
Connective Tissue Growth Factor in Idiopathic Pulmonary Fibrosis: Breaking the Bridge


{kind=link}
{kind=link}
Abstract
:1. Introduction
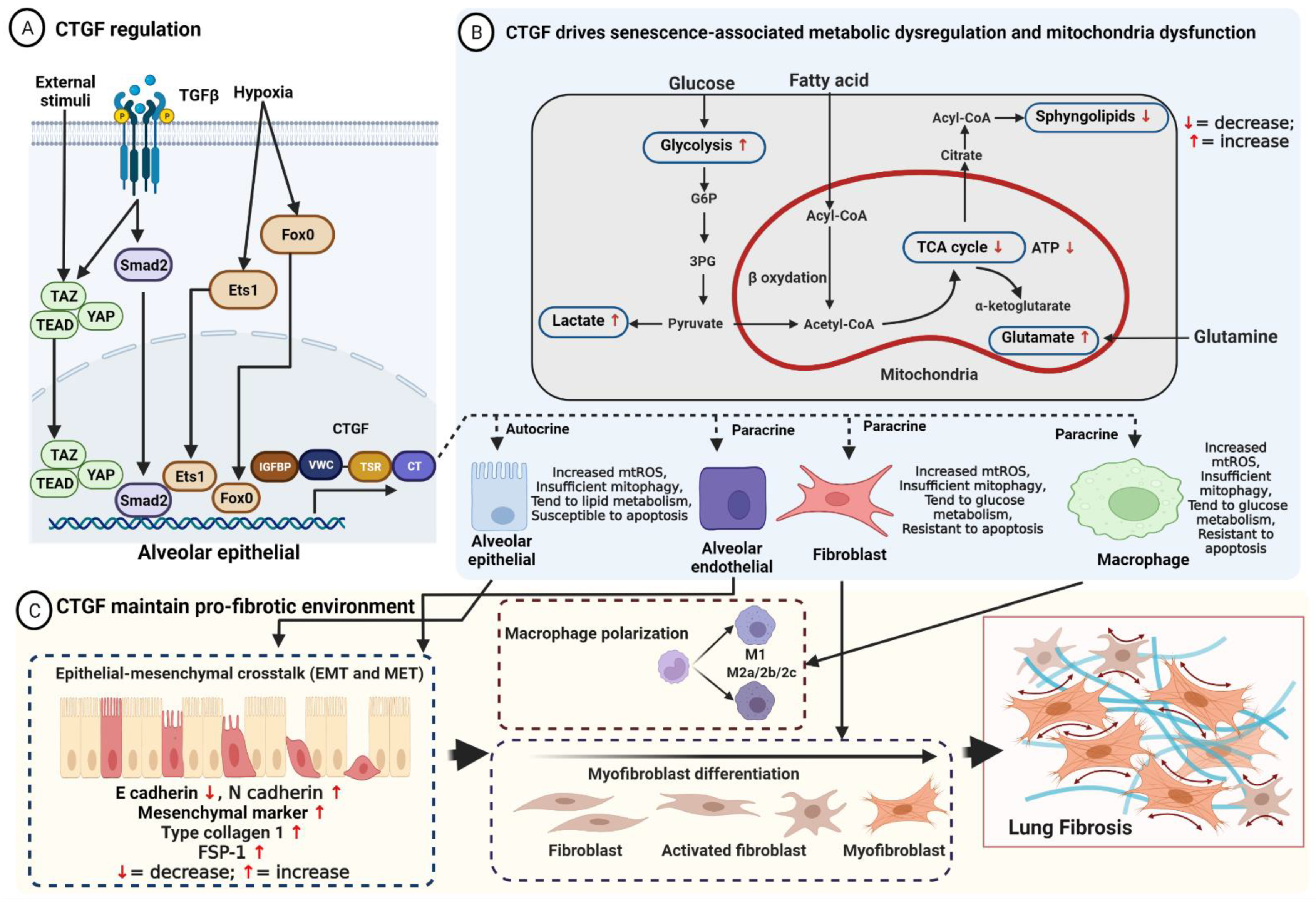
2. Structure, Regulation, and Function of CTGF
3. CTGF Maintains the Pro-Fibrotic Environment in IPF
3.1. Activated Alveolar Epithelial Cells Initiate a Cycle of Fibrosis through CTGF
3.2. CTGF Stimulates the Differentiation of Lung Fibroblasts
3.3. CTGF Modulates Dysfunction of Macrophage Polarization
3.4. CTGF Increases Endothelial Growth
3.5. Fibrocyte Differentiation Involved in CTGF
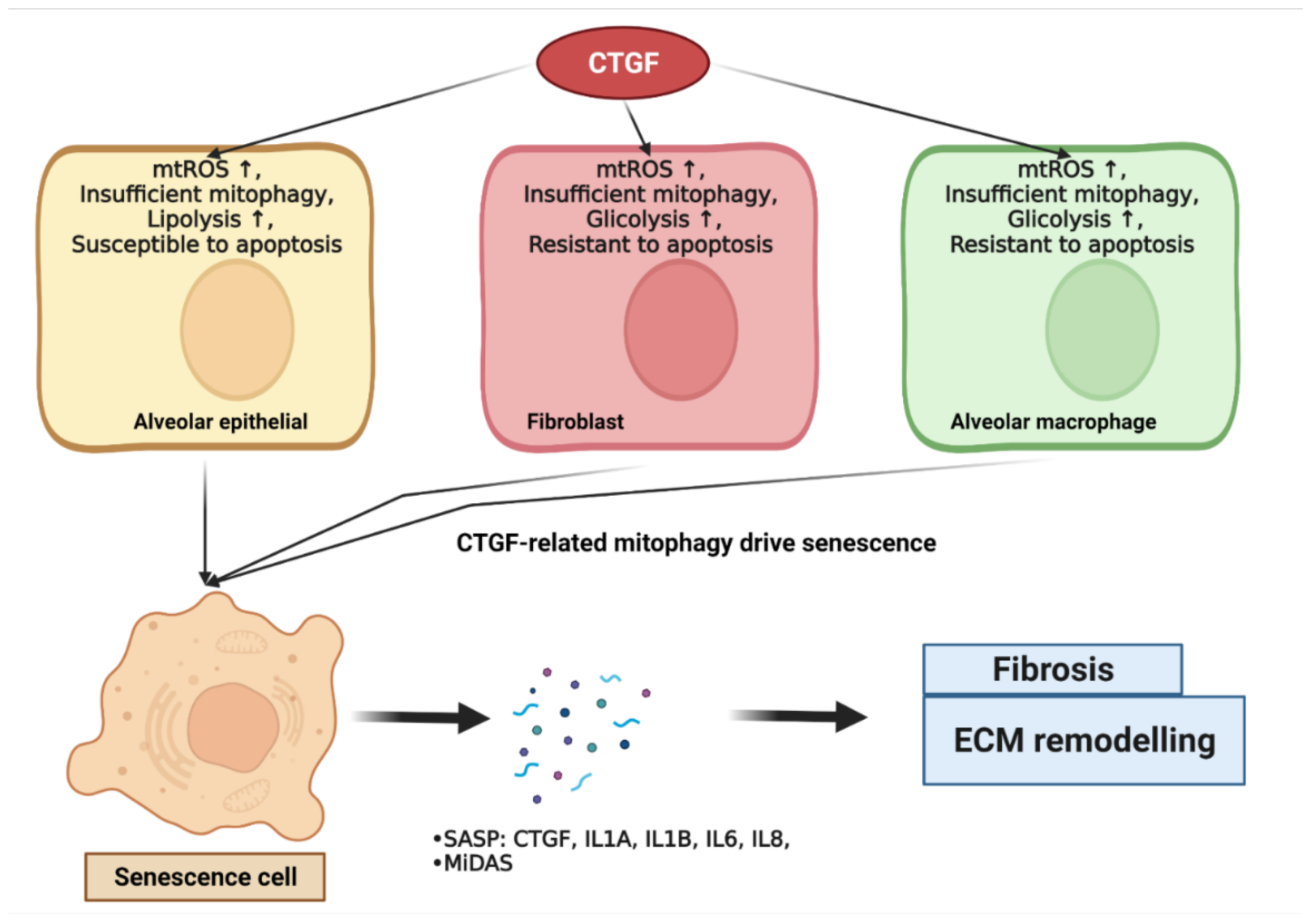
4. CTGF Drives Senescence in IPF
4.1. CTGF Influences Cellular Mitochondria Bioenergetics
4.1.1. Alveolar Epithelial Mitochondria Dysfunction
4.1.2. Lung Fibroblast Mitochondria Dysfunction
4.1.3. Macrophage Mitochondria Dysfunction
4.2. CTGF Regulates Cellular Metabolic Dysregulation
4.2.1. Glucose Metabolism
4.2.2. Lipid Metabolism
4.2.3. Glutamine Metabolism
4.3. CTGF Promotes Mitochondria–Metabolic-Dysfunction-Related Cellular Senescence
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rendin, L.E.; Löfdahl, A.; Xiong, L.; Michaliková, B.; Zhou, X.; Dellgren, G.; Westergren-Thorsson, G. Altered ECM niche in IPF affect structural remodeling. Eur. Respir. J. 2020, 56, 3376. [Google Scholar]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45. [Google Scholar] [CrossRef] [Green Version]
- Paolocci, G.; Folletti, I.; Torén, K.; Ekström, M.; Dell’Omo, M.; Muzi, G.; Murgia, N. Occupational risk factors for idiopathic pulmonary fibrosis in Southern Europe: A case-control study. BMC Pulm. Med. 2018, 18, 75. [Google Scholar] [CrossRef] [Green Version]
- Walters, G.I. Occupational exposures and idiopathic pulmonary fibrosis. Curr. Opin. Allergy Clin. Immunol. 2020, 20, 103–111. [Google Scholar] [CrossRef]
- Hall-Glenn, F.; Lyons, K.M. Roles for CCN2 in normal physiological processes. Cell. Mol. Life Sci. 2011, 68, 3209–3217. [Google Scholar] [CrossRef] [Green Version]
- Moussad, E.E.D.A.; Brigstock, D.R. Connective tissue growth factor: What’s in a name? Mol. Genet. Metab. 2000, 71, 276–292. [Google Scholar] [CrossRef]
- Ungvari, Z.; Valcarcel-Ares, M.N.; Tarantini, S.; Yabluchanskiy, A.; Fülöp, G.A.; Kiss, T.; Csiszar, A. Connective tissue growth factor (CTGF) in age-related vascular pathologies. GeroScience 2017, 39, 491–498. [Google Scholar] [CrossRef]
- Phanish, M.K.; Winn, S.K.; Dockrell, M.E.C. Connective Tissue Growth Factor-(CTGF, CCN2)—A Marker, Mediator and Therapeutic Target for Renal Fibrosis. Nephron Exp. Nephrol. 2010, 114, e83–e92. [Google Scholar] [CrossRef]
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The CCN family of proteins: Structure-function relationships. Trends Biochem. Sci. 2008, 33, 461–473. [Google Scholar] [CrossRef]
- Jun, J.-I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi-Wen, X.; Leask, A.; Abraham, D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008, 19, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Gressner, A.M. Connective tissue growth factor: A fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008, 28, 1065–1079. [Google Scholar] [CrossRef]
- Kubota, S.; Takigawa, M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin. Sci. 2014, 128, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Preisser, F.; Giehl, K.; Rehm, M.; Goppelt-Struebe, M. Inhibitors of oxygen sensing prolyl hydroxylases regulate nuclear localization of the transcription factors Smad2 and YAP/TAZ involved in CTGF synthesis. Biochim. Biophys. Acta 2016, 1863, 2027–2036. [Google Scholar] [CrossRef]
- Van Beek, J.P.; Kennedy, L.; Rockel, J.S.; Bernier, S.M.; Leask, A. The induction of CCN2 by TGFbeta1 involves Ets-1. Arthritis Res. Ther. 2006, 8, R36. [Google Scholar] [CrossRef] [Green Version]
- Samarin, J.; Cicha, I.; Goppelt-Struebe, M. Cell type-specific regulation of CCN2 protein expression by PI3K-AKT-FoxO signaling. J. Cell Commun. Signal. 2009, 3, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Bu, Y.; Jiang, S.; Chang, F.; Jia, F.; Xiao, X.; Song, G.; Zhang, M.; Ning, P.; Jia, Q. CCN2-MAPK-Id-1 loop feedback amplification is involved in maintaining stemness in oxaliplatin-resistant hepatocellular carcinoma. Hepatol. Int. 2019, 13, 440–453. [Google Scholar] [CrossRef] [Green Version]
- Ihn, H. Pathogenesis of fibrosis: Role of TGF-beta and CTGF. Curr. Opin. Rheumatol. 2002, 14, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Yeger, H.; Perbal, B. CCN family of proteins: Critical modulators of the tumor cell microenvironment. J. Cell Commun. Signal. 2016, 10, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.F.; Goldschmeding, R.; Schlingemann, R.O. The Angio-Fibrotic Switch of VEGF and CTGF in Proliferative Diabetic Retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef]
- Zhao, Z.; Ho, L.; Wang, J.; Qin, W.; Festa, E.D.; Mobbs, C.; Hof, P.; Rocher, A.; Masur, S.; Haroutunian, V.; et al. Connective tissue growth factor (CTGF) expression in the brain is a downstream effector of insulin resistance- associated promotion of Alzheimer’s disease beta-amyloid neuropathology. FASEB J. 2005, 19, 2081–2082. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, K.; Makino, T.; Stawski, L.; Lipson, K.E.; Leask, A.; Trojanowska, M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res. Ther. 2017, 19, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Chaqour, B. Cysteine-rich protein 61 (CCN1) and connective tissue growth factor (CCN2) at the crosshairs of ocular neovascular and fibrovascular disease therapy. J. Cell Commun. Signal. 2013, 7, 253. [Google Scholar] [CrossRef] [Green Version]
- Hou, N.; Wen, Y.; Yuan, X.; Xu, H.; Wang, X.; Li, F.; Ye, B. Activation of Yap1/Taz signaling in ischemic heart disease and dilated cardiomyopathy. Exp. Mol. Pathol. 2017, 103, 267–275. [Google Scholar] [CrossRef]
- Accornero, F.; van Berlo, J.H.; Correll, R.N.; Elrod, J.W.; Sargent, M.A.; York, A.; Rabinowitz, J.E.; Leask, A.; Molkentin, J.D. Genetic Analysis of Connective Tissue Growth Factor as an Effector of Transforming Growth Factor β Signaling and Cardiac Remodeling. Mol. Cell. Biol. 2015, 35, 2154–2164. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; Ten Dijke, P. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plantier, L.; Renaud, H.; Respaud, R.; Marchand-Adam, S.; Crestani, B. Transcriptome of Cultured Lung Fibroblasts in Idiopathic Pulmonary Fibrosis: Meta-Analysis of Publically Available Microarray Datasets Reveals Repression of Inflammation and Immunity Pathways. Int. J. Mol. Sci. 2016, 17, 2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, M.; King, T.E.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Kage, H.; Borok, Z. EMT and Interstitial Lung Disease: A Mysterious Relationship. Curr. Opin. Pulm. Med. 2012, 18, 517. [Google Scholar] [CrossRef]
- Kolb, M.; Borensztajn, K.; Crestani, B.; Kolb, M. Idiopathic Pulmonary Fibrosis: From Epithelial Injury to Biomarkers—Insights from the Bench Side. Respiration 2013, 86, 441–452. [Google Scholar]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Zhou, Y.; Li, J.; Wickens, L.; Conforti, F.; Rattu, A.; Ibrahim, F.M.; Alzetani, A.; Marshall, B.G.; Fletcher, S.V.; et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 2021, 297, 101096. [Google Scholar] [CrossRef]
- Xu, X.; Dai, H.; Wang, C. Epithelium-dependent profibrotic milieu in the pathogenesis of idiopathic pulmonary fibrosis: Current status and future directions. Clin. Respir. J. 2016, 10, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabeyrin, M.; Thivolet, F.; Ferretti, G.R.; Chalabreysse, L.; Jankowski, A.; Cottin, V.; Pison, C.; Cordier, J.-F.; Lantuejoul, S. Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations. Ann. Diagn. Pathol. 2015, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Vanstapel, A.; Goldschmeding, R.; Broekhuizen, R.; Nguyen, T.; Sacreas, A.; Kaes, J.; Heigl, T.; Verleden, S.E.; De Zutter, A.; Verleden, G.; et al. Connective Tissue Growth Factor Is Overexpressed in Explant Lung Tissue and Broncho-Alveolar Lavage in Transplant-Related Pulmonary Fibrosis. Front. Immunol. 2021, 12, 661761. [Google Scholar] [CrossRef]
- Ponticos, M.; Holmes, A.M.; Shi-wen, X.; Leoni, P.; Khan, K.; Rajkumar, V.S.; Hoyles, R.K.; Bou-Gharios, G.; Black, C.M.; Denton, C.P.; et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009, 60, 2142–2155. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wan, X.; Geng, J.; Li, F.; Yang, T.; Dai, H. Rapamycin regulates connective tissue growth factor expression of lung epithelial cells via phosphoinositide 3-kinase. Exp. Biol. Med. 2013, 238, 1082–1094. [Google Scholar] [CrossRef]
- Kono, M.; Nakamura, Y.; Suda, T.; Kato, M.; Kaida, Y.; Hashimoto, D.; Inui, N.; Hamada, E.; Miyazaki, O.; Kurashita, S.; et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin. Chim. Acta 2011, 412, 2211–2215. [Google Scholar] [CrossRef]
- Yanagihara, T.; Tsubouchi, K.; Gholiof, M.; Chong, S.G.; Lipson, K.E.; Zhou, Q.; Scallan, C.; Upagupta, C.; Tikkanen, J.; Keshavjee, S.; et al. Connective-Tissue Growth Factor Contributes to TGF-β1-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 260–270. [Google Scholar] [CrossRef]
- Morishima, Y.; Nomura, A.; Uchida, Y.; Noguchi, Y.; Sakamoto, T.; Ishii, Y.; Goto, Y.; Masuyama, K.; Zhang, M.J.; Hirano, K.; et al. Triggering the induction of myofibroblast and fibrogenesis by airway epithelial shedding. Am. J. Respir. Cell Mol. Biol. 2001, 24, 1–11. [Google Scholar] [CrossRef]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.H.; Yamauchi, K.; Uzuki, M.; Nakanishi, T.; Takigawa, M.; Inoue, H.; Sawai, T. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur. Respir. J. 2001, 17, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Velikoff, M.; Canalis, E.; Horowitz, J.C.; Kim, K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L786–L796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Yu, Q.; Lin, H.; Wang, Z.; Fu, G.; Lei, L.; Shi, Y.; Zhang, L.; Qin, L.; Liu, Y. The Role of CTGF in Inflammatory Responses Induced by Silica Particles in Human Bronchial Epithelial Cells. Lung 2019, 197, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Dong, N.; Fang, X.; Wang, X. Regulatory mechanisms of TGF-β1-induced fibrogenesis of human alveolar epithelial cells. J. Cell. Mol. Med. 2016, 20, 2183–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnylal, S.; Xu, S.; Jones, H.; Tam, A.; Sreeram, V.R.; Ponticos, M.; Norman, J.; Agrawal, P.; Abraham, D.; de Crombrugghe, B. Connective tissue growth factor causes EMT-like cell fate changes in vivo and in vitro. J. Cell Sci. 2013, 126, 2164–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafieian, M.; Chen, S.; Wu, S. Integrin-linked kinase mediates CTGF-induced epithelial to mesenchymal transition in alveolar type II epithelial cells. Pediatr. Res. 2015, 77, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Lin, C.H.; Chen, J.Y.; Li, C.H.; Liu, Y.T.; Chen, B.C. Induction of Connective Tissue Growth Factor Expression by Hypoxia in Human Lung Fibroblasts via the MEKK1/MEK1/ERK1/GLI-1/GLI-2 and AP-1 Pathways. PLoS ONE 2016, 11, e0160593. [Google Scholar] [CrossRef]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-β1-Induced EMT In Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-C.; Bai, K.-J.; Cheng, W.-H.; Chen, J.-Y.; Lin, C.-H.; Wen, H.-C.; Chen, B.-C. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. Int. J. Mol. Sci. 2020, 21, 9084. [Google Scholar] [CrossRef]
- Madala, S.K.; Schmidt, S.; Davidson, C.; Ikegami, M.; Wert, S.; Hardie, W.D. MEK-ERK Pathway Modulation Ameliorates Pulmonary Fibrosis Associated with Epidermal Growth Factor Receptor Activation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 380–388. [Google Scholar] [CrossRef]
- Zhang, C.; Meng, X.; Zhu, Z.; Liu, J.; Deng, A. Connective tissue growth factor regulates the key events in tubular epithelial to myofibroblast transition in vitro. Cell Biol. Int. 2004, 28, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Gore-Hyer, E.; Shegogue, D.; Markiewicz, M.; Lo, S.; Hazen-Martin, D.; Greene, E.L.; Grotendorst, G.; Trojanowska, M. TGF-β and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am. J. Physiol. Physiol. 2002, 283, F707–F716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.F. Origin of Myofibroblasts in Lung Fibrosis. Curr. Tissue Microenviron. Rep. 2020, 1, 155–162. [Google Scholar] [CrossRef]
- Moore, M.W.; Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Garrett, Q.; Khaw, P.T.; Blalock, T.D.; Schultz, G.S.; Grotendorst, G.R.; Daniels, J.T. Involvement of CTGF in TGF-beta1-stimulation of myofibroblast differentiation and collagen matrix contraction in the presence of mechanical stress. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1109–1116. [Google Scholar] [CrossRef]
- Folger, P.A.; Zekaria, D.; Grotendorst, G.; Masur, S.K. Transforming growth factor-beta-stimulated connective tissue growth factor expression during corneal myofibroblast differentiation. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2534–2541. [Google Scholar]
- Yang, Z.; Sun, Z.; Liu, H.; Ren, Y.; Shao, D.; Zhang, W.; Lin, J.; Wolfram, J.; Wang, F.; Nie, S. Connective tissue growth factor stimulates the proliferation, migration and differentiation of lung fibroblasts during paraquat-induced pulmonary fibrosis. Mol. Med. Rep. 2015, 12, 1091–1097. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-C.; Wu, S.-B.; Kau, H.-C.; Wei, Y.-H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chen, Y.; Suo, L.; Chen, H.; Zhu, L.; Wan, G.; Han, X. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 2019, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Tam, A.Y.Y.; Horwell, A.L.; Trinder, S.L.; Khan, K.; Xu, S.; Ong, V.; Denton, C.P.; Norman, J.T.; Holmes, A.M.; Bou-Gharios, G.; et al. Selective deletion of connective tissue growth factor attenuates experimentally-induced pulmonary fibrosis and pulmonary arterial hypertension. Int. J. Biochem. Cell Biol. 2021, 134, 105961. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Oxidative Stress Induced by Reactive Oxygen Species (ROS) and NADPH Oxidase 4 (NOX4) in the Pathogenesis of the Fibrotic Process in Systemic Sclerosis: A Promising Therapeutic Target. J. Clin. Med. 2021, 10, 4791. [Google Scholar] [CrossRef]
- Shibata, S.; Ishiyama, J. Secreted protein acidic and rich in cysteine (SPARC) is upregulated by transforming growth factor (TGF)-β and is required for TGF-β-induced hydrogen peroxide production in fibroblasts. Fibrogenesis Tissue Repair 2013, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-C.; Lai, S.; Guo, X.; Zhang, X.; de Crombrugghe, B.; Sonnylal, S.; Arnett, F.C.; Zhou, X. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res. Ther. 2010, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Sonnylal, S.; Arnett, F.C.; De Crombrugghe, B.; Zhou, X. Attenuation of expression of extracellular matrix genes with siRNAs to Sparc and Ctgf in skin fibroblasts of CTGF transgenic mice. Int. J. Immunopathol. Pharmacol. 2011, 24, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457. [Google Scholar] [CrossRef]
- Riley, K.G.; Pasek, R.C.; Maulis, M.F.; Dunn, J.C.; Bolus, W.R.; Kendall, P.L.; Hasty, A.H.; Gannon, M. Macrophages are essential for CTGF-mediated adult β-cell proliferation after injury. Mol. Metab. 2015, 4, 584–591. [Google Scholar] [CrossRef]
- Wang, T.-T.; Yuan, J.-H.; Ma, J.-Z.; Yang, W.-J.; Liu, X.-N.; Yin, Y.-P.; Liu, Y.; Pan, W.; Sun, S.-H. CTGF secreted by mesenchymal-like hepatocellular carcinoma cells plays a role in the polarization of macrophages in hepatocellular carcinoma progression. Biomed. Pharmacother. 2017, 95, 111–119. [Google Scholar] [CrossRef]
- Zhang, S.-M.; Wei, C.-Y.; Wang, Q.; Wang, L.; Lu, L.; Qi, F.-Z. M2-polarized macrophages mediate wound healing by regulating connective tissue growth factor via AKT, ERK1/2, and STAT3 signaling pathways. Mol. Biol. Rep. 2021, 48, 6443–6456. [Google Scholar] [CrossRef]
- Bickelhaupt, S.; Erbel, C.; Timke, C.; Wirkner, U.; Dadrich, M.; Flechsig, P.; Tietz, A.; Pföhler, J.; Gross, W.; Peschke, P.; et al. Effects of CTGF Blockade on Attenuation and Reversal of Radiation-Induced Pulmonary Fibrosis. J. Natl. Cancer Inst. 2017, 109, djw339. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Jimenez, S.A. Protein kinase Cδ and c-Abl kinase are required for transforming growth factor β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011, 63, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Zhang, J.; Miao, Y.; He, M.; Kang, J.; Huang, H.-Y.; Chou, C.-H.; Huang, T.-S.; Hong, H.-C.; Su, S.-H.; et al. Role of endothelial cells in pulmonary fibrosis via SREBP2 activation. JCI Insight 2021, 6, e125635. [Google Scholar] [CrossRef]
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021, 12, 327. [Google Scholar] [CrossRef]
- Brigstock, D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002, 5, 153–165. [Google Scholar] [CrossRef]
- Pi, L.; Fu, C.; Lu, Y.; Zhou, J.; Jorgensen, M.; Shenoy, V.; Lipson, K.E.; Scott, E.W.; Bryant, A.J. Vascular Endothelial Cell-Specific Connective Tissue Growth Factor (CTGF) Is Necessary for Development of Chronic Hypoxia-Induced Pulmonary Hypertension. Front. Physiol. 2018, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-C.; Chuang, S.-M.; Hsu, C.-J.; Tsai, C.-H.; Wang, S.-W.; Tang, C.-H. CTGF increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing miR-210 expression. Cell Death Dis. 2014, 5, e1485. [Google Scholar] [CrossRef] [Green Version]
- Kinashi, H.; Falke, L.L.; Nguyen, T.Q.; Bovenschen, N.; Aten, J.; Leask, A.; Ito, Y.; Goldschmeding, R. Connective tissue growth factor regulates fibrosis-associated renal lymphangiogenesis. Kidney Int. 2017, 92, 850–863. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Inui, N.; Hakamata, A.; Suzuki, Y.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; Watanabe, H.; Suda, T. Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir. Res. 2018, 19, 127. [Google Scholar] [CrossRef]
- Heukels, P.; van Hulst, J.A.C.; van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; van den Toorn, L.M.; Boomars, K.A.T.; Wijsenbeek, M.S.; Hoogsteden, H.; von der Thüsen, J.H.; et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 90. [Google Scholar] [CrossRef]
- Rosin, N.L.; Falkenham, A.; Sopel, M.J.; Lee, T.D.G.; Légaré, J.-F. Regulation and Role of Connective Tissue Growth Factor in AngII-Induced Myocardial Fibrosis. Am. J. Pathol. 2013, 182, 714–726. [Google Scholar] [CrossRef]
- Weng, C.-M.; Chen, B.-C.; Wang, C.-H.; Feng, P.-H.; Lee, M.-J.; Huang, C.-D.; Kuo, H.-P.; Lin, C.-H. The Endothelin A Receptor Mediates Fibrocyte Differentiation in Chronic Obstructive Asthma. The Involvement of Connective Tissue Growth Factor. Am. J. Respir. Crit. Care Med. 2013, 188, 298–308. [Google Scholar] [CrossRef]
- Wang, T.-Y.; Lo, Y.-L.; Wang, C.-H.; Kuo, H.-P. Increased CTGF expression of circulating fibrocytes in asthmatic patients with severe OSA—The role of HIF-1a and HDAC7. Eur. Respir. J. 2018, 52, PA2186. [Google Scholar]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421. [Google Scholar] [CrossRef]
- Blokland, K.E.C.; Waters, D.W.; Schuliga, M.; Read, J.; Pouwels, S.D.; Grainge, C.L.; Jaffar, J.; Westall, G.; Mutsaers, S.E.; Prêle, C.M.; et al. Senescence of IPF Lung Fibroblasts Disrupt Alveolar Epithelial Cell Proliferation and Promote Migration in Wound Healing. Pharmaceutics 2020, 12, 389. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial dysfunction in pulmonary fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef]
- Tur, J.; Pereira-Lopes, S.; Vico, T.; Marín, E.A.; Muñoz, J.P.; Hernández-Alvarez, M.; Cardona, P.-J.; Zorzano, A.; Lloberas, J.; Celada, A. Mitofusin 2 in Macrophages Links Mitochondrial ROS Production, Cytokine Release, Phagocytosis, Autophagy, and Bactericidal Activity. Cell Rep. 2020, 32, 108079. [Google Scholar] [CrossRef]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Xin, Y.; Wu, W.; Qu, J.; Wang, X.; Lei, S.; Yuan, L.; Liu, X. Inhibition of Mitofusin-2 Promotes Cardiac Fibroblast Activation via the PERK/ATF4 Pathway and Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2019, 2019, 3649808. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.H.; Yang, C.C.; Chan, D.C.; Wu, C.T.; Chen, L.P.; Huang, J.W.; Hung, K.Y.; Chiang, C.K. Chemical chaperon 4-phenylbutyrate protects against the endoplasmic reticulum stress-mediated renal fibrosis in vivo and in vitro. Oncotarget 2016, 7, 22116–22127. [Google Scholar] [CrossRef] [Green Version]
- Kosmider, B.; Lin, C.-R.; Karim, L.; Tomar, D.; Vlasenko, L.; Marchetti, N.; Bolla, S.; Madesh, M.; Criner, G.J.; Bahmed, K. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. eBioMedicine 2019, 46, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [Green Version]
- Watts, K.L.; Spiteri, M.A. Connective tissue growth factor expression and induction by transforming growth factor-β is abrogated by simvastatin via a Rho signaling mechanism. Am. J. Physiol. Cell. Mol. Physiol. 2004, 287, L1323–L1332. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Mihajlovic, M.; Valentijn, F.; Nguyen, T.Q.; Goldschmeding, R.; Masereeuw, R. A Human Conditionally Immortalized Proximal Tubule Epithelial Cell Line as a Novel Model for Studying Senescence and Response to Senolytics. Front. Pharmacol. 2022, 13, 791612. [Google Scholar] [CrossRef] [PubMed]
- Souza-Neto, F.V.; Jiménez-González, S.; Delgado-Valero, B.; Jurado-López, R.; Genty, M.; Romero-Miranda, A.; Rodríguez, C.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants 2021, 10, 1274. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; He, C.; Carter, A.B. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020, 33, 101426. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. CCN2 induces cellular senescence in fibroblasts. J. Cell Commun. Signal. 2017, 11, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.H.; Deng, Y.T.; Hsieh, Y.P.; Wu, K.J.; Kuo, M.Y.P. NADPH Oxidase 4 Mediates TGFβ1-induced CCN2 in Gingival Fibroblasts. J. Dent. Res. 2015, 94, 976–982. [Google Scholar] [CrossRef]
- Kim, K.-H.; Park, G.-T.; Lim, Y.-B.; Rue, S.-W.; Jung, J.-C.; Sonn, J.-K.; Bae, Y.-S.; Park, J.-W.; Lee, Y.-S. Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor-β-mediated signaling pathway. Biochem. Biophys. Res. Commun. 2004, 318, 819–825. [Google Scholar] [CrossRef]
- Blokland, K.E.C.; Habibie, H.; Borghuis, T.; Teitsma, G.J.; Schuliga, M.; Melgert, B.N.; Knight, D.A.; Brandsma, C.-A.; Pouwels, S.D.; Burgess, J.K. Regulation of Cellular Senescence Is Independent from Profibrotic Fibroblast-Deposited ECM. Cells 2021, 10, 1628. [Google Scholar] [CrossRef]
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284. [Google Scholar] [CrossRef] [Green Version]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef]
- Xie, N.; Cui, H.; Ge, J.; Banerjee, S.; Guo, S.; Dubey, S.; Abraham, E.; Liu, R.M.; Liu, G. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L834–L844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Dennery, P.A.; Yao, H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including BPD, COPD, and pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L544–L554. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Lee, S.B.; Lee, J.M.; Kim, H.M.; Hong, J.Y.; Lee, W.J.; Choi, C.W.; Shin, H.K.; Kim, D.J.; Koh, E.S.; et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J. Proteome Res. 2016, 15, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Yin, L.; Archer, S.; Lu, C.; Zhao, G.; Yao, Y.; Wu, L.; Hsin, M.; Waddell, T.K.; Keshavjee, S.; et al. Metabolic heterogeneity of idiopathic pulmonary fibrosis: A metabolomic study. BMJ Open Respir. Res. 2017, 4, e000183. [Google Scholar] [CrossRef] [Green Version]
- Summer, R.; Mora, A.L. Lipid metabolism: A new player in the conundrum of lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 61, 669–670. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Mutlu, G.M. Metabolic requirements of pulmonary fibrosis: Role of fibroblast metabolism. FEBS J. 2021, 288, 6331–6352. [Google Scholar] [CrossRef]
- Ogger, P.P.; Byrne, A.J. Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol. 2021, 14, 282–295. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M. Aerobic Glycolysis and the Warburg Effect. An Unexplored Realm in the Search for Fibrosis Therapies? Am. J. Respir. Crit. Care Med. 2015, 192, 1407–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, L.; Ruppert, C.; Ochs, M. Tissue remodelling in pulmonary fibrosis. Cell Tissue Res. 2017, 367, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Acuña, M.J.; Cabrera, D.; Goldschmeding, R.; Brandan, E. The pro-fibrotic connective tissue growth factor (CTGF/CCN2) correlates with the number of necrotic-regenerative foci in dystrophic muscle. J. Cell Commun. Signal. 2018, 12, 413–421. [Google Scholar] [CrossRef] [Green Version]
- El-Chemaly, S.; Malide, D.; Yao, J.; Nathan, S.D.; Rosas, I.O.; Gahl, W.A.; Moss, J.; Gochuico, B.R. Glucose transporter-1 distribution in fibrotic lung disease: Association with [18F]-2-fluoro-2-deoxyglucose-PET scan uptake, inflammation, and neovascularization. Chest 2013, 143, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Moon, J.S.; Lee, C.M.; Choi, A.M.K.; Stout-Delgado, H.W. Glucose transporter 1-dependent glycolysis is increased during aging-related lung fibrosis, and phloretin inhibits lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 521–531. [Google Scholar] [CrossRef]
- Heilig, C.W.; Deb, D.K.; Abdul, A.; Riaz, H.; James, L.R.; Salameh, J.; Nahman, N.S. GLUT1 regulation of the pro-sclerotic mediators of diabetic nephropathy. Am. J. Nephrol. 2013, 38, 39–49. [Google Scholar] [CrossRef]
- Kim, H.; Son, S.; Ko, Y.; Shin, I. CTGF regulates cell proliferation, migration, and glucose metabolism through activation of FAK signaling in triple-negative breast cancer. Oncogene 2021, 40, 2667–2681. [Google Scholar] [CrossRef]
- James, L.R.; Le, C.; Doherty, H.; Kim, H.; Maeda, N. Connective Tissue Growth Factor (CTGF) Expression Modulates Response to High Glucose. PLoS ONE 2013, 8, e70441. [Google Scholar] [CrossRef]
- Ha, Y.M.; Lee, D.H.; Kim, M.; Kang, Y.J. High glucose induces connective tissue growth factor expression and extracellular matrix accumulation in rat aorta vascular smooth muscle cells via extracellular signal-regulated kinase 1/2. Korean J. Physiol. Pharmacol. 2013, 17, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Shibusawa, Y.; Negishi, I.; Tabata, Y.; Ishikawa, O. Mouse model of dermal fibrosis induced by one-time injection of bleomycin-poly(L-lactic acid) microspheres. Rheumatology 2008, 47, 454–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schruf, E.; Schroeder, V.; Kuttruff, C.A.; Weigle, S.; Krell, M.; Benz, M.; Bretschneider, T.; Holweg, A.; Schuler, M.; Frick, M.; et al. Human lung fibroblast-to-myofibroblast transformation is not driven by an LDH5-dependent metabolic shift towards aerobic glycolysis. Respir. Res. 2019, 20, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, D.K.; Chen, Y.; Sun, J.; Wang, Y.; Li, Y.C. ATP-citrate lyase is essential for high glucose-induced histone hyperacetylation and fibrogenic gene upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 2017, 313, F423–F429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Tu, Z.; Shi, Y.; Zhang, L.; Li, Q.; Wang, W.; Ye, F.; Wang, J.; Bu, H. Interference of Cyclosporine on Glucose Metabolism: Potential Role in Chronic Transplantation Kidney Fibrosis. Transplant. Proc. 2006, 38, 2065–2068. [Google Scholar] [CrossRef]
- Yan, F.; Wen, Z.; Wang, R.; Luo, W.; Du, Y.; Wang, W.; Chen, X. Identification of the lipid biomarkers from plasma in idiopathic pulmonary fibrosis by Lipidomics. BMC Pulm. Med. 2017, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; Dubois, G.; Pyne, S. Role of sphingosine 1-phosphate and lysophosphatidic acid in fibrosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 228–238. [Google Scholar] [CrossRef]
- Shea, B.S.; Tager, A.M. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, M.; Nakerakanti, S.S.; Kapanadze, B.; Ghatnekar, A.; Trojanowska, M. Connective tissue growth factor (CTGF/CCN2) mediates angiogenic effect of S1P in human dermal microvascular endothelial cells. Microcirculation 2011, 18, 1–11. [Google Scholar] [CrossRef]
- Eckes, T.; Patyna, S.; Koch, A.; Oftring, A.; Gauer, S.; Obermüller, N.; Schwalm, S.; Schaefer, L.; Chun, J.; Gröne, H.-J.; et al. Sphingosine 1-Phosphate Receptor 5 (S1P5) Knockout Ameliorates Adenine-Induced Nephropathy. Int. J. Mol. Sci. 2022, 23, 3952. [Google Scholar] [CrossRef]
- Murakami, K.; Kohno, M.; Kadoya, M.; Nagahara, H.; Fujii, W.; Seno, T.; Yamamoto, A.; Oda, R.; Fujiwara, H.; Kubo, T.; et al. Knock out of S1P3 receptor signaling attenuates inflammation and fibrosis in bleomycin-induced lung Injury Mice Model. PLoS ONE 2014, 9, e106792. [Google Scholar]
- Sobel, K.; Menyhart, K.; Killer, N.; Renault, B.; Bauer, Y.; Studer, R.; Steiner, B.; Bolli, M.H.; Nayler, O.; Gatfield, J. Sphingosine 1-Phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2and S1P 3 receptors and smad-independent signaling. J. Biol. Chem. 2013, 288, 14839–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.-C.; Wang, E.Y.; Yi, Y.; Thakur, A.; Tsai, S.-H.; Hoodless, P.A. S1P Stimulates Proliferation by Upregulating CTGF Expression through S1PR2-Mediated YAP Activation. Mol. Cancer Res. 2018, 16, 1543–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wünsche, C.; Koch, A.; Goldschmeding, R.; Schwalm, S.; Meyer Zu Heringdorf, D.; Huwiler, A.; Pfeilschifter, J. Transforming growth factor β2 (TGF-β2)-induced connective tissue growth factor (CTGF) expression requires sphingosine 1-phosphate receptor 5 (S1P5) in human mesangial cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; He, X.; Zeng, M. The role of S1P and the related signaling pathway in the development of tissue fibrosis. Front. Pharmacol. 2019, 9, 1504. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine kinase 1/s1p signaling contributes to pulmonary fibrosis by activating hippo/yap pathway and mitochondrial reactive oxygen species in lung fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Chun, J.; Duffield, J.S.; Wada, T.; Luster, A.D.; Tager, A.M. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013, 27, 1830–1846. [Google Scholar] [CrossRef] [Green Version]
- Riquelme-Guzmán, C.; Contreras, O.; Brandan, E. Expression of CTGF/CCN2 in response to LPA is stimulated by fibrotic extracellular matrix via the integrin/FAK axis. Am. J. Physiol. Physiol. 2018, 314, C415–C427. [Google Scholar] [CrossRef]
- Yu, Z.-L.; Li, D.-Q.; Huang, X.-Y.; Xing, X.; Yu, R.-Q.; Li, Z.; Li, Z.-B. Lysophosphatidic acid upregulates connective tissue growth factor expression in osteoblasts through the GPCR/PKC and PKA pathways. Int. J. Mol. Med. 2016, 37, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Liu, Y.; Dai, H.; Wang, C. Fatty Acid Metabolism and Idiopathic Pulmonary Fibrosis. Front. Physiol. 2022, 12, 794629. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, K.M.; Song, H.E.; Choi, K.H.; Hwang, J.J.; Yoo, H.J.; Song, J.W. The role of free fatty acids in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 50, OA4635. [Google Scholar]
- Deng, Y.; Matsui, Y.; Pan, W.; Li, Q.; Lai, Z.-C. Yap1 plays a protective role in suppressing free fatty acid-induced apoptosis and promoting beta-cell survival. Protein Cell 2016, 7, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; McLennan, S.V.; Allen, T.J.; Tsoutsman, T.; Semsarian, C.; Twigg, S.M. Adverse effects of high glucose and free fatty acid on cardiomyocytes are mediated by connective tissue growth factor. Am. J. Physiol. Cell Physiol. 2009, 297, C1490–C1500. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.J.; Kloppenburg, M.; Toes, R.E.M.; Ioan-Facsinay, A. Fatty acids, lipid mediators, and T-cell function. Front. Immunol. 2014, 5, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid mediators regulate pulmonary fibrosis: Potential mechanisms and signaling pathways. Int. J. Mol. Sci. 2020, 21, 4257. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Gillissen, A.; Buhl, R.; Hoyt, R.F.; Hubbard, R.C.; Ozaki, T.; Rennard, S.I.; Crystal, R.G. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am. Rev. Respir. Dis. 1991, 144, 1080–1084. [Google Scholar] [CrossRef]
- Black, S.A.J.; Palamakumbura, A.H.; Stan, M.; Trackman, P.C. Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: Interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase. J. Biol. Chem. 2007, 282, 15416–15429. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Sheng, W.; Michkov, A.; Sriarm, K.; Sun, R.; Dvorkin-Gheva, A.; Insel, P.A.; Janssen, L.J. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca2+ signaling. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L810–L821. [Google Scholar] [CrossRef]
- Aihara, K.; Handa, T.; Oga, T.; Watanabe, K.; Tanizawa, K.; Ikezoe, K.; Taguchi, Y.; Sato, H.; Chin, K.; Nagai, S.; et al. Clinical Relevance of Plasma Prostaglandin F2α Metabolite Concentrations in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2013, 8, e66017. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, T.D.; Smith, E.R. A Metabolic Reprogramming of Glycolysis and Glutamine Metabolism Is a Requisite for Renal Fibrogenesis—Why and How? Front. Physiol. 2021, 12, 645857. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; O’Leary, E.M.; Witt, L.J.; Tian, Y.; Gökalp, G.A.; Meliton, A.Y.; Dulin, N.O.; Mutlu, G.M. Glutamine metabolism is required for collagen protein synthesis in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 597–606. [Google Scholar] [CrossRef]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.; Lee, S.; Caesar, J.A.; Pruchenko, S.; Leask, A.; Knowles, J.A.; Sinon, J.; Chaqour, B. A CTGF-YAP Regulatory Pathway Is Essential for Angiogenesis and Barriergenesis in the Retina. iScience 2020, 23, 101184. [Google Scholar] [CrossRef] [PubMed]
- Dillinger, A.E.; Kuespert, S.; Froemel, F.; Tamm, E.R.; Fuchshofer, R. CCN2/CTGF promotor activity in the developing and adult mouse eye. Cell Tissue Res. 2021, 384, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.A.; Coelho Aguiar, J.M.; Kahn, S.A.; Reis, A.H.; Dubois, L.G.; Romão, L.F.; Ferreira, L.S.S.; Chneiweiss, H.; Moura Neto, V.; Abreu, J.G. Connective-Tissue Growth Factor (CTGF/CCN2) Induces Astrogenesis and Fibronectin Expression of Embryonic Neural Cells In Vitro. PLoS ONE 2015, 10, e0133689. [Google Scholar] [CrossRef]
- Weiss, M.; Reinehr, S.; Mueller-Buehl, A.M.; Doerner, J.D.; Fuchshofer, R.; Stute, G.; Dick, H.B.; Joachim, S.C. Activation of Apoptosis in a βB1-CTGF Transgenic Mouse Model. Int. J. Mol. Sci. 2021, 22, 1997. [Google Scholar] [CrossRef]
- Bai, L.; Bernard, K.; Tang, X.; Hu, M.; Horowitz, J.C.; Thannickal, V.J.; Sanders, Y.Y. Glutaminolysis epigenetically regulates antiapoptotic gene expression in idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 60, 49–57. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Blokland, K.E.C.; Pouwels, S.D.; Schuliga, M.; Knight, D.A.; Burgess, J.K. Regulation of cellular senescence by extracellular matrix during chronic fibrotic diseases. Clin. Sci. 2020, 134, 2681–2706. [Google Scholar] [CrossRef]
- Jang, J.-H.; Chand, H.S.; Bruse, S.; Doyle-Eisele, M.; Royer, C.; McDonald, J.; Qualls, C.; Klingelhutz, A.J.; Lin, Y.; Mallampalli, R.; et al. Connective Tissue Growth Factor Promotes Pulmonary Epithelial Cell Senescence and Is Associated with COPD Severity. COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 228–237. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Duan, Y.; Li, Y.; Sun, Y.; Sun, H.; Yu, X.; Gao, X.; Zhang, C.; Zhang, H.; et al. Metabolic Regulation: A Potential Strategy for Rescuing Stem Cell Senescence. Stem Cell Rev. Rep. 2022; online ahead of print. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Effendi, W.I.; Nagano, T. Connective Tissue Growth Factor in Idiopathic Pulmonary Fibrosis: Breaking the Bridge. Int. J. Mol. Sci. 2022, 23, 6064. https://doi.org/10.3390/ijms23116064
Effendi WI, Nagano T. Connective Tissue Growth Factor in Idiopathic Pulmonary Fibrosis: Breaking the Bridge. International Journal of Molecular Sciences. 2022; 23(11):6064. https://doi.org/10.3390/ijms23116064
Chicago/Turabian StyleEffendi, Wiwin Is, and Tatsuya Nagano. 2022. "Connective Tissue Growth Factor in Idiopathic Pulmonary Fibrosis: Breaking the Bridge" International Journal of Molecular Sciences 23, no. 11: 6064. https://doi.org/10.3390/ijms23116064
APA StyleEffendi, W. I., & Nagano, T. (2022). Connective Tissue Growth Factor in Idiopathic Pulmonary Fibrosis: Breaking the Bridge. International Journal of Molecular Sciences, 23(11), 6064. https://doi.org/10.3390/ijms23116064