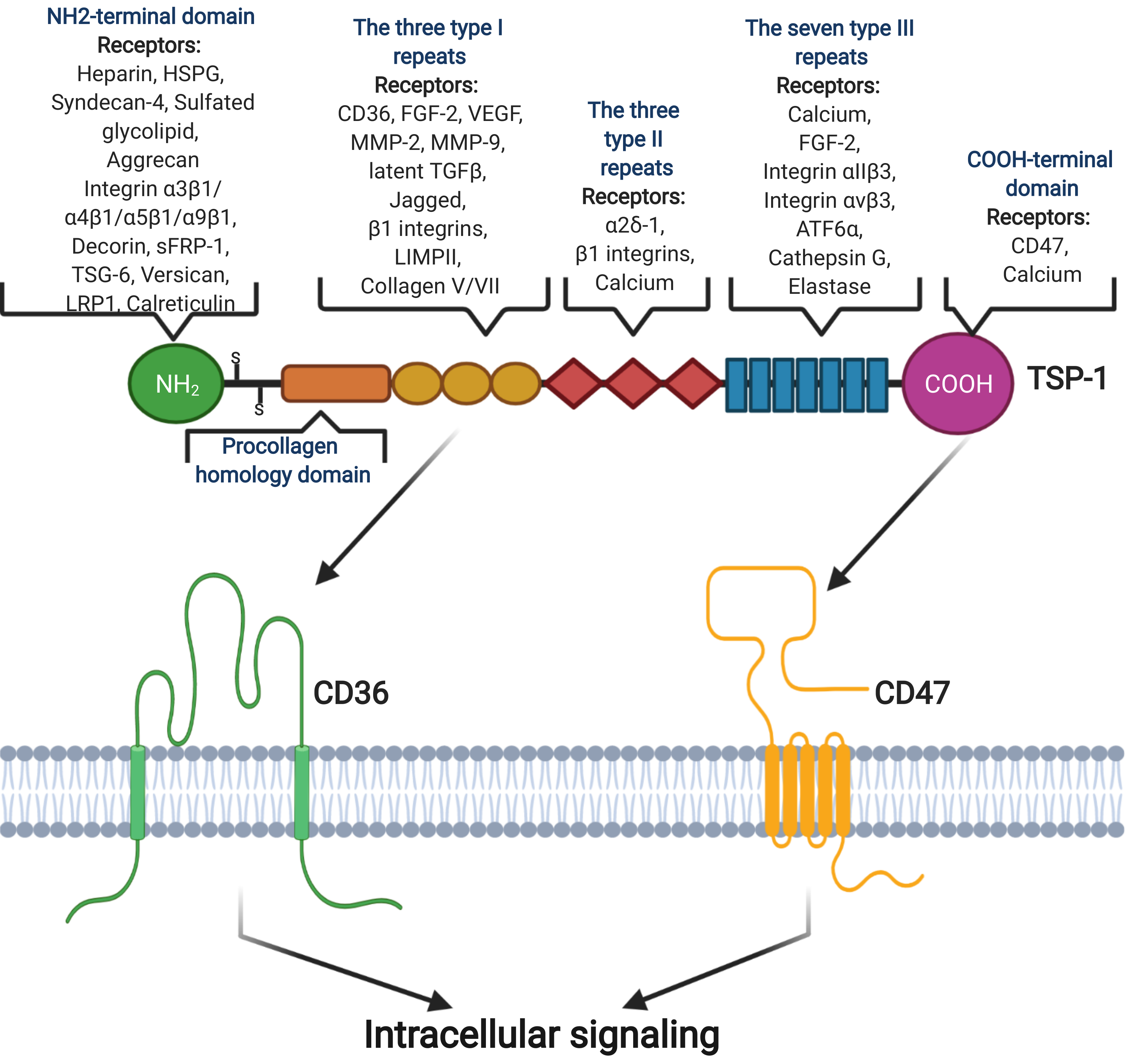
2.3. Thrombospondin-1
2.4. CD47
3. CD36 and Thrombospondin-1 in Glioblastoma
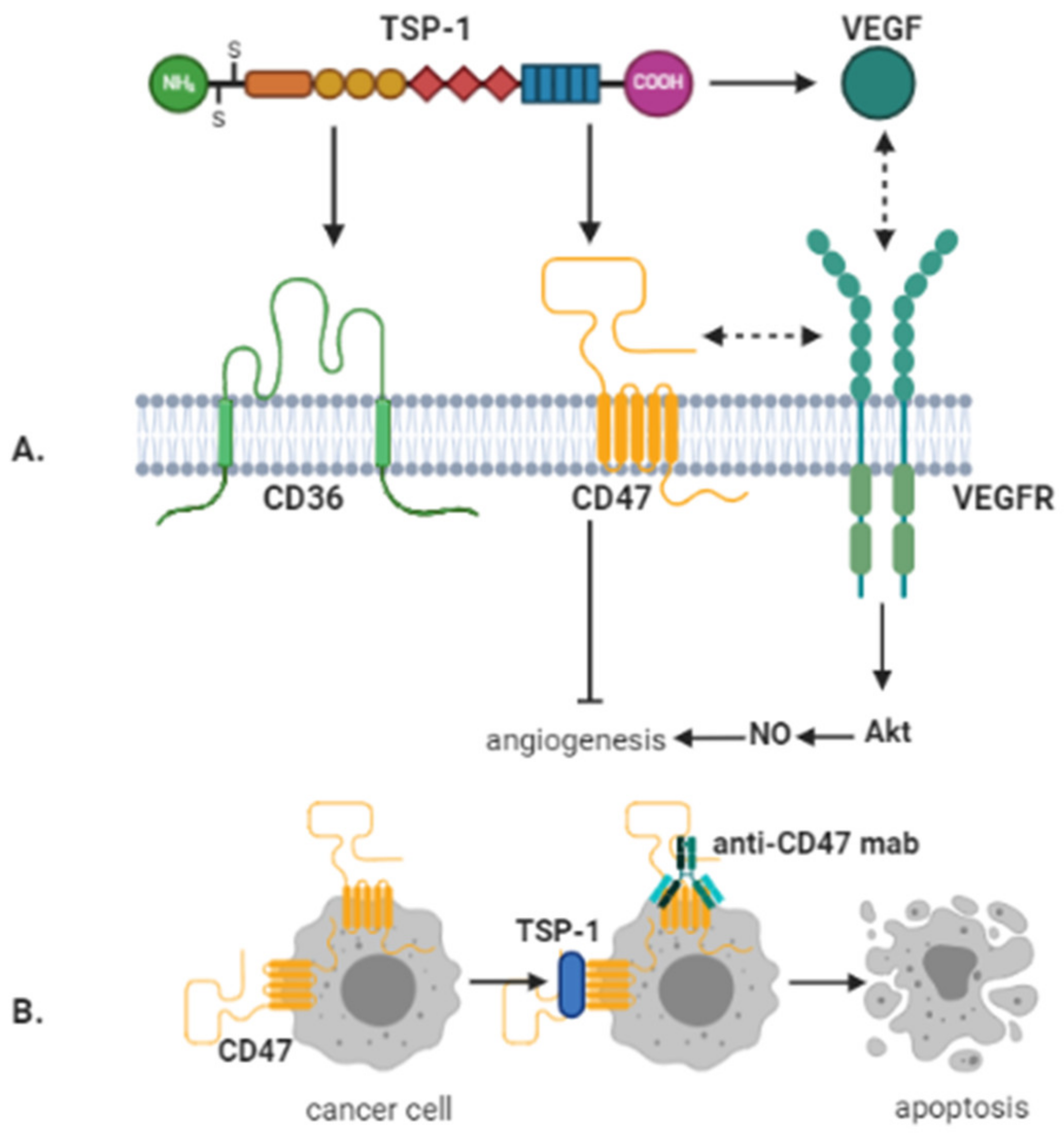
4. CD47 and Thrombospondin-1 in Glioblastoma
5. Signaling Pathways Activated by CD36 and CD47 Binding to TSP-1 in Glioblastoma
6. Therapeutic Targeting of TSP-1/CD36/CD47
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inda, M.M.; Bonavia, R.; Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef]
- Qazi, M.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.; Albulescu, R.; Codrici, E.; Popescu, I.D.; Mihai, S.; Enciu, A.M.; Cruceru, M.L.; Popa, A.C.; Neagu, A.I.; Necula, L.G.; et al. Circulating biomarker panels for targeted therapy in brain tumors. Future Oncol. 2015, 11, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Costello, J.F. Epigenetic mechanisms in glioblastoma multiforme. Semin. Cancer Biol. 2009, 19, 188–197. [Google Scholar] [CrossRef]
- Pop, S.; Enciu, A.M.; Necula, L.G.; Tanase, C. Long non-coding RNAs in brain tumours: Focus on recent epigenetic findings in glioma. J. Cell. Mol. Med. 2018, 22, 4597–4610. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncology 2016, 18, 160–172. [Google Scholar] [CrossRef]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Yu, D.; Xuan, Q.; Zhang, C.; Hu, C.; Li, Y.; Zhao, X.; Liu, S.; Ren, F.; Zhang, Y.; Zhou, L.; et al. Metabolic Alterations Related to Glioma Grading Based on Metabolomics and Lipidomics Analyses. Metabolites 2020, 10, 478. [Google Scholar] [CrossRef]
- Elsherbiny, M.E.; Emara, M.; Godbout, R. Interaction of brain fatty acid-binding protein with the polyunsaturated fatty acid environment as a potential determinant of poor prognosis in malignant glioma. Prog. Lipid Res. 2013, 52, 562–570. [Google Scholar] [CrossRef]
- Tanase, C.P.; Enciu, A.M.; Mihai, S.; Neagu, A.I.; Calenic, B.; Cruceru, M.L. Anti-cancer Therapies in High Grade Gliomas. Curr. Proteom. 2013, 10, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.P.; Cruceru, M.L.; Enciu, A.-M.; Popa, A.C.; Albulescu, R.; Neagu, M.; Constantinescu, S.N. Signal transduction molecule patterns indicating potential glioblastoma therapy approaches. Onco Targets Ther. 2013, 6, 1737–1749. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Tremblay, B.L.; Guenard, F.; Rudkowska, I.; Lemieux, S.; Couture, P.; Vohl, M.C. Epigenetic changes in blood leukocytes following an omega-3 fatty acid supplementation. Clin. Epigenetics 2017, 9, 43. [Google Scholar] [CrossRef]
- Pop, S.; Enciu, A.M.; Tarcomnicu, I.; Gille, E.; Tanase, C. Phytochemicals in cancer prevention: Modulating epigenetic alterations of DNA methylation. Phytochem. Rev. 2019, 18, 1005–1024. [Google Scholar] [CrossRef]
- Martinez, M.; Mougan, I. Fatty acid composition of human brain phospholipids during normal development. J. Neurochem. 1998, 71, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Hillard, C.J.; Spector, A.A.; Watkins, P.A. Brain uptake and utilization of fatty acids, lipids and lipoproteins: Application to neurological disorders. J. Mol. Neurosci. 2007, 33, 2–11. [Google Scholar] [CrossRef]
- Watkins, P.A.; Hamilton, J.A.; Leaf, A.; Spector, A.A.; Moore, S.A.; Anderson, R.E.; Moser, H.W.; Noetzel, M.J.; Katz, R. Brain uptake and utilization of fatty acids: Applications to peroxisomal biogenesis diseases. J. Mol. Neurosci. 2001, 16, 87–92; discussion 151–157. [Google Scholar] [CrossRef]
- Yehuda, S.; Rabinovitz, S.; Mostofsky, D.I. Essential fatty acids are mediators of brain biochemistry and cognitive functions. J. Neurosci. Res. 1999, 56, 565–570. [Google Scholar] [CrossRef]
- Ioghen, O.; Chițoiu, L.; Gherghiceanu, M.; Ceafalan, L.C.; Hinescu, M.E.; Majewska, A. CD36—A novel molecular target in the neurovascular unit. Eur. J. Neurosci. 2021, 53, 2500–2510. [Google Scholar] [CrossRef]
- Wilkinson, B.; Koenigsknecht-Talboo, J.; Grommes, C.; Lee, C.Y.; Landreth, G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J. Biol. Chem. 2006, 281, 20842–20850. [Google Scholar] [CrossRef]
- Ramchandani, D.; Mittal, V. Thrombospondin in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 133–147. [Google Scholar] [PubMed]
- Codrici, E.; Enciu, A.M.; Popescu, I.D.; Mihai, S.; Tanase, C. Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Targets. Stem Cells Int. 2016, 2016, 5728438. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2020, 599, 1745–1757. [Google Scholar] [CrossRef]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef]
- Zaidi, N.; Royaux, I.; Swinnen, J.V.; Smans, K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol. Cancer Ther. 2012, 11, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.M.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Nathoo, N.; Barnett, G.H.; Golubic, M. The eicosanoid cascade: Possible role in gliomas and meningiomas. J. Clin. Pathol. 2004, 57, 6–13. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Laye, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Moore, S.A. Polyunsaturated fatty acid synthesis and release by brain-derived cells in vitro. J. Mol. Neurosci. 2001, 16, 195–200; discussion 215–221. [Google Scholar] [CrossRef]
- Marks, F.; Muller-Decker, K.; Furstenberger, G. A causal relationship between unscheduled eicosanoid signaling and tumor development: Cancer chemoprevention by inhibitors of arachidonic acid metabolism. Toxicology 2000, 153, 11–26. [Google Scholar] [CrossRef]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Hayashi, K.; Takenaka, K.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Ordering of ceramide formation, caspase activation, and Bax/Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Differ. 2000, 7, 761–772. [Google Scholar] [CrossRef]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Influence of Bax or Bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene 2000, 19, 3508–3520. [Google Scholar] [CrossRef]
- Taib, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef]
- Sperry, J.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.O.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; Christofk, H.; et al. Glioblastoma Utilizes Fatty Acids and Ketone Bodies for Growth Allowing Progression during Ketogenic Diet Therapy. iScience 2020, 23, 101453. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M. Sterols in the central nervous system. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef]
- Nygren, C.; von Holst, H.; Mansson, J.E.; Fredman, P. Increased levels of cholesterol esters in glioma tissue and surrounding areas of human brain. Br. J. Neurosurg. 1997, 11, 216–220. [Google Scholar]
- Iannelli, F.; Lombardi, R.; Milone, M.R.; Pucci, B.; De Rienzo, S.; Budillon, A.; Bruzzese, F. Targeting Mevalonate Pathway in Cancer Treatment: Repurposing of Statins. Recent Pat. Anticancer. Drug. Discov. 2018, 13, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Toriumi, D.M.; Cozzens, J.; Michael, M.A.; Ossoff, R.H. Arachnoid cyst manifested as an ethmoid mass with cerebrospinal fluid rhinorrhea. Otolaryngol. Head Neck Surg. 1987, 97, 406–408. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgoz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.-K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289.e5. [Google Scholar] [CrossRef]
- Bi, J.; Mischel, P.S. Acyl-CoA-Binding Protein Fuels Gliomagenesis. Cell Metab. 2019, 30, 229–230. [Google Scholar] [CrossRef]
- Dahan, P.; Gala, J.M.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Moyal, E.C.-J.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Auffinger, B.; Guo, D.; Hasan, T.; Deheeger, M.; Tobias, A.L.; Kim, J.Y.; Atashi, F.; Zhang, L.; Lesniak, M.S.; et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol. Cancer Ther. 2016, 15, 3064–3076. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Massari, M.S.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.; Miska, J.; Shireman, J.; Park, C.H.; Muroski, M.; Lesniak, M.S.; Ahmed, U. Temozolomide Treatment Increases Fatty Acid Uptake in Glioblastoma Stem Cells. Cancers 2020, 12, 3126. [Google Scholar] [CrossRef] [PubMed]
- Pirmoradi, L.; Seyfizadeh, N.; Ghavami, S.; Zeki, A.A.; Shojaei, S. Targeting cholesterol metabolism in glioblastoma: A new therapeutic approach in cancer therapy. J. Investig. Med. 2019, 67, 715–719. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Ji, S.; Zhang, P.; Bhuiyan, R.H.; Ohkawa, Y.; Tajima, O.; Hashimoto, N.; Furukawa, K. Glycolipids: Essential regulator of neuro-inflammation, metabolism and gliomagenesis. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2479–2484. [Google Scholar] [CrossRef]
- Clemetson, K.J.; Pfueller, S.L.; Luscher, E.F.; Jenkins, C.S. Isolation of the membrane glycoproteins of human blood platelets by lectin affinity chromatography. Biochim. Biophys. Acta 1977, 464, 493–508. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Tajima, O.; Hashimoto, N.; Furukawa, K. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Bidlingmaier, S.; Zhu, X.; Liu, B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J. Mol. Med. 2008, 86, 1025–1032. [Google Scholar] [CrossRef]
- Shakya, S.; Gromovsky, A.D.; Hale, J.S.; Knudsen, A.M.; Prager, B.; Wallace, L.C.; Penalva, L.O.F.; Brown, H.A.; Kristensen, B.W.; Rich, J.N.; et al. Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Acta Neuropathol. Commun. 2021, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Gregoire, H.; Roncali, L.; Rousseau, A.; Cherel, M.; Delneste, Y.; Jeannin, P.; Hindré, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Kindy, M.S. Oxidized high-density lipoprotein induces neuron death. Exp. Neurol. 2000, 161, 621–630. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Hale, J.S.; Li, M.; Sinyuk, M.; Jahnen-Dechent, W.; Lathia, J.D.; Silverstein, R.L. Context dependent role of the CD36—thrombospondin—histidine-rich glycoprotein axis in tumor angiogenesis and growth. PLoS ONE 2012, 7, e40033. [Google Scholar] [CrossRef]
- Kaur, B.; Cork, S.M.; Sandberg, E.M.; Devi, N.S.; Zhang, Z.; Klenotic, P.A.; Febbraio, M.; Shim, H.; Mao, H.; Tucker-Burden, C.; et al. Vasculostatin inhibits intracranial glioma growth and negatively regulates in vivo angiogenesis through a CD36-dependent mechanism. Cancer Res. 2009, 69, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Resovi, A.; Pinessi, D.; Chiorino, G.; Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014, 37, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Dee, Z.; Pidcock, K.; Gutierrez, L.S. Thrombospondin-1: Multiple paths to inflammation. Mediat. Inflamm. 2011, 2011, 296069. [Google Scholar] [CrossRef]
- Wang, P.; Zeng, Z.; Lin, C.; Wang, J.; Xu, W.; Ma, W.; Xiang, Q.; Liu, H.; Liu, S.-L. Thrombospondin-1 as a Potential Therapeutic Target: Multiple Roles in Cancers. Curr. Pharm. Des. 2020, 26, 2116–2136. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.S.; Outeiro-Bernstein, M.A.; Juliano, L.; Vardiero, F.; Nader, H.B.; Woods, A.; Legrand, C.; Morandi, V. Syndecan-4 contributes to endothelial tubulogenesis through interactions with two motifs inside the pro-angiogenic N-terminal domain of thrombospondin-1. J. Cell. Physiol. 2008, 214, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O’Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar] [CrossRef]
- Margosio, B.; Rusnati, M.; Bonezzi, K.; Cordes, B.L.; Annis, D.S.; Urbinati, C.; Giavazzi, R.; Presta, M.; Ribatti, D.; Mosher, D.F.; et al. Fibroblast growth factor-2 binding to the thrombospondin-1 type III repeats, a novel antiangiogenic domain. Int. J. Biochem. Cell Biol. 2008, 40, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Daubon, T.; Leon, C.; Clarke, K.; Andrique, L.; Salabert, L.; Darbo, E.; Pineau, R.; Guérit, S.; Maitre, M.; Dedieu, S.; et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat. Commun. 2019, 10, 1146. [Google Scholar] [CrossRef]
- Lin, T.N.; Kim, G.M.; Chen, J.J.; Cheung, W.M.; He, Y.Y.; Hsu, C.Y. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke 2003, 34, 177–186. [Google Scholar] [CrossRef]
- Kragh, M.; Quistorff, B.; Tenan, M.; Van Meir, E.G.; Kristjansen, P.E. Overexpression of thrombospondin-1 reduces growth and vascular index but not perfusion in glioblastoma. Cancer Res. 2002, 62, 1191–1195. [Google Scholar] [PubMed]
- Tsutsui, T.; Kawahara, H.; Kimura, R.; Dong, Y.; Jiapaer, S.; Sabit, H.; Zhang, J.; Yoshida, T.; Nakada, M.; Hanayama, R. Glioma-derived extracellular vesicles promote tumor progression by conveying WT1. Carcinogenesis 2020, 41, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Tenan, M.; Fulci, G.; Albertoni, M.; Diserens, A.C.; Hamou, M.F.; El Atifi-Borel, M.; Feige, J.-J.; Pepper, M.; Van Meir, E.G. Thrombospondin-1 is downregulated by anoxia and suppresses tumorigenicity of human glioblastoma cells. J. Exp. Med. 2000, 191, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Nakagawa, K.; Saito, M.; Kohno, S.; Nagato, S.; Furukawa, K.; Kumon, Y.; Hamada, K.; Ohnishi, T. Introduction of wild-type p53 enhances thrombospondin-1 expression in human glioma cells. Cancer Lett. 2003, 191, 109–119. [Google Scholar] [CrossRef]
- Choi, S.H.; Tamura, K.; Khajuria, R.K.; Bhere, D.; Nesterenko, I.; Lawler, J.; Shah, K. Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol. Ther. 2015, 23, 235–243. [Google Scholar] [CrossRef] [PubMed]
- De Fraipont, F.; Keramidas, M.; El Atifi, M.; Chambaz, E.M.; Berger, F.; Feige, J.J. Expression of the thrombospondin 1 fragment 167-569 in C6 glioma cells stimulates tumorigenicity despite reduced neovascularization. Oncogene 2004, 23, 3642–3649. [Google Scholar] [CrossRef]
- Elstner, A.; Stockhammer, F.; Nguyen-Dobinsky, T.N.; Nguyen, Q.L.; Pilgermann, I.; Gill, A.; Guhr, A.; Zhang, T.; von Eckardstein, K.; Picht, T.; et al. Identification of diagnostic serum protein profiles of glioblastoma patients. J. Neuro-Oncol. 2011, 102, 71–80. [Google Scholar] [CrossRef]
- Qi, C.; Lei, L.; Hu, J.; Wang, G.; Liu, J.; Ou, S. Thrombospondin-1 is a prognostic biomarker and is correlated with tumor immune microenvironment in glioblastoma. Oncol. Lett. 2021, 21, 22. [Google Scholar] [CrossRef]
- Soto-Pantoja, D.R.; Kaur, S.; Roberts, D.D. CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kuznetsova, S.A.; Pendrak, M.L.; Sipes, J.M.; Romeo, M.J.; Li, Z.; Zhang, L.; Roberts, D.D. Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. J. Biol. Chem. 2011, 286, 14991–15002. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, A.; Cai, Y.; et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [PubMed]
- Liu, X.; Kwon, H.; Li, Z.; Fu, Y.X. Is CD47 an innate immune checkpoint for tumor evasion? J. Hematol. Oncol. 2017, 10, 12. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Kern, N.; Vale, R.D. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity 2020, 53, 290–302.e6. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef]
- Von Roemeling, C.A.; Wang, Y.; Qie, Y.; Yuan, H.; Zhao, H.; Liu, X.; Yang, Z.; Yang, M.; Deng, W.; Bruno, K.A.; et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat. Commun. 2020, 11, 1508. [Google Scholar] [CrossRef]
- Gholamin, S.; Youssef, O.A.; Rafat, M.; Esparza, R.; Kahn, S.; Shahin, M.; Giaccia, A.J.; E Graves, E.; Weissman, I.; Mitra, S.; et al. Irradiation or temozolomide chemotherapy enhances anti-CD47 treatment of glioblastoma. Innate Immun. 2020, 26, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Gu, Y.; Jin, K.; Fang, H.; Chen, Y.; Cao, Y.; Liu, X.; Lv, K.; He, X.; Lin, C.; et al. CD47 expression in gastric cancer clinical correlates and association with macrophage infiltration. Cancer Immunol. Immunother. 2021, 70, 1831–1840. [Google Scholar] [CrossRef]
- Walz, D.A. Thrombospondin as a mediator of cancer cell adhesion in metastasis. Cancer Metastasis Rev. 1992, 11, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Krutzsch, H.C.; Inman, J.K.; Roberts, D.D. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res. 1997, 57, 1735–1742. [Google Scholar]
- Kuijlen, J.M.A.; Bremer, E.; Mooij, J.J.A.; den Dunnen, W.F.A.; Helfrich, W. Review: On TRAIL for malignant glioma therapy? Neuropathol. Appl. Neurobiol. 2010, 36, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef]
- Chu, L.-Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef]
- Johnson, L.D.; Goubran, H.A.; Kotb, R.R. Histidine rich glycoprotein and cancer: A multi-faceted relationship. Anticancer. Res. 2014, 34, 593–603. [Google Scholar]
- Jones, C.; Kärrlander, M.; Lindberg, N.; Olofsson, T.; Kastemar, M.; Olsson, A.-K.; Uhrbom, L. Histidine-Rich Glycoprotein Can Prevent Development of Mouse Experimental Glioblastoma. PLoS ONE 2009, 4, e8536. [Google Scholar]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Zhang, X.; Lawler, J. Thrombospondin-based antiangiogenic therapy. Microvasc. Res. 2007, 74, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Roberts, D.D. CD47 applies the brakes to angiogenesis via vascular endothelial growth factor receptor-2. Cell Cycle 2011, 10, 10–12. [Google Scholar] [CrossRef]
- Falero-Perez, J.; Song, Y.S.; Zhao, Y.; Teixeira, L.; Sorenson, C.M.; Sheibani, N. Cyp1b1 expression impacts the angiogenic and inflammatory properties of liver sinusoidal endothelial cells. PLoS ONE 2018, 13, e0206756. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Martin-Manso, G.; Pendrak, M.L.; Garfield, S.H.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem. 2010, 285, 38923–38932. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Martin-Manso, G.; Maxhimer, J.B.; Roberts, D.D. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nat. Rev. Cancer 2009, 9, 182–194. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Shiva, S.; Gladwin, M. Thrombospondin-1-CD47 blockade and exogenous nitrite enhance ischemic tissue survival, blood flow and angiogenesis via coupled NO-cGMP pathway activation. Nitric Oxide 2009, 21, 52–62. [Google Scholar] [CrossRef]
- Stein, E.V.; Miller, T.W.; Ivins-O’Keefe, K.; Kaur, S.; Roberts, D.D. Secreted Thrombospondin-1 Regulates Macrophage Interleukin-1beta Production and Activation through CD47. Sci. Rep. 2016, 6, 19684. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Annis, D.S.; Pendrak, M.L.; Ptaszynska, M.; Frazier, W.A.; Mosher, D.F.; Roberts, D.D. Differential interactions of thrombospondin-1, -2, and -4 with CD47 and effects on cGMP signaling and ischemic injury responses. J. Biol. Chem. 2009, 284, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Frazier, W.A.; Roberts, D.D. Thrombospondin-1: A physiological regulator of nitric oxide signaling. Cell. Mol. Life Sci. 2008, 65, 728–742. [Google Scholar] [CrossRef]
- Zhang, X.; Kazerounian, S.; Duquette, M.; Perruzzi, C.; Nagy, J.A.; Dvorak, H.F.; Parangi, S.; Lawler, J. Thrombospondin-1 modulates vascular endothelial growth factor activity at the receptor level. FASEB J. 2009, 23, 3368–3376. [Google Scholar] [CrossRef]
- Dawson, D.W.; Pearce, S.F.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N.P. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef]
- Oldenborg, P.A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol. 2013, 2013, 614619. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Naganuma, H.; Satoh, E.; Nagasaka, M.; Isoe, S.; Nakano, S.; Nukui, H. Secretion of Transforming Growth Factor-β1 and -β2 by Malignant Glioma Cells. Neurol. Med.-Chir. 1995, 35, 423–430. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Ridnour, L.A.; Dimitry, J.; Frazier, W.A.; Wink, D.A.; Roberts, D.D. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J. Biol. Chem. 2006, 281, 26069–26080. [Google Scholar] [CrossRef]
- Kaur, S.; Chang, T.; Singh, S.P.; Lim, L.; Mannan, P.; Garfield, S.H.; Pendrak, M.L.; Soto-Pantoja, D.R.; Rosenberg, A.Z.; Jin, S.; et al. CD47 signaling regulates the immunosuppressive activity of VEGF in T cells. J. Immunol. 2014, 193, 3914–3924. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef]
- Gupta, K.; Gupta, P.; Wild, R.; Ramakrishnan, S.; Hebbel, R.P. Binding and displacement of vascular endothelial growth factor (VEGF) by thrombospondin: Effect on human microvascular endothelial cell proliferation and angiogenesis. Angiogenesis 1999, 3, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Jia, Y.; Fukuyama, J.; Switzer, C.H.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J. Biol. Chem. 2007, 282, 15404–15415. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Rev. Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef]
- Van Deventer, S.; Arp, A.B.; van Spriel, A.B. Dynamic Plasma Membrane Organization: A Complex Symphony. Trends Cell Biol. 2021, 31, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Sanchez-Madrid, F.; Del Hoyo, G.M. Function and Dynamics of Tetraspanins during Antigen Recognition and Immunological Synapse Formation. Front. Immunol. 2015, 6, 653. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Savill, J. Proinflammatory cytokines potentiate thrombospondin-mediated phagocytosis of neutrophils undergoing apoptosis. J. Immunol. 1995, 154, 2366–2374. [Google Scholar]
- Primo, L.; Ferrandi, C.; Roca, C.; Marchio, S.; di Blasio, L.; Alessio, M.; Bussolino, F. Identification of CD36 molecular features required for its in vitro angiostatic activity. FASEB J. 2005, 19, 1713–1715. [Google Scholar] [CrossRef]
- Jeanne, A.; Schneider, C.; Martiny, L.; Dedieu, S. Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front. Pharmacol. 2015, 6, 252. [Google Scholar] [CrossRef] [PubMed]
- Enciu, A.-M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? BioMed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef]
- Kaur, S.; Cicalese, K.V.; Banerjee, R.; Roberts, D.D. Preclinical and clinical development of therapeutic antibodies targeting functions of CD47 in the tumor microenvironment. Antib. Ther. 2020, 3, 179–192. [Google Scholar] [CrossRef]
- Barazi, H.O.; Li, Z.; Cashel, J.A.; Krutzsch, H.C.; Annis, D.S.; Mosher, D.F.; Roberts, D.D. Regulation of integrin function by CD47 ligands. Differential effects on alpha vbeta 3 and alpha 4beta1 integrin-mediated adhesion. J. Biol. Chem. 2002, 277, 42859–42866. [Google Scholar] [CrossRef]
- Tulasne, D.; Judd, B.A.; Johansen, M.; Asazuma, N.; Best, D.; Brown, E.J.; Kahn, M.; Koretzky, G.A.; Watson, S.P. C-terminal peptide of thrombospondin-1 induces platelet aggregation through the Fc receptor gamma-chain-associated signaling pathway and by agglutination. Blood 2001, 98, 3346–3352. [Google Scholar] [CrossRef]
- Kalas, W.; Swiderek, E.; Switalska, M.; Wietrzyk, J.; Rak, J.; Strzadala, L. Thrombospondin-1 receptor mediates autophagy of RAS-expressing cancer cells and triggers tumour growth inhibition. Anticancer. Res. 2013, 33, 1429–1438. [Google Scholar]
- Leclair, P.; Lim, C.J. CD47-independent effects mediated by the TSP-derived 4N1K peptide. PLoS ONE 2014, 9, e98358. [Google Scholar] [CrossRef]
- Jeanne, A.; Sick, E.; Devy, J.; Floquet, N.; Belloy, N.; Theret, L.; Boulagnon-Rombi, C.; Diebold, M.-D.; Dauchez, M.; Martiny, L.; et al. Identification of TAX2 peptide as a new unpredicted anti-cancer agent. Oncotarget 2015, 6, 17981–18000. [Google Scholar] [CrossRef]
- Jeanne, A.; Sarazin, T.; Charle, M.; Moali, C.; Fichel, C.; Boulagnon-Rombi, C.; Callewaert, M.; Andry, M.-C.; Diesis, E.; Delolme, F.; et al. Targeting Ovarian Carcinoma with TSP-1:CD47 Antagonist TAX2 Activates Anti-Tumor Immunity. Cancers 2021, 13, 5019. [Google Scholar] [CrossRef]
- Taraboletti, G.; Rusnati, M.; Ragona, L.; Colombo, G. Targeting tumor angiogenesis with TSP-1-based compounds: Rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget 2010, 1, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Margosio, B.; Ragona, L.; Neves, M.; Bonifacio, S.; Annis, D.S.; Stravalaci, M.; Tomaselli, S.; Giavazzi, R.; Rusnati, M.; et al. Non-peptidic thrombospondin-1 mimics as fibroblast growth factor-2 inhibitors: An integrated strategy for the development of new antiangiogenic compounds. J. Biol. Chem. 2010, 285, 8733–8742. [Google Scholar] [CrossRef] [PubMed]
- Pagano, K.; Torella, R.; Foglieni, C.; Bugatti, A.; Tomaselli, S.; Zetta, L.; Presta, M.; Rusnati, M.; Taraboletti, G.; Colombo, G.; et al. Direct and allosteric inhibition of the FGF2/HSPGs/FGFR1 ternary complex formation by an antiangiogenic, thrombospondin-1-mimic small molecule. PLoS ONE 2012, 7, e36990. [Google Scholar] [CrossRef] [PubMed]
- Meli, M.; Pagano, K.; Ragona, L.; Colombo, G. Investigating the dynamic aspects of drug-protein recognition through a combination of MD and NMR analyses: Implications for the development of protein-protein interaction inhibitors. PLoS ONE 2014, 9, e97153. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Harb, W.; Patnaik, A.; Ulahannan, S.; Mahdi, H.; Ahluwalia, M.; Patel, M.; Dowlati, A.; Bullock, A.; Wen, P.; et al. 374 A first-in-human Phase 1/2 open label trial evaluating the safety, pharmacology, and preliminary efficacy of VT1021 in subjects with advanced solid tumors. J. ImmunoTherapy Cancer 2020, 8 (Suppl. 3), A399. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, H.; Yu, J.; Tian, W.; Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.C.; Guo, N.; Sockolosky, J.T.; Ring, A.M.; Weiskopf, K.; Ozkan, E.; Mori, Y.; Weissman, I.L.; Garcia, K.C. “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein alpha (SIRPalpha) antagonists that enhance antibody-dependent cellular phagocytosis. J. Biol. Chem. 2015, 290, 12650–12663. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| TSP-1 Concentration | Ligand | Signaling Pathway/Proteins | Effect |
|---|---|---|---|
| 10–100 pM | CD47 | NO/cGMP/cGK | Increased endothelial cell adhesion [125,126,131,135] |
| eNOS/NO/sGC | Anti-angiogenic effect/inhibits NO production [127,131] | ||
| 100–150 pM | CD47 | VEFGR2 direct binding | Anti-angiogenic effect [125,138] |
| 1–10 nM | CD36 | NO/cGMP/cGK Src kinasesFyn/p38 phosphorylation | Inhibits NO production Anti-angiogenic effect [126] Endothelial cell apoptosis [139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.-A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604. https://doi.org/10.3390/ijms23020604
Tanase C, Enciu AM, Codrici E, Popescu ID, Dudau M, Dobri AM, Pop S, Mihai S, Gheorghișan-Gălățeanu A-A, Hinescu ME. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? International Journal of Molecular Sciences. 2022; 23(2):604. https://doi.org/10.3390/ijms23020604
Chicago/Turabian StyleTanase, Cristiana, Ana Maria Enciu, Elena Codrici, Ionela Daniela Popescu, Maria Dudau, Ana Maria Dobri, Sevinci Pop, Simona Mihai, Ancuța-Augustina Gheorghișan-Gălățeanu, and Mihail Eugen Hinescu. 2022. "Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately?" International Journal of Molecular Sciences 23, no. 2: 604. https://doi.org/10.3390/ijms23020604
APA StyleTanase, C., Enciu, A. M., Codrici, E., Popescu, I. D., Dudau, M., Dobri, A. M., Pop, S., Mihai, S., Gheorghișan-Gălățeanu, A.-A., & Hinescu, M. E. (2022). Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? International Journal of Molecular Sciences, 23(2), 604. https://doi.org/10.3390/ijms23020604