
Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by CYS59TYR BOLA3 Mutation

 ,
,  and
and 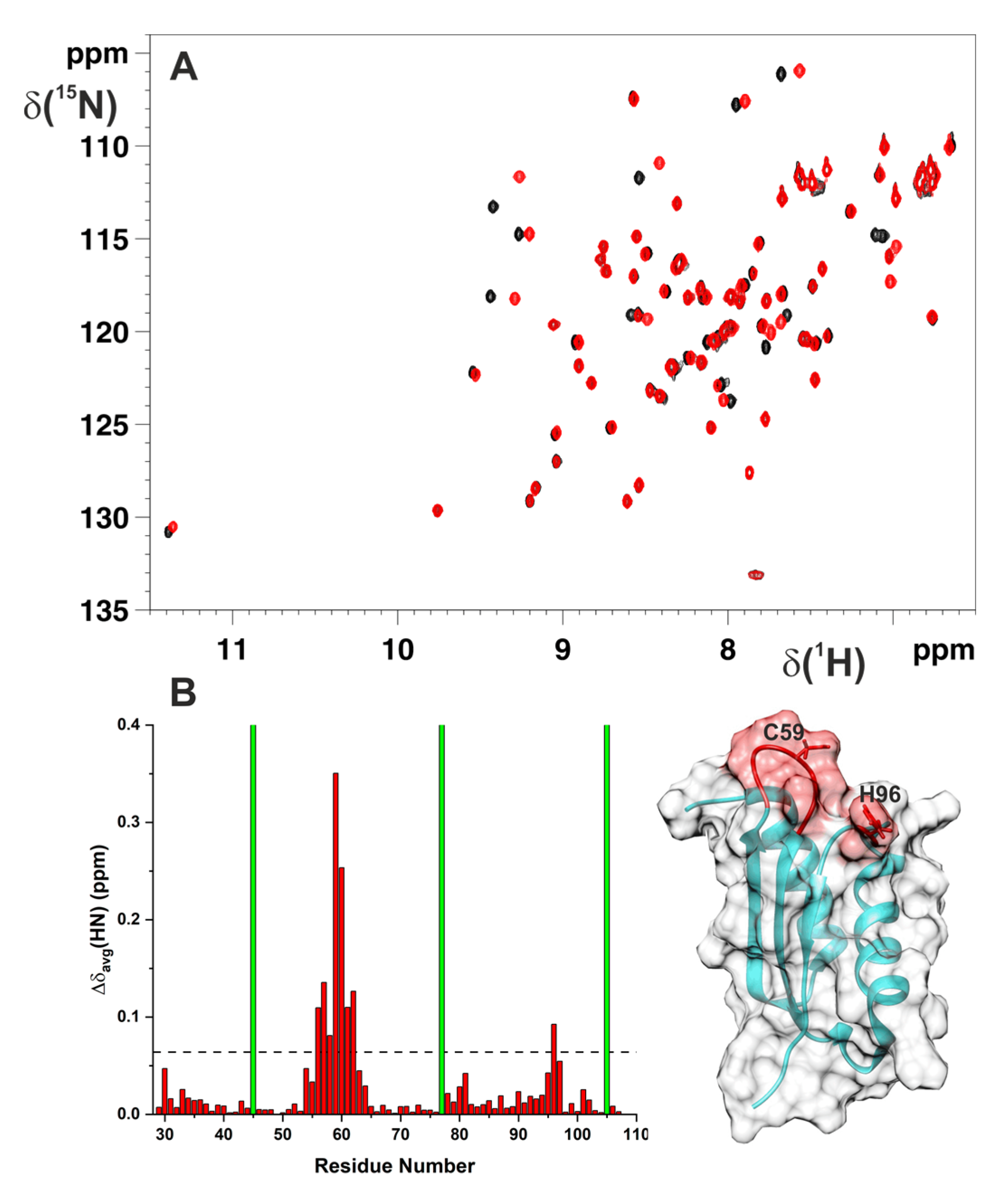{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cys59Tyr Mutation Structurally Perturbed Only the Fe–S Cluster-Binding Region of BOLA3
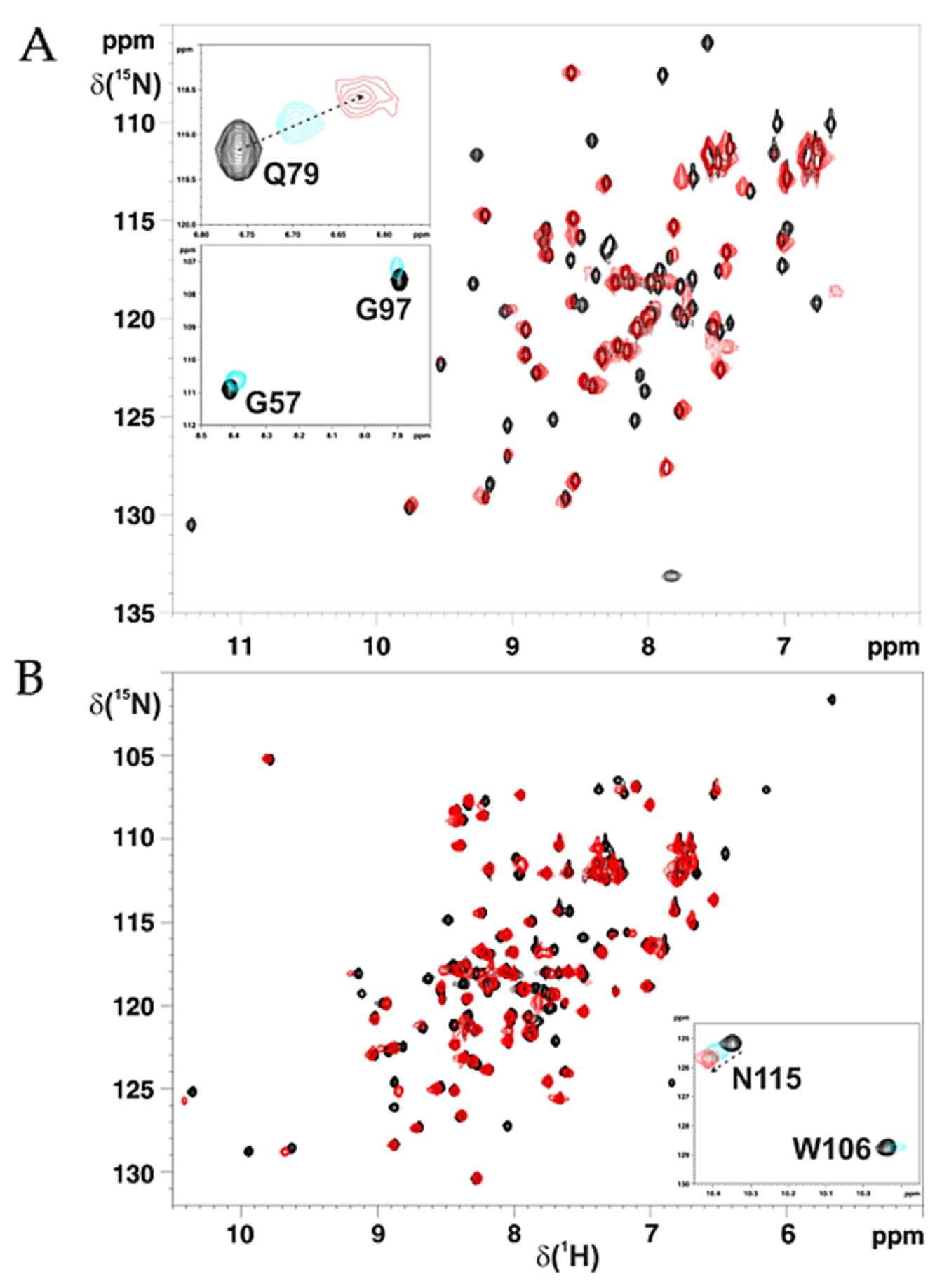
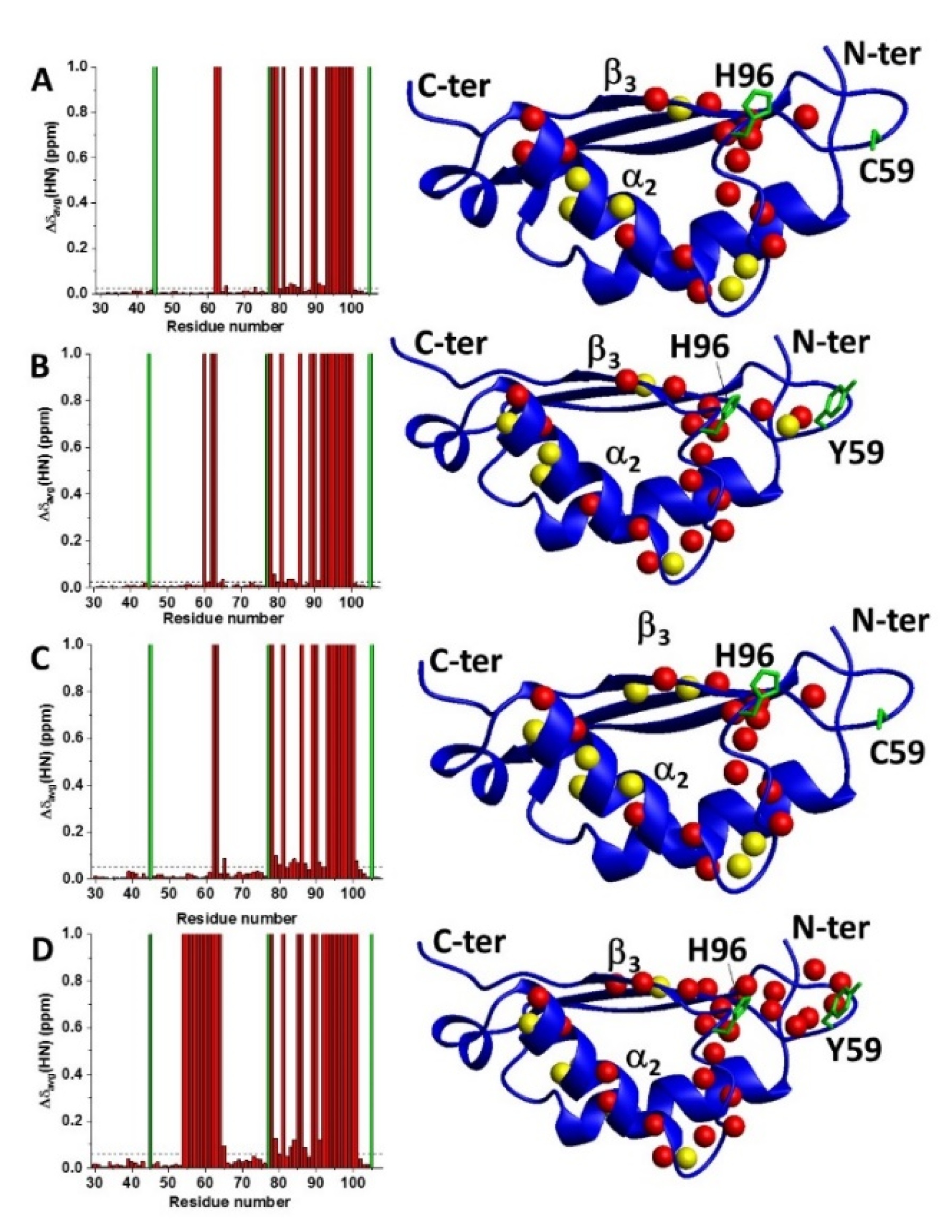
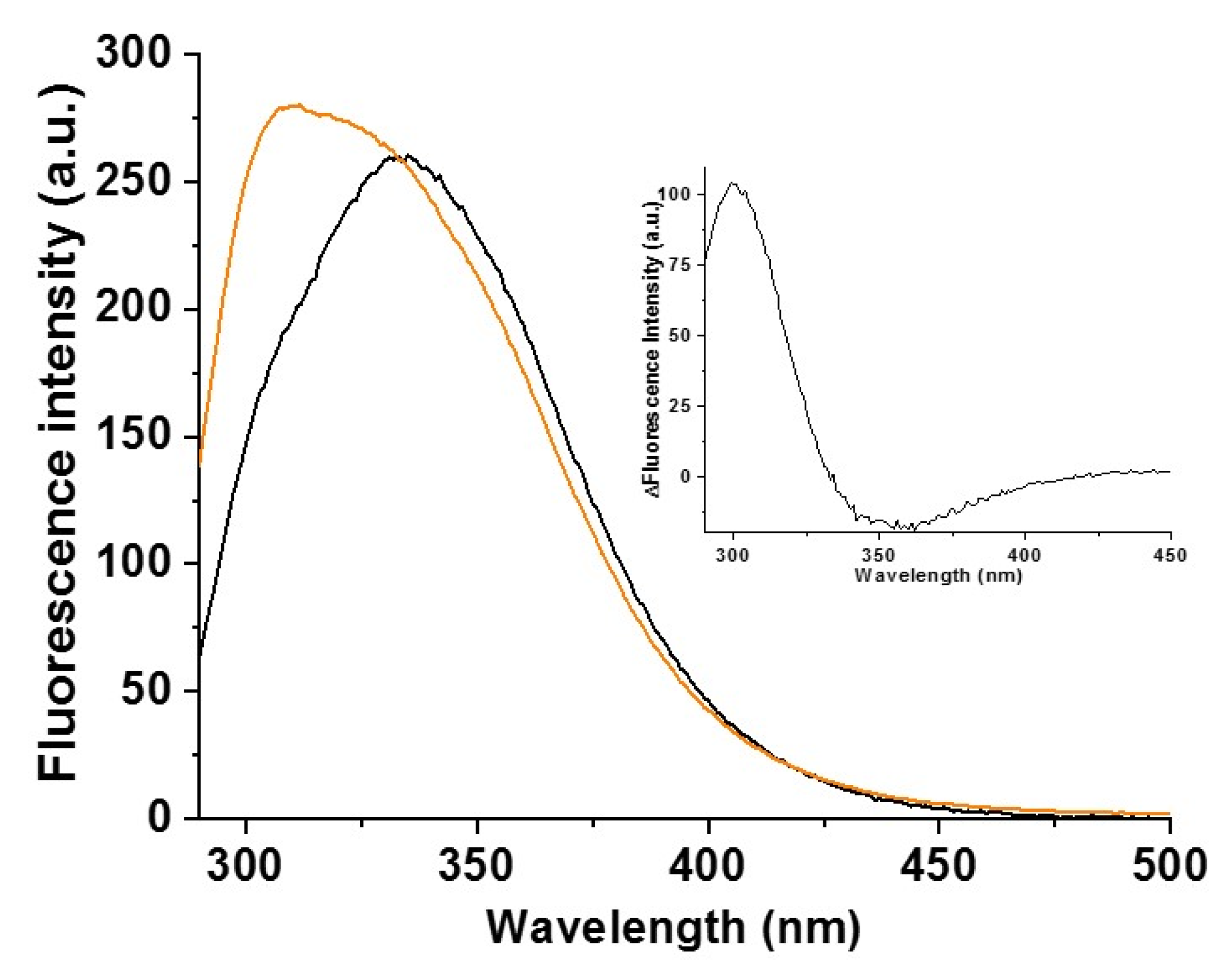
2.2. Cys59Tyr BOLA3 Mutation Modified the Interaction of BOLA3 with Apo GLRX5
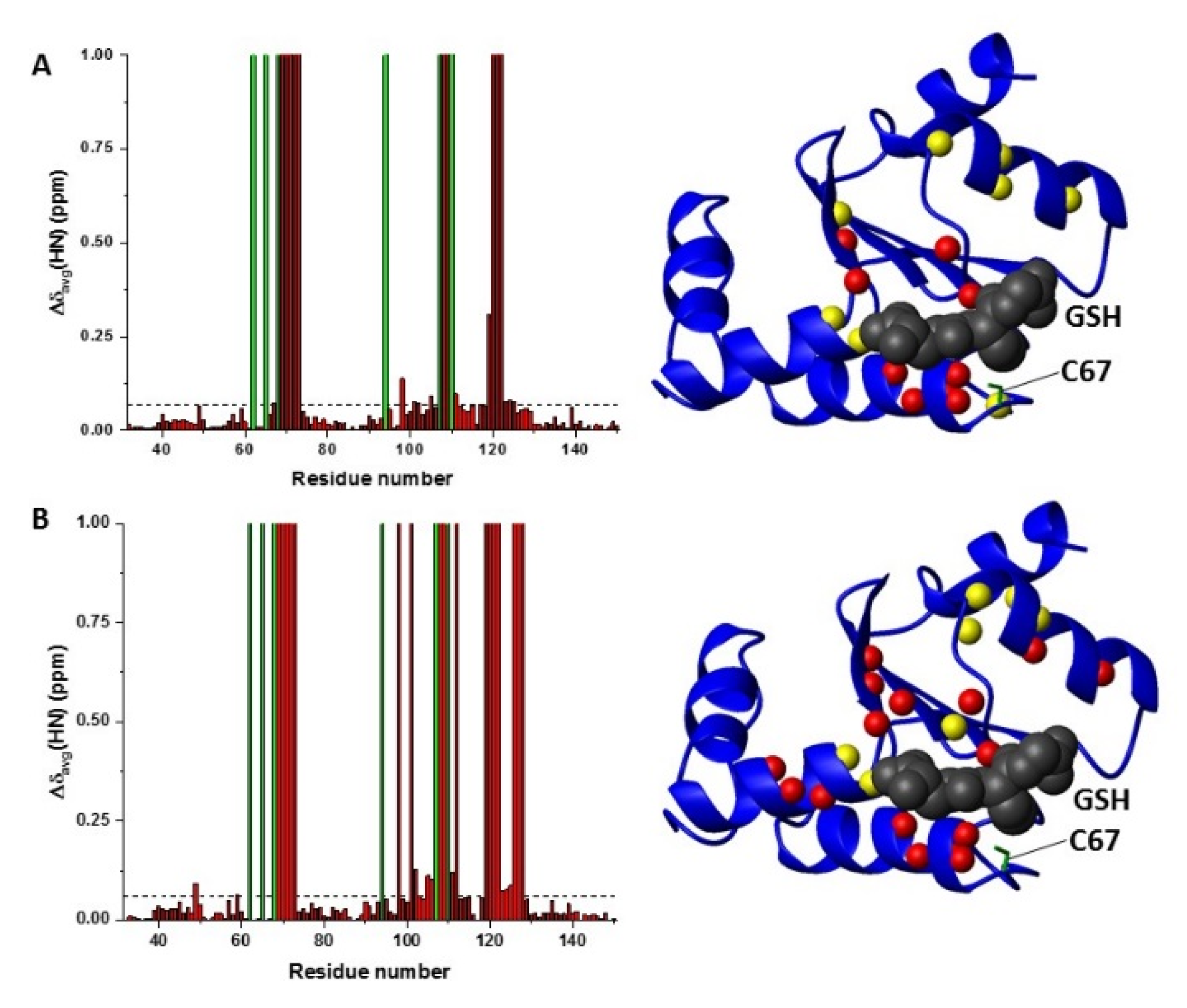
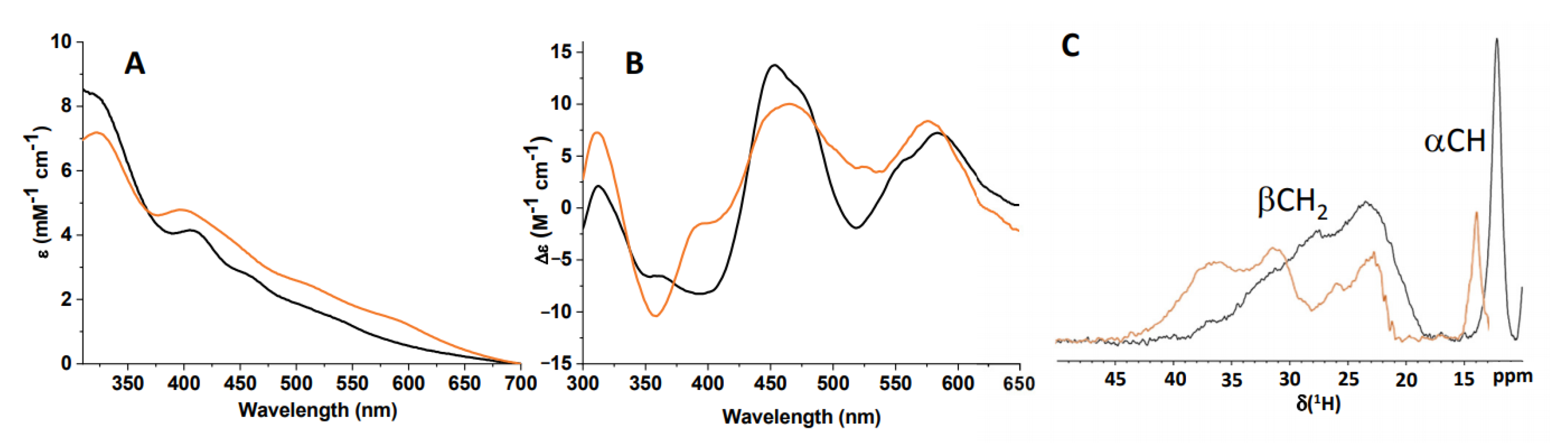
2.3. C59Y BOLA3–GLRX5 Complex Bound a [2Fe–2S]2+ Cluster
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. In Vitro Chemical Reconstitution of the Hetero-Complexes
4.3. NMR Spectroscopy to Characterize Proteins and Hetero-Complexes in Their Apo Forms
4.4. Spectroscopic Methods to Characterize Fe–S Cluster-Binding and Transfer
4.5. Data-Driven Biomolecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seyda, A.; Newbold, R.F.; Hudson, T.J.; Verner, A.; MacKay, N.; Winter, S.; Feigenbaum, A.; Malaney, S.; Gonzalez-Halphen, D.; Cuthbert, A.P.; et al. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14–2p13. Am. J. Hum. Genet. 2001, 68, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Rouault, T.A. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem. Sci. 2020, 45, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Wachnowsky, C.; Fidai, I.; Cowan, J.A. Iron-sulfur cluster biosynthesis and trafficking—Impact on human disease conditions. Metallomics 2018, 10, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.M.; Janer, A.; Levandovskiy, V.; MacKay, N.; Rouault, T.A.; Tong, W.H.; Ogilvie, I.; Shoubridge, E.A.; Robinson, B.H. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 2011, 89, 486–495. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; Del, T.M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef]
- Baker, P.R.; Friederich, M.W.; Swanson, M.A.; Shaikh, T.; Bhattacharya, K.; Scharer, G.H.; Aicher, J.; Creadon-Swindell, G.; Geiger, E.; MacLean, K.N.; et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 2014, 137, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Ajit, B.N.; Vanlander, A.V.; Wilbrecht, C.; Van der Aa, N.; Smet, J.; De, P.B.; Vandeweyer, G.; Kooy, F.; Eyskens, F.; De, L.E.; et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum. Mol. Genet. 2013, 22, 2590–2602. [Google Scholar] [CrossRef]
- Shukla, A.; Hebbar, M.; Srivastava, A.; Kadavigere, R.; Upadhyai, P.; Kanthi, A.; Brandau, O.; Bielas, S.; Girisha, K.M. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J. Hum. Genet. 2017, 62, 723–727. [Google Scholar] [CrossRef]
- Alfadhel, M. Multiple Mitochondrial Dysfunctions Syndrome 4 Due to ISCA2 Gene Defects: A Review. Child. Neurol. Open 2019, 6. [Google Scholar] [CrossRef]
- Haack, T.B.; Rolinski, B.; Haberberger, B.; Zimmermann, F.; Schum, J.; Strecker, V.; Graf, E.; Athing, U.; Hoppen, T.; Wittig, I.; et al. Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J. Inherit. Metab. Dis. 2013, 36, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lebigot, E.; Gaignard, P.; Dorboz, I.; Slama, A.; Rio, M.; de Lonlay, P.; Heron, B.; Sabourdy, F.; Boespflug-Tanguy, O.; Cardoso, A.; et al. Impact of mutations within the [Fe-S] cluster or the lipoic acid biosynthesis pathways on mitochondrial protein expression profiles in fibroblasts from patients. Mol. Genet. Metab. 2017, 122, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Bindu, P.S.; Sonam, K.; Chiplunkar, S.; Govindaraj, P.; Nagappa, M.; Vekhande, C.C.; Aravinda, H.R.; Ponmalar, J.J.; Mahadevan, A.; Gayathri, N.; et al. Mitochondrial leukoencephalopathies: A border zone between acquired and inherited white matter disorders in children? Mult. Scler. Relat. Disord. 2018, 20, 84–92. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- Nikam, R.M.; Gripp, K.W.; Choudhary, A.K.; Kandula, V. Imaging phenotype of multiple mitochondrial dysfunction syndrome 2, a rare BOLA3-associated leukodystrophy. Am. J. Med. Genet. A 2018, 176, 2787–2790. [Google Scholar] [CrossRef]
- Khoza, S.; Ngqaneka, T.; Magwebu, Z.E.; Chauke, C.G. Nonketotic hyperglycinemia in captive-bred Vervet monkeys (Chlorocebus aethiops) with cataracts. J. Med. Primatol. 2019, 48, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Meldau, S.; Fratter, C.; Bhengu, L.N.; Sergeant, K.; Khan, K.; Riordan, G.T.; Berman, P.A.M. Pitfalls of relying on genetic testing only to diagnose inherited metabolic disorders in non-western populations—5 cases of pyruvate dehydrogenase deficiency from South Africa. Mol. Genet. Metab. Rep. 2020, 24, 100629. [Google Scholar] [CrossRef]
- Stutterd, C.A.; Lake, N.J.; Peters, H.; Lockhart, P.J.; Taft, R.J.; van der Knaap, M.S.; Vanderver, A.; Thorburn, D.R.; Simons, C.; Leventer, R.J. Severe Leukoencephalopathy with Clinical Recovery Caused by Recessive BOLA3 Mutations. JIMD Rep. 2019, 43, 63–70. [Google Scholar]
- Nishioka, M.; Inaba, Y.; Motobayashi, M.; Hara, Y.; Numata, R.; Amano, Y.; Shingu, K.; Yamamoto, Y.; Murayama, K.; Ohtake, A.; et al. An infant case of diffuse cerebrospinal lesions and cardiomyopathy caused by a BOLA3 mutation. Brain Dev. 2018, 40, 484–488. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Meldau, S.; Owen, E.P.; Khan, K.; Riordan, G.T. Mitochondrial molecular genetic results in a South African cohort: Divergent mitochondrial and nuclear DNA findings. J. Clin. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nasta, V.; Giachetti, A.; Ciofi-Baffoni, S.; Banci, L. Structural insights into the molecular function of human (2Fe-2S) BOLA1-GRX5 and (2Fe-2S) BOLA3-GRX5 complexes. Biochim. Biophys. Acta 2017, 1861, 2119–2131. [Google Scholar] [CrossRef]
- Uzarska, M.A.; Nasta, V.; Weiler, B.D.; Spantgar, F.; Ciofi-Baffoni, S.; Saviello, M.; Gonnelli, L.; Muhlenhoff, U.; Banci, L.; Lill, R. Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron-sulfur proteins. Elife 2016, 5, e16673. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Rao, B.; Wachnowsky, C.; Cowan, J.A. Cluster exchange reactivity of [2Fe-2S] cluster-bridged complexes of BOLA3 with monothiol glutaredoxins. Metallomics 2018, 10, 1282–1290. [Google Scholar] [CrossRef]
- Jia, M.; Sen, S.; Wachnowsky, C.; Fidai, I.; Cowan, J.A.; Wysocki, V. Characterization of [2Fe-2S]-Cluster-Bridged Protein Complexes and Reaction Intermediates by use of Robust Native Mass Spectrometric Methods. Angew. Chem. Int. Ed. (Engl.) 2020, 59, 6724–6728. [Google Scholar] [CrossRef]
- Nasta, V.; Suraci, D.; Gourdoupis, S.; Ciofi-Baffoni, S.; Banci, L. A pathway for assembling [4Fe-4S]2+ clusters in mitochondrial iron-sulfur protein biogenesis. FEBS J. 2020, 287, 2312–2327. [Google Scholar] [CrossRef]
- Tong, W.H.; Jameson, G.N.; Huynh, B.H.; Rouault, T.A. Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc. Natl. Acad. Sci. USA 2003, 100, 9762–9767. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Na, U.; Vashisht, A.; Weiler, B.D.; Lill, R.; Wohlschlegel, J.A.; Winge, D.R. Role of Nfu1 and Bol3 in iron-sulfur cluster transfer to mitochondrial clients. Elife 2016, 5, e15991. [Google Scholar] [CrossRef]
- Suraci, D.; Saudino, G.; Nasta, V.; Ciofi-Baffoni, S.; Banci, L. ISCA1 orchestrates ISCA2 and NFU1 in the maturation of human mitochondrial [4Fe-4S] proteins. J. Mol. Biol. 2021, 433, 166924. [Google Scholar] [CrossRef] [PubMed]
- Ahting, U.; Mayr, J.A.; Vanlander, A.V.; Hardy, S.A.; Santra, S.; Makowski, C.; Alston, C.L.; Zimmermann, F.A.; Abela, L.; Plecko, B.; et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front. Genet. 2015, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Tonduti, D.; Dorboz, I.; Imbard, A.; Slama, A.; Boutron, A.; Pichard, S.; Elmaleh, M.; Vallee, L.; Benoist, J.F.; Ogier, H.; et al. New spastic paraplegia phenotype associated to mutation of NFU1. Orphanet J. Rare Dis 2015, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, F.; Ardissone, A.; Lamantea, E.; Garavaglia, B.; Zeviani, M.; Farina, L.; Ghezzi, D.; Moroni, I. Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Front. Genet. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.S.; Bonvin, A.M. Clustering biomolecular complexes by residue contacts similarity. Proteins 2012, 80, 1810–1817. [Google Scholar] [CrossRef]
- Noodleman, L.; Baerens, E.J. Electronic structure, magnetic properties, ESR and optical spectra for 2-Fe ferredoxin models by LCAO-Xa valence bond theory. J. Am. Chem. Soc. 1984, 106, 2316–2327. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef]
- Freibert, S.A.; Weiler, B.D.; Bill, E.; Pierik, A.J.; Muhlenhoff, U.; Lill, R. Biochemical Reconstitution and Spectroscopic Analysis of Iron-Sulfur Proteins. Methods Enzym. 2018, 599, 197–226. [Google Scholar]
- Mapolelo, D.T.; Zhang, B.; Naik, S.G.; Huynh, B.H.; Johnson, M.K. Spectroscopic and functional characterization of iron-sulfur cluster-bound forms of Azotobacter vinelandii (Nif)IscA. Biochemistry 2012, 51, 8071–8084. [Google Scholar] [CrossRef]
- Banci, L.; Camponeschi, F.; Ciofi-Baffoni, S.; Piccioli, M. The NMR contribution to protein-protein networking in Fe-S protein maturation. J. Biol. Inorg. Chem. 2018, 23, 687. [Google Scholar] [CrossRef]
- Xia, B.; Jenk, D.; LeMaster, D.M.; Westler, W.M.; Markley, J.L. Electron-nuclear interactions in two prototypical [2Fe-2S] proteins: Selective (chiral) deuteration and analysis of 1H and 2H NMR signals from the alpha and beta hydrogens of cysteinyl residues that ligate the iron in the active sites of human ferredoxin and Anabaena 7120 vegetative ferredoxin. Arch. Biochem. Biophys. 2000, 373, 328–334. [Google Scholar]
- Banci, L.; Bertini, I.; Luchinat, C. The 1H NMR parameters of magnetically coupled dimers—The Fe2S2 proteins as an example. Bioinorg. Chem. 1990, 72, 113–136. [Google Scholar]
- Cleland, W.E.J.; Averill, B.A. Effects of phenoxide ligation on iron-sulfur clusters. 2. Preparation and properties of [Fe2S2(OAr)4]2- ions. Inorg. Chem. 1984, 23, 4192–4197. [Google Scholar] [CrossRef]
- Fujinaga, J.; Gaillard, J.; Meyer, J. Mutated forms of a [2Fe-2S] ferredoxin with serine ligands to the iron-sulfur cluster. Biochem. Biophys. Res. Commun. 1993, 194, 104–111. [Google Scholar] [CrossRef]
- Nicolet, Y.; Rohac, R.; Martin, L.; Fontecilla-Camps, J.C. X-ray snapshots of possible intermediates in the time course of synthesis and degradation of protein-bound Fe4S4 clusters. Proc. Natl. Acad. Sci. USA 2013, 110, 7188–7192. [Google Scholar] [CrossRef]
- Owens, C.P.; Katz, F.E.; Carter, C.H.; Oswald, V.F.; Tezcan, F.A. Tyrosine-Coordinated P-Cluster in G. diazotrophicus Nitrogenase: Evidence for the Importance of O-Based Ligands in Conformationally Gated Electron Transfer. J. Am. Chem. Soc. 2016, 138, 10124–10127. [Google Scholar] [CrossRef]
- O’Neil, J.D.; Hofmann, T. Tyrosine and tyrosinate fluorescence of pig intestinal Ca2+-binding protein. Biochem. J. 1987, 243, 611–615. [Google Scholar] [CrossRef]
- Szabo, A.G.; Lynn, K.R.; Krajcarski, D.T.; Rayner, D.M. Tyrosinate fluorescence maxima at 345 nm in proteins lacking tryptophan at pH 7. FEBS Lett. 1978, 94, 249–252. [Google Scholar] [CrossRef]
- Kierdaszuk, B. Fluorescence anisotropy of tyrosinate anion using one-, two- and three-photon excitation: Tyrosinate anion fluorescence. J. Fluoresc. 2013, 23, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Pundak, S.; Roche, R.S. Tyrosine and tyrosinate fluorescence of bovine testes calmodulin: Calcium and pH dependence. Biochemistry 1984, 23, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Cornog, J.L., Jr.; Adams, W.R. The fluorescence of tyrosine in alkaline solution. Biochim. Biophys. Acta 1963, 66, 356–365. [Google Scholar] [CrossRef]
- Teale, F.W.; Weber, G. Ultraviolet fluorescence of the aromatic amino acids. Biochem. J. 1957, 65, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Pokalsky, C.; Wick, P.; Harms, E.; Lytle, F.E.; Van Etten, R.L. Fluorescence resolution of the intrinsic tryptophan residues of bovine protein tyrosyl phosphatase. J. Biol. Chem. 1995, 270, 3809–3815. [Google Scholar] [CrossRef] [PubMed]
- Ghisaidoobe, A.B.; Chung, S.J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Förster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of frequency-domain fluorescence spectroscopy and applications to cell membranes. Sub-Cell. Biochem. 1988, 13, 89–126. [Google Scholar]
- Reibarkh, M.; Malia, T.J.; Wagner, G. NMR distinction of single- and multiple-mode binding of small-molecule protein ligands. J. Am. Chem. Soc. 2006, 128, 2160–2161. [Google Scholar] [CrossRef] [PubMed]
- Schilder, J.; Ubbink, M. Formation of transient protein complexes. Curr. Opin. Struct. Biol. 2013, 23, 911–918. [Google Scholar] [CrossRef]
- Jain, A.; Singh, A.; Maio, N.; Rouault, T.A. Assembly of the [4Fe-4S] cluster of NFU1 requires the coordinated donation of two [2Fe-2S] clusters from the scaffold proteins, ISCU2 and ISCA1. Hum. Mol. Genet. 2020, 29, 3165–3182. [Google Scholar] [CrossRef]
- Banci, L.; Brancaccio, D.; Ciofi-Baffoni, S.; Del Conte, R.; Gadepalli, R.; Mikolajczyk, M.; Neri, S.; Piccioli, M.; Winkelmann, J. [2Fe-2S] cluster transfer in iron-sulfur protein biogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 6203–6208. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Camponeschi, F.; Ciofi-Baffoni, S.; Muzzioli, R. Elucidating the molecular function of human BOLA2 in GRX3-Dependent anamorsin maturation pathway. J. Am. Chem. Soc. 2015, 137, 16133–16134. [Google Scholar] [CrossRef]
- Gao, G.; Williams, J.G.; Campbell, S.L. Protein-protein interaction analysis by nuclear magnetic resonance spectroscopy. Methods Mol. Biol. 2004, 261, 79–92. [Google Scholar] [PubMed]
- Inubushi, T.; Becker, E.D. Efficient detection of paramagnetically shifted NMR resonances by optimizing the WEFT pulse sequence. J. Magn. Reson. 1983, 51, 128–133. [Google Scholar] [CrossRef]
- van Zundert, G.C.; Rodrigues, J.P.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Roos, A.K.; Montano, S.J.; Sengupta, R.; Filippakopoulos, P.; Guo, K.; von Delft, F.; Holmgren, A.; Oppermann, U.; Kavanagh, K.L. The crystal structure of human GLRX5: Iron-sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochem. J. 2011, 433, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12, 12th ed.; University of California: San Francisco, CA, USA, 2012. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saudino, G.; Suraci, D.; Nasta, V.; Ciofi-Baffoni, S.; Banci, L. Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by CYS59TYR BOLA3 Mutation. Int. J. Mol. Sci. 2021, 22, 4848. https://doi.org/10.3390/ijms22094848
Saudino G, Suraci D, Nasta V, Ciofi-Baffoni S, Banci L. Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by CYS59TYR BOLA3 Mutation. International Journal of Molecular Sciences. 2021; 22(9):4848. https://doi.org/10.3390/ijms22094848
Chicago/Turabian StyleSaudino, Giovanni, Dafne Suraci, Veronica Nasta, Simone Ciofi-Baffoni, and Lucia Banci. 2021. "Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by CYS59TYR BOLA3 Mutation" International Journal of Molecular Sciences 22, no. 9: 4848. https://doi.org/10.3390/ijms22094848
APA StyleSaudino, G., Suraci, D., Nasta, V., Ciofi-Baffoni, S., & Banci, L. (2021). Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by CYS59TYR BOLA3 Mutation. International Journal of Molecular Sciences, 22(9), 4848. https://doi.org/10.3390/ijms22094848