
Role of ABCA7 in Human Health and in Alzheimer’s Disease



Abstract
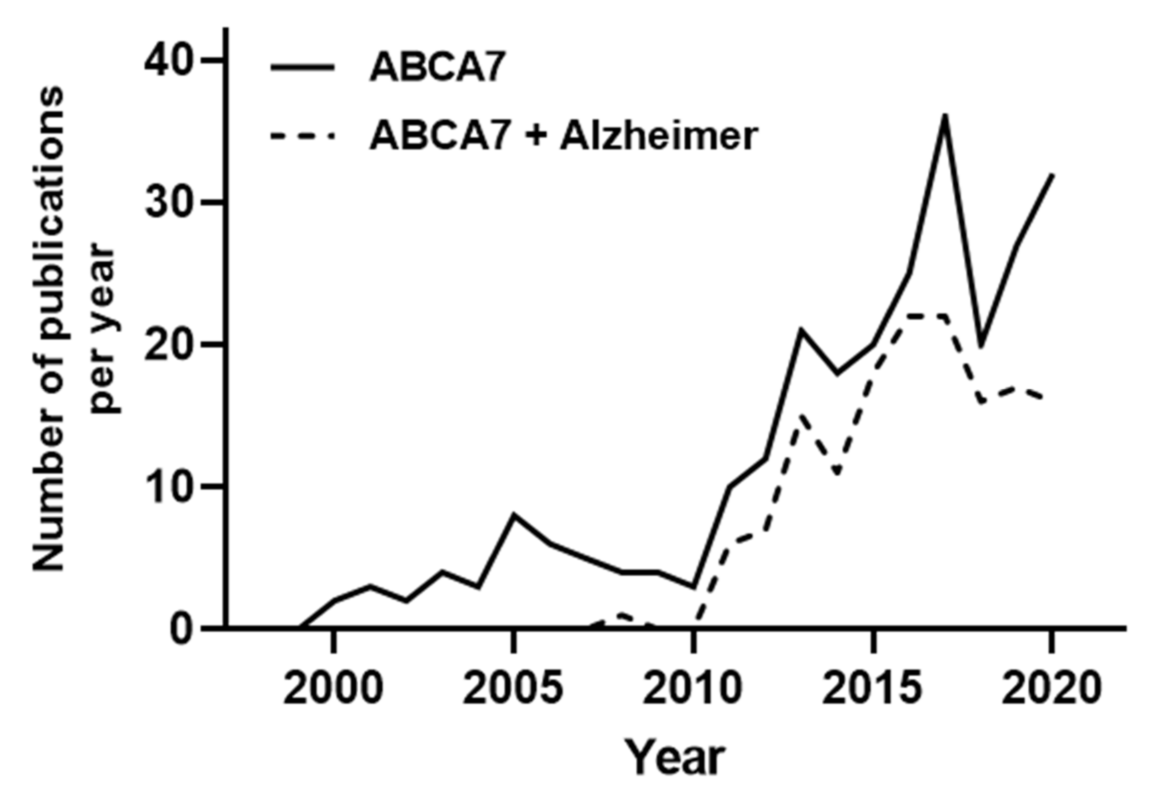
1. Introduction
2. ABCA7 Expression Pattern and Structure
2.1. Tissue Localization of ABCA7
2.2. ABCA7 Alternative Splicing
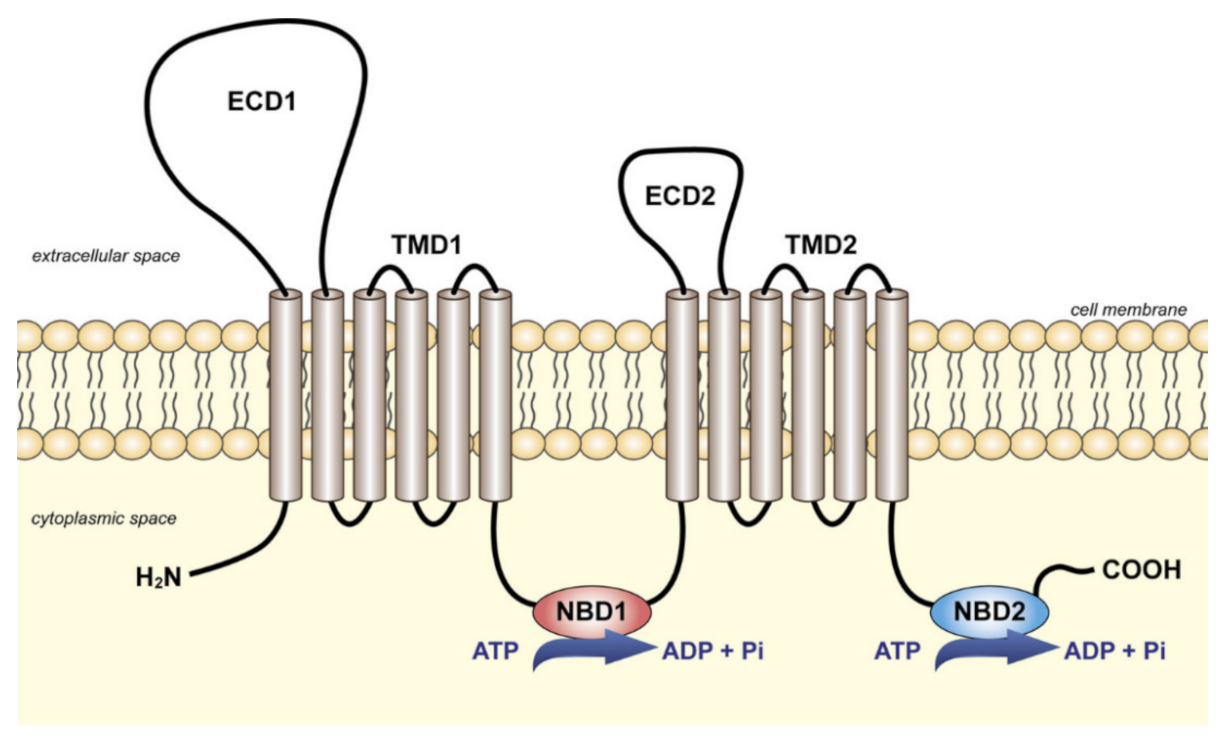
2.3. ABCA7 Structure
3. ABCA7 and the Lipid Metabolism
3.1. Role of ABCA7 in Lipid Release and Trafficking
3.2. Role of ABCA7 in Phagocytosis and Immune Response
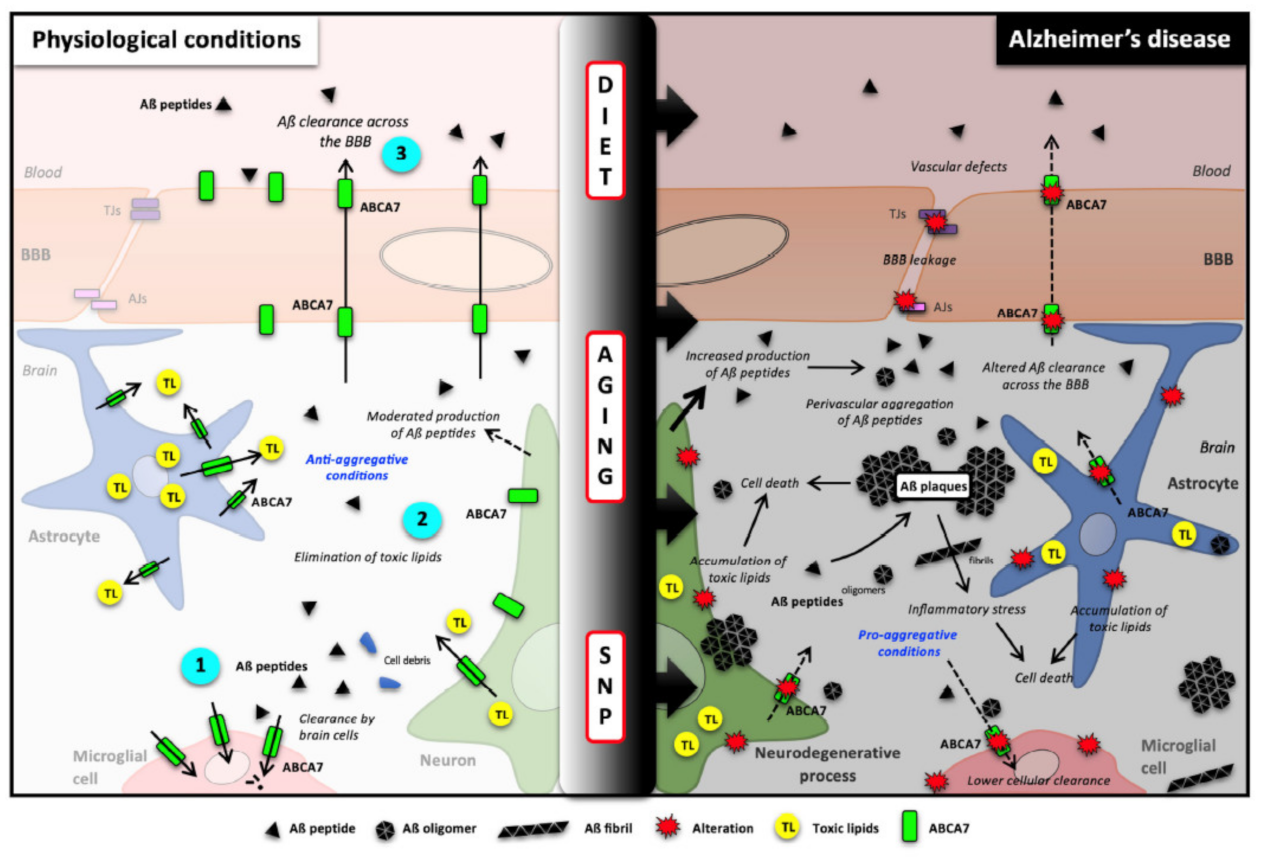
4. Roles of ABCA7 in Brain Functioning and in Alzheimer’s Disease (AD)
4.1. AD Pathology
4.2. ABCA7 in Brain Functions
4.3. Impact of ABCA7 Depletion in Aβ Burden in Animals and Cells
4.4. Evidence in AD Patients
5. ABCA7 and Cancers
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D.; et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. ABC Family Transporters. Adv. Exp. Med. Biol. 2019, 1141, 13–100. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, C.J.; Tocchetti, G.N.; Rigalli, J.P.; Mottino, A.D. Acute regulation of apical ABC transporters in the gut. Potential influence on drug bioavailability. Pharmacol. Res. 2021, 163, 105251. [Google Scholar] [CrossRef] [PubMed]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef] [PubMed]
- Muriithi, W.; Macharia, L.W.; Heming, C.P.; Echevarria, J.L.; Nyachieo, A.; Filho, P.N.; Neto, V.M. ABC transporters and the hallmarks of cancer: Roles in cancer aggressiveness beyond multidrug resistance. Cancer Biol. Med. 2020, 17, 253–269. [Google Scholar] [CrossRef]
- Schmitz, G.; Liebisch, G.; Langmann, T. Lipidomic strategies to study structural and functional defects of ABC-transporters in cellular lipid trafficking. FEBS Lett. 2006, 580, 5597–5610. [Google Scholar] [CrossRef]
- Gosselet, F.; Saint-Pol, J.; Candela, P.; Fenart, L. Amyloid-beta Peptides, Alzheimer’s Disease and the Blood-brain Barrier. Curr. Alzheimer Res. 2013, 10, 1015–1033. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 895–897. [Google Scholar] [CrossRef]
- Pereira, C.D.; Martins, F.; Wiltfang, J.; da Cruz, E.S.O.A.B.; Rebelo, S. ABC Transporters Are Key Players in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 463–485. [Google Scholar] [CrossRef]
- Pahnke, J.; Walker, L.C.; Scheffler, K.; Krohn, M. Alzheimer’s disease and blood-brain barrier function-Why have anti-beta-amyloid therapies failed to prevent dementia progression? Neurosci. Biobehav. Rev. 2009, 33, 1099–1108. [Google Scholar] [CrossRef]
- Kaminski, W.E.; Orso, E.; Diederich, W.; Klucken, J.; Drobnik, W.; Schmitz, G. Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem. Biophys. Res. Commun. 2000, 273, 532–538. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Sasaki, M.; Shoji, A.; Kubo, Y.; Nada, S.; Yamaguchi, A. Cloning of rat ABCA7 and its preferential expression in platelets. Biochem. Biophys. Res. Commun. 2003, 304, 777–782. [Google Scholar] [CrossRef]
- Broccardo, C.; Osorio, J.; Luciani, M.F.; Schriml, L.M.; Prades, C.; Shulenin, S.; Arnould, I.; Naudin, L.; Lafargue, C.; Rosier, M.; et al. Comparative analysis of the promoter structure and genomic organization of the human and mouse ABCA7 gene encoding a novel ABCA transporter. Cytogenet. Cell Genet. 2001, 92, 264–270. [Google Scholar] [CrossRef]
- Heng, T.S.; Painter, M.W. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Kim, W.S.; Fitzgerald, M.L.; Kang, K.; Okuhira, K.; Bell, S.A.; Manning, J.J.; Koehn, S.L.; Lu, N.; Moore, K.J.; Freeman, M.W. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J. Biol. Chem. 2005, 280, 3989–3995. [Google Scholar] [CrossRef]
- Gosselet, F.; Candela, P.; Sevin, E.; Berezowski, V.; Cecchelli, R.; Fenart, L. Transcriptional profiles of receptors and transporters involved in brain cholesterol homeostasis at the blood-brain barrier: Use of an in vitro model. Brain Res. 2009, 1249, 34–42. [Google Scholar] [CrossRef]
- Tomioka, M.; Toda, Y.; Mañucat, N.B.; Akatsu, H.; Fukumoto, M.; Kono, N.; Arai, H.; Kioka, N.; Ueda, K. Lysophosphatidylcholine export by human ABCA7. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 658–665. [Google Scholar] [CrossRef]
- Wang, N.; Lan, D.; Gerbod-Giannone, M.; Linsel-Nitschke, P.; Jehle, A.W.; Chen, W.; Martinez, L.O.; Tall, A.R. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J. Biol. Chem. 2003, 278, 42906–42912. [Google Scholar] [CrossRef]
- Lamartiniere, Y.; Boucau, M.C.; Dehouck, L.; Krohn, M.; Pahnke, J.; Candela, P.; Gosselet, F.; Fenart, L. ABCA7 Downregulation Modifies Cellular Cholesterol Homeostasis and Decreases Amyloid-beta Peptide Efflux in an in vitro Model of the Blood-Brain Barrier. J. Alzheimers Dis. 2018, 64, 1195–1211. [Google Scholar] [CrossRef]
- Ikeda, Y.; Abe-Dohmae, S.; Munehira, Y.; Aoki, R.; Kawamoto, S.; Furuya, A.; Shitara, K.; Amachi, T.; Kioka, N.; Matsuo, M.; et al. Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem. Biophys. Res. Commun. 2003, 311, 313–318. [Google Scholar] [CrossRef]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; St George-Hyslop, P.; Fraser, P.E. ATP-binding Cassette Transporter A7 (ABCA7) Loss of Function Alters Alzheimer Amyloid Processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef]
- Lyssenko, N.N.; Praticò, D. ABCA7 and the altered lipidostasis hypothesis of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 164–174. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Allen, M.; Lincoln, S.J.; Corda, M.; Watzlawik, J.O.; Carrasquillo, M.M.; Reddy, J.S.; Burgess, J.D.; Nguyen, T.; Malphrus, K.; Petersen, R.C.; et al. ABCA7 loss-of-function variants, expression, and neurologic disease risk. Neurol. Genet. 2017, 3, e126. [Google Scholar] [CrossRef]
- Pasello, M.; Giudice, A.M.; Scotlandi, K. The ABC subfamily A transporters: Multifaceted players with incipient potentialities in cancer. Semin. Cancer Biol. 2020, 60, 57–71. [Google Scholar] [CrossRef]
- Ye, Z.; Lu, Y.; Wu, T. The impact of ATP-binding cassette transporters on metabolic diseases. Nutr. Metab. 2020, 17, 61. [Google Scholar] [CrossRef]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e1210. [Google Scholar] [CrossRef]
- Attie, A.D.; Kastelein, J.P.; Hayden, M.R. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J. Lipid Res. 2001, 42, 1717–1726. [Google Scholar] [CrossRef]
- Schmitz, G.; Kaminski, W.E.; Porsch-Ozcürümez, M.; Klucken, J.; Orsó, E.; Bodzioch, M.; Büchler, C.; Drobnik, W. ATP-binding cassette transporter A1 (ABCA1) in macrophages: A dual function in inflammation and lipid metabolism? Pathobiol. J. Immunopathol. Mol. Cell. Biol. 1999, 67, 236–240. [Google Scholar] [CrossRef]
- Jacobo-Albavera, L.; Domínguez-Pérez, M.; Medina-Leyte, D.J.; González-Garrido, A.; Villarreal-Molina, T. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int. J. Mol. Sci. 2021, 22, 1593. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.J.; Denis, M.; Genest, J. Cellular physiology of cholesterol efflux in vascular endothelial cells. Circulation 2004, 110, 2881–2888. [Google Scholar] [CrossRef] [PubMed]
- Singaraja, R.R.; Brunham, L.R.; Visscher, H.; Kastelein, J.J.; Hayden, M.R. Efflux and atherosclerosis: The clinical and biochemical impact of variations in the ABCA1 gene. Arter. Thromb. Vasc. Biol. 2003, 23, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Iatan, I.; Alrasadi, K.; Ruel, I.; Alwaili, K.; Genest, J. Effect of ABCA1 mutations on risk for myocardial infarction. Curr. Atheroscler. Rep. 2008, 10, 413–426. [Google Scholar] [CrossRef]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef]
- Meurs, I.; Calpe-Berdiel, L.; Habets, K.L.; Zhao, Y.; Korporaal, S.J.; Mommaas, A.M.; Josselin, E.; Hildebrand, R.B.; Ye, D.; Out, R.; et al. Effects of deletion of macrophage ABCA7 on lipid metabolism and the development of atherosclerosis in the presence and absence of ABCA1. PLoS ONE 2012, 7, e30984. [Google Scholar] [CrossRef]
- Iwamoto, N.; Abe-Dohmae, S.; Sato, R.; Yokoyama, S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 2006, 47, 1915–1927. [Google Scholar] [CrossRef]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Fitzgerald, M.L.; Yokoyama, S. Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J. Lipid Res. 2010, 51, 2591–2599. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Vandenhaute, E.; Boucau, M.C.; Candela, P.; Dehouck, L.; Cecchelli, R.; Dehouck, M.P.; Fenart, L.; Gosselet, F. Brain Pericytes ABCA1 Expression Mediates Cholesterol Efflux but not Cellular Amyloid-beta Peptide Accumulation. J. Alzheimers Dis. 2012, 30, 489–503. [Google Scholar] [CrossRef]
- Kielar, D.; Kaminski, W.E.; Liebisch, G.; Piehler, A.; Wenzel, J.J.; Möhle, C.; Heimerl, S.; Langmann, T.; Friedrich, S.O.; Böttcher, A.; et al. Adenosine triphosphate binding cassette (ABC) transporters are expressed and regulated during terminal keratinocyte differentiation: A potential role for ABCA7 in epidermal lipid reorganization. J. Investig. Dermatol. 2003, 121, 465–474. [Google Scholar] [CrossRef]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef]
- Su, H.P.; Nakada-Tsukui, K.; Tosello-Trampont, A.C.; Li, Y.; Bu, G.; Henson, P.M.; Ravichandran, K.S. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J. Biol. Chem. 2002, 277, 11772–11779. [Google Scholar] [CrossRef]
- Association, A.s. Alzheimer’s Disease Facts and Figures Alzheimer’s Dementia 2019. Annu. Rep. 2019, 15, 321–387. [Google Scholar]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Martins, I.J.; Berger, T.; Sharman, M.J.; Verdile, G.; Fuller, S.J.; Martins, R.N. Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 111, 1275–1308. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Candela, P.; Boucau, M.C.; Fenart, L.; Gosselet, F. Oxysterols decrease apical-to-basolateral transport of Abeta peptides via an ABCB1-mediated process in an in vitro Blood-brain barrier model constituted of bovine brain capillary endothelial cells. Brain Res. 2013, 1517, 1–15. [Google Scholar] [CrossRef]
- Hofrichter, J.; Krohn, M.; Schumacher, T.; Lange, C.; Feistel, B.; Walbroel, B.; Heinze, H.J.; Crockett, S.; Sharbel, T.F.; Pahnke, J. Reduced Alzheimer’s disease pathology by St. John’s Wort treatment is independent of hyperforin and facilitated by ABCC1 and microglia activation in mice. Curr. Alzheimer Res. 2013, 10, 1057–1069. [Google Scholar] [CrossRef]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced Brain Amyloid-beta Clearance by Rifampicin and Caffeine as a Possible Protective Mechanism Against Alzheimer’s Disease. J. Alzheimers Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Alfen, F.; et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Hartman, R.E.; Bales, K.R.; Paul, S.M.; Holtzman, D.M. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2005, 280, 43236–43242. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Kim, J.; Li, A.; Knoten, A.; Jain, S.; Hirsch-Reinshagen, V.; Wellington, C.L.; Bales, K.R.; et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118, 671–682. [Google Scholar] [CrossRef]
- Hirsch-Reinshagen, V.; Maia, L.F.; Burgess, B.L.; Blain, J.F.; Naus, K.E.; McIsaac, S.A.; Parkinson, P.F.; Chan, J.Y.; Tansley, G.H.; Hayden, M.R.; et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 2005, 280, 43243–43256. [Google Scholar] [CrossRef]
- Gosselet, F. The Mysterious Link between Cholesterol and Alzheimer’s Disease: Is the Blood-Brain Barrier a Suspect? J. Alzheimer’s Dis. Parkinsonism 2011, 1, 2161-0460. [Google Scholar] [CrossRef]
- Aikawa, T.; Holm, M.L.; Kanekiyo, T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018, 8, 27. [Google Scholar] [CrossRef]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, H.; Liu, B.; Wang, T.; Han, Z.; Ji, X. rs4147929 variant minor allele increases ABCA7 gene expression and ABCA7 shows increased gene expression in Alzheimer’s disease patients compared with controls. Acta Neuropathol. 2020, 139, 937–940. [Google Scholar] [CrossRef]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e221–220.e229. [Google Scholar] [CrossRef]
- Cukier, H.N.; Kunkle, B.W.; Vardarajan, B.N.; Rolati, S.; Hamilton-Nelson, K.L.; Kohli, M.A.; Whitehead, P.L.; Dombroski, B.A.; Van Booven, D.; Lang, R.; et al. ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurology. Genet. 2016, 2, e79. [Google Scholar] [CrossRef]
- De Roeck, A.; Van den Bossche, T.; van der Zee, J.; Verheijen, J.; De Coster, W.; Van Dongen, J.; Dillen, L.; Baradaran-Heravi, Y.; Heeman, B.; Sanchez-Valle, R.; et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease. Acta Neuropathol. 2017, 134, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Chen, K.; Keenan, B.T.; Chibnik, L.B.; Fleisher, A.; Thiyyagura, P.; Roontiva, A.; McCabe, C.; Patsopoulos, N.A.; Corneveaux, J.J.; et al. Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol. 2013, 70, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.C.; Zong, Y.; Wang, H.F.; Li, J.Q.; Cao, X.P.; Tan, L. ABCA7 genotype altered Aβ levels in cerebrospinal fluid in Alzheimer’s disease without dementia. Ann. Transl. Med. 2018, 6, 437. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, L.M.; Goukasian, N.; Porat, S.; Hwang, K.S.; Eastman, J.A.; Hurtz, S.; Wang, B.; Vang, N.; Sears, R.; Klein, E.; et al. Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiol. Aging 2016, 39, 82–89. [Google Scholar] [CrossRef]
- Karch, C.M.; Jeng, A.T.; Nowotny, P.; Cady, J.; Cruchaga, C.; Goate, A.M. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE 2012, 7, e50976. [Google Scholar] [CrossRef]
- Engelman, C.D.; Koscik, R.L.; Jonaitis, E.M.; Okonkwo, O.C.; Hermann, B.P.; La Rue, A.; Sager, M.A. Interaction between two cholesterol metabolism genes influences memory: Findings from the Wisconsin Registry for Alzheimer’s Prevention. J. Alzheimers Dis. 2013, 36, 749–757. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Khan, Q.; Murray, M.E.; Krishnan, S.; Aakre, J.; Pankratz, V.S.; Nguyen, T.; Ma, L.; Bisceglio, G.; Petersen, R.C.; et al. Late-onset Alzheimer disease genetic variants in posterior cortical atrophy and posterior AD. Neurology 2014, 82, 1455–1462. [Google Scholar] [CrossRef]
- Schott, J.M.; Crutch, S.J.; Carrasquillo, M.M.; Uphill, J.; Shakespeare, T.J.; Ryan, N.S.; Yong, K.X.; Lehmann, M.; Ertekin-Taner, N.; Graff-Radford, N.R.; et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 862–871. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Crook, J.E.; Pedraza, O.; Thomas, C.S.; Pankratz, V.S.; Allen, M.; Nguyen, T.; Malphrus, K.G.; Ma, L.; Bisceglio, G.D.; et al. Late-onset Alzheimer’s risk variants in memory decline, incident mild cognitive impairment, and Alzheimer’s disease. Neurobiol. Aging 2015, 36, 60–67. [Google Scholar] [CrossRef]
- Nettiksimmons, J.; Tranah, G.; Evans, D.S.; Yokoyama, J.S.; Yaffe, K. Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age 2016, 38, 41. [Google Scholar] [CrossRef]
- Andrews, S.J.; Das, D.; Anstey, K.J.; Easteal, S. Late Onset Alzheimer’s Disease Risk Variants in Cognitive Decline: The PATH Through Life Study. J. Alzheimers Dis. 2017, 57, 423–436. [Google Scholar] [CrossRef]
- Roshchupkin, G.V.; Adams, H.H.; van der Lee, S.J.; Vernooij, M.W.; van Duijn, C.M.; Uitterlinden, A.G.; van der Lugt, A.; Hofman, A.; Niessen, W.J.; Ikram, M.A. Fine-mapping the effects of Alzheimer’s disease risk loci on brain morphology. Neurobiol. Aging 2016, 48, 204–211. [Google Scholar] [CrossRef]
- Wachinger, C.; Nho, K.; Saykin, A.J.; Reuter, M.; Rieckmann, A. A Longitudinal Imaging Genetics Study of Neuroanatomical Asymmetry in Alzheimer’s Disease. Biol. Psychiatry 2018, 84, 522–530. [Google Scholar] [CrossRef]
- Monsell, S.E.; Mock, C.; Fardo, D.W.; Bertelsen, S.; Cairns, N.J.; Roe, C.M.; Ellingson, S.R.; Morris, J.C.; Goate, A.M.; Kukull, W.A. Genetic Comparison of Symptomatic and Asymptomatic Persons With Alzheimer Disease Neuropathology. Alzheimer Dis. Assoc. Disord. 2017, 31, 232–238. [Google Scholar] [CrossRef]
- Hughes, T.M.; Lopez, O.L.; Evans, R.W.; Kamboh, M.I.; Williamson, J.D.; Klunk, W.E.; Mathis, C.A.; Price, J.C.; Cohen, A.D.; Snitz, B.E.; et al. Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol. Aging 2014, 35, 802–807. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Risacher, S.L.; Duran, T.; Stage, E.C.; Goukasian, N.; West, J.D.; Do, T.M.; Grotts, J.; Wilhalme, H.; Nho, K.; et al. Associations of the Top 20 Alzheimer Disease Risk Variants With Brain Amyloidosis. JAMA Neurol. 2018, 75, 328–341. [Google Scholar] [CrossRef]
- Stage, E.; Duran, T.; Risacher, S.L.; Goukasian, N.; Do, T.M.; West, J.D.; Wilhalme, H.; Nho, K.; Phillips, M.; Elashoff, D.; et al. The effect of the top 20 Alzheimer disease risk genes on gray-matter density and FDG PET brain metabolism. Alzheimer’s Dement. 2016, 5, 53–66. [Google Scholar] [CrossRef]
- Sinha, N.; Reagh, Z.M.; Tustison, N.J.; Berg, C.N.; Shaw, A.; Myers, C.E.; Hill, D.; Yassa, M.A.; Gluck, M.A. ABCA7 risk variant in healthy older African Americans is associated with a functionally isolated entorhinal cortex mediating deficient generalization of prior discrimination training. Hippocampus 2019, 29, 527–538. [Google Scholar] [CrossRef]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef]
- Kim, W.S.; Guillemin, G.J.; Glaros, E.N.; Lim, C.K.; Garner, B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport 2006, 17, 891–896. [Google Scholar] [CrossRef]
- Fu, Y.; Hsiao, J.H.; Paxinos, G.; Halliday, G.M.; Kim, W.S. ABCA7 Mediates Phagocytic Clearance of Amyloid-beta in the Brain. J. Alzheimers Dis. 2016, 54, 569–584. [Google Scholar] [CrossRef]
- Chan, S.L.; Kim, W.S.; Kwok, J.B.; Hill, A.F.; Cappai, R.; Rye, K.A.; Garner, B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 2008, 106, 793–804. [Google Scholar] [CrossRef]
- Sakae, N.; Liu, C.C.; Shinohara, M.; Frisch-Daiello, J.; Ma, L.; Yamazaki, Y.; Tachibana, M.; Younkin, L.; Kurti, A.; Carrasquillo, M.M.; et al. ABCA7 Deficiency Accelerates Amyloid-beta Generation and Alzheimer’s Neuronal Pathology. J. Neurosci. 2016, 36, 3848–3859. [Google Scholar] [CrossRef]
- Logge, W.; Cheng, D.; Chesworth, R.; Bhatia, S.; Garner, B.; Kim, W.S.; Karl, T. Role of Abca7 in mouse behaviours relevant to neurodegenerative diseases. PLoS ONE 2012, 7, e45959. [Google Scholar] [CrossRef]
- Li, H.; Karl, T.; Garner, B. Abca7 deletion does not affect adult neurogenesis in the mouse. Biosci. Rep. 2016, 36, 1–6. [Google Scholar] [CrossRef]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef]
- Aikawa, T.; Ren, Y.; Yamazaki, Y.; Tachibana, M.; Johnson, M.R.; Anderson, C.T.; Martens, Y.A.; Holm, M.L.; Asmann, Y.W.; Saito, T.; et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 23790–23796. [Google Scholar] [CrossRef]
- Vacher, M.; Porter, T.; Villemagne, V.L.; Milicic, L.; Peretti, M.; Fowler, C.; Martins, R.; Rainey-Smith, S.; Ames, D.; Masters, C.L.; et al. Validation of a priori candidate Alzheimer’s disease SNPs with brain amyloid-beta deposition. Sci. Rep. 2019, 9, 17069. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Zou, F.; Chai, H.S.; Younkin, C.S.; Crook, J.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; Nair, A.A.; Middha, S.; et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 2012, 79, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Wellens, S.; Dehouck, L.; Chandrasekaran, V.; Singh, P.; Loiola, R.A.; Sevin, E.; Exner, T.; Jennings, P.; Gosselet, F.; Culot, M. Evaluation of a human iPSC-derived BBB model for repeated dose toxicity testing with cyclosporine A as model compound. Toxicol. In Vitro 2021, 73, 105112. [Google Scholar] [CrossRef] [PubMed]
- Wurm, J.; Konttinen, H.; Andressen, C.; Malm, T.; Spittau, B. Microglia Development and Maturation and Its Implications for Induction of Microglia-Like Cells from Human iPSCs. Int. J. Mol. Sci. 2021, 22, 3088. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Q.; Zhou, J.; Zhang, S. ATP-binding cassette transporter A7 accelerates epithelial-to-mesenchymal transition in ovarian cancer cells by upregulating the transforming growth factor-β signaling pathway. Oncol. Lett. 2018, 16, 5868–5874. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Shui, C.; Fang, X.; Peng, Y.; Qin, L. miR-197-3p reduces epithelial-mesenchymal transition by targeting ABCA7 in ovarian cancer cells. 3 Biotech 2020, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.C.; Rosario, E.R.; Kreimer, S.; Villamagna, A.; Gentzschein, E.; Stanczyk, F.Z.; Pike, C.J. Sex differences in β-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res. 2010, 1366, 233–245. [Google Scholar] [CrossRef]
- Schäfer, S.; Wirths, O.; Multhaup, G.; Bayer, T.A. Gender dependent APP processing in a transgenic mouse model of Alzheimer’s disease. J. Neural Transm. 2007, 114, 387–394. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variation | Interpretation | Reported Significant Effect of the Risk Allele | |
|---|---|---|---|
| Common Risk-Increasing Variants | Amyloid and Tau pathology | Brain Morphology and Clinical Symptoms | |
| rs3764650 | Intronic GWAS SNP, low predicted functional effect |
| |
| rs4147929 | Intronic GWAS SNP, low predicted functional effect | ||
| rs3752246 | Missense GWAS SNP, predicted benign | ||
| rs115550680 | Intronic GWAS SNP, low predicted functional effect | ||
| rs78117248 | Intronic GWAS SNP, low predicted functional effect | ||
| rs142076058 | Loss-of-function | ||
| ABCA7 VNTR expansions | Reduced ABCA7 expression, loss of exon 19 encoding an ATP-binding domain | ||
| Common Protective Variants | Amyloid and Tau pathology | Brain Morphology and Clinical Symptoms | |
| rs72973581 | Missense variant | ||
| CpG Methylation | Amyloid and Tau pathology | Brain Morphology and Clinical Symptoms | |
| cg02308560 | Hypermethylation in AD, effect on ABCA7 unknown | ||
| cg24402332 | |||
| cg04587220 |
| ||
| Rare Variants | Amyloid and Tau pathology | Brain Morphology and Clinical Symptoms | |
| Missense and PTC variants | Loss-of-function for PTC variants. Unclear for missense variants. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603. https://doi.org/10.3390/ijms22094603
Dib S, Pahnke J, Gosselet F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(9):4603. https://doi.org/10.3390/ijms22094603
Chicago/Turabian StyleDib, Shiraz, Jens Pahnke, and Fabien Gosselet. 2021. "Role of ABCA7 in Human Health and in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 9: 4603. https://doi.org/10.3390/ijms22094603
APA StyleDib, S., Pahnke, J., & Gosselet, F. (2021). Role of ABCA7 in Human Health and in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(9), 4603. https://doi.org/10.3390/ijms22094603