
A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma

 , , , , , ,
, , , , , ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Selection of Drugs for Screening in Patient-Derived In Vitro Models
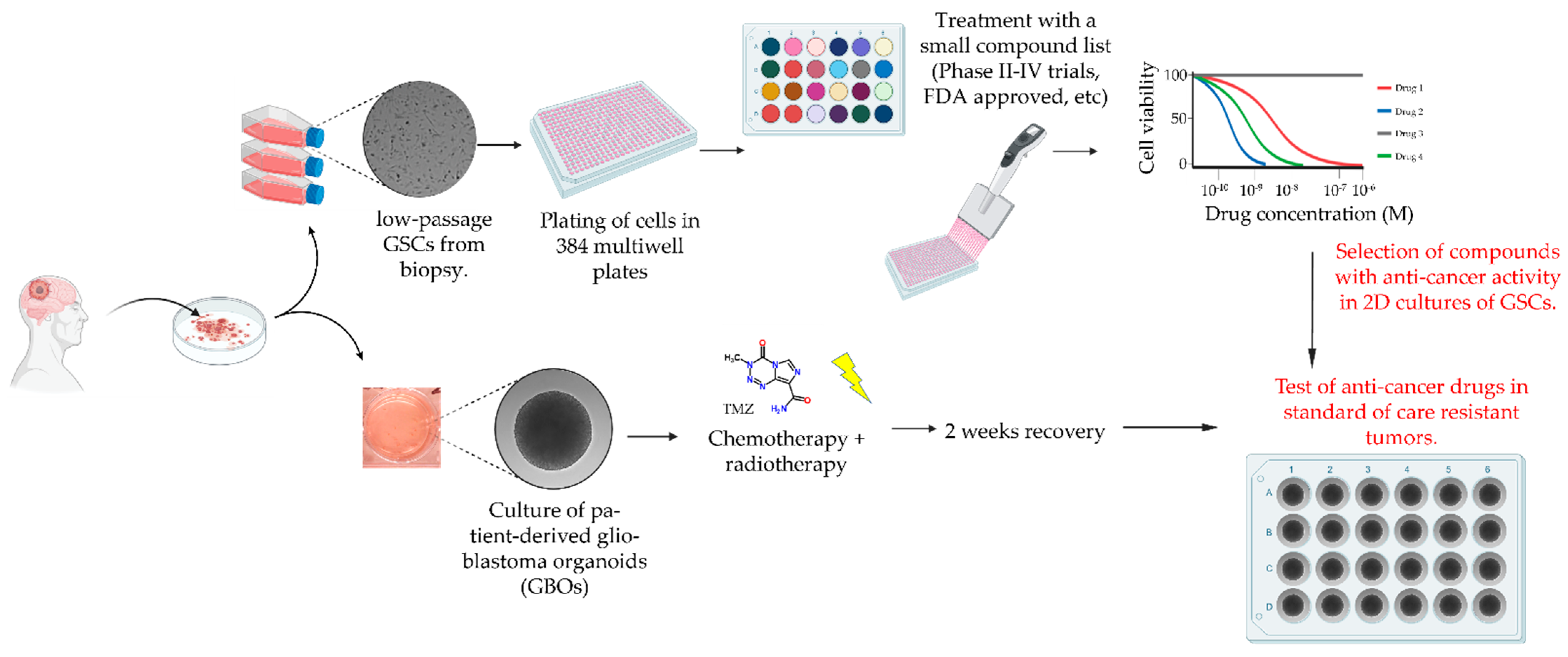
2.2. Drug Screening Pipeline
2.3. Drug Screening in 2D Cultures of Patient-Derived GSCs
2.4. Effect of Drug Treatment in 2D and 3D In Vitro Models of Primary and Standard of Care Resistant Glioblastoma
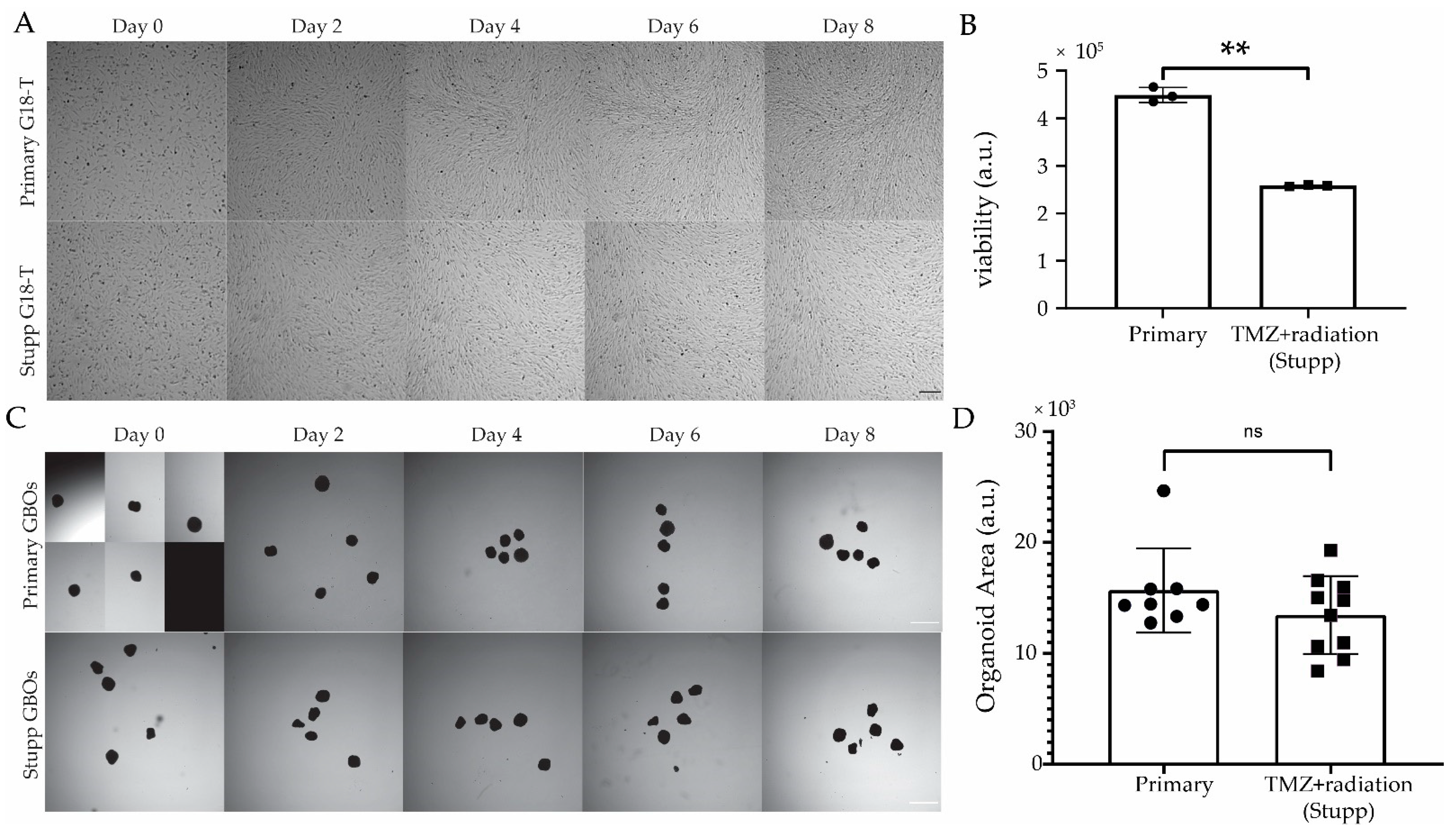
2.4.1. Generation of TMZ+radiation (“Stupp”) Resistant 2D and 3D Patient-Derived In Vitro Models
2.4.2. Response of Stupp Treated G18-T Cell Line and GBO to Selected Drugs
3. Discussion
3.1. Drug Screening Using a Combination of 2D and 3D Patient-Derived In Vitro Models for Glioblastoma
3.2. Drug Inhibitors Have Varying Effects on Different Patient-Derived GSC Cultures
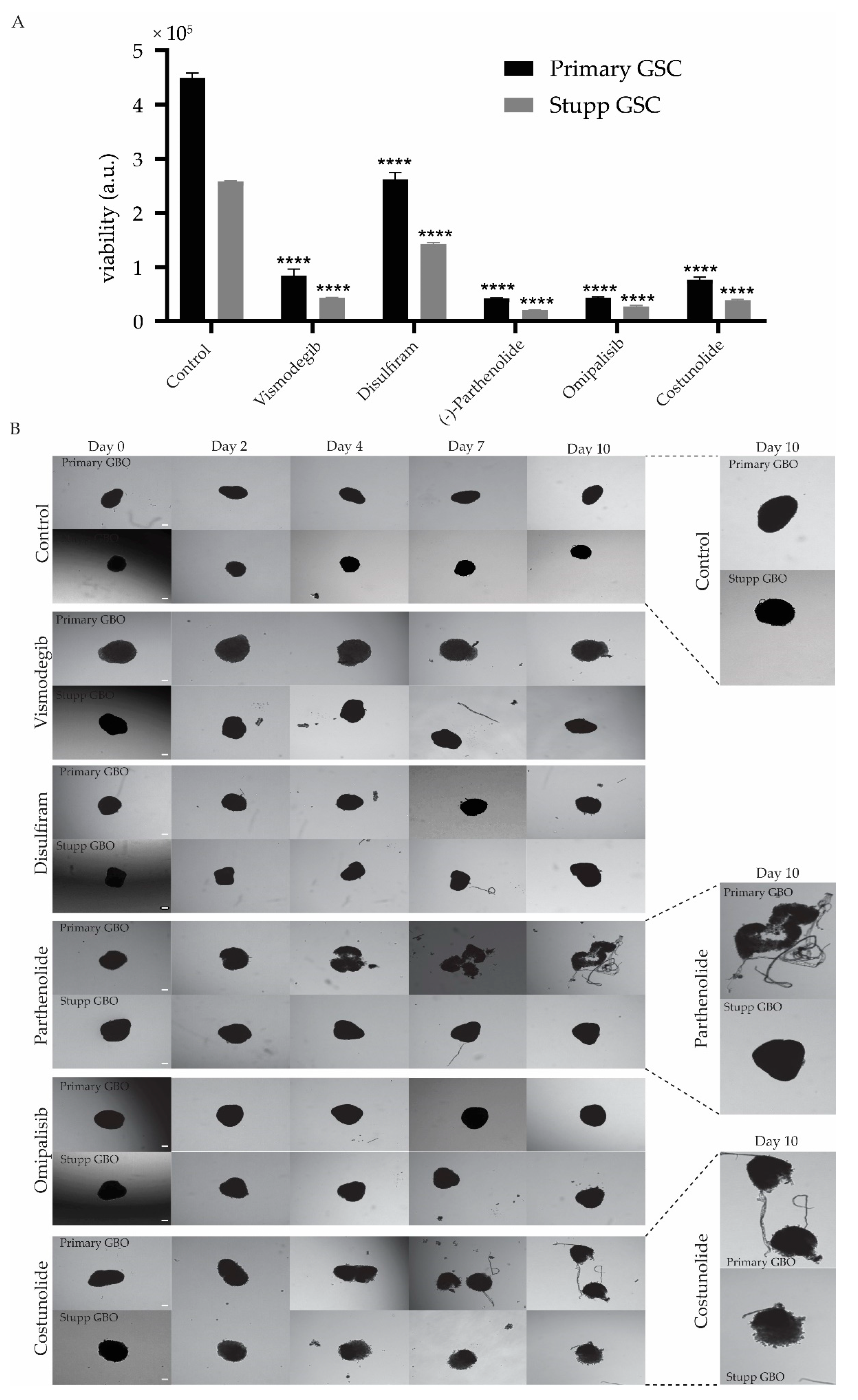
3.3. Response of Stupp Treated 2D and 3D Cultures to Vismodegib, Disulfiram, Parthenolide, Omipalisib and Costunolide
4. Materials and Methods
4.1. Drug Library
4.2. Glioblastoma Cell Culture
4.3. Seeding of Tumor Cells into 384-Well Plates
4.4. Serial Drug Dilutions and Addition of Drugs to Cells Using Automated Liquid Handler Robot
4.5. Cell Viability Assay
4.6. Generation of Glioblastoma Organoids (GBOs)
4.7. TMZ+Radiation Treatment (Stupp Protocol) “In a Dish”
4.8. Treatment with Selected Drugs
4.9. Statistical Analysis
4.10. Artwork
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | two dimensional |
| 3D | three dimensional |
| ATP | adenosine triphosphate |
| CAR-T | chimeric antigen receptor T cells |
| CSC | cancer stem cell |
| DPBS | Dulbecco’s Phosphate Buffered Saline |
| EGFR | epidermal growth factor receptor |
| FDA | U.S. Food & Drug Administration |
| GBO | glioblastoma explant organoid |
| GSC | glioma stem cell |
| IC50 | half maximal inhibitory concentration |
| IDH1 | isocitrate dehydrogenase 1 |
| MET | mesenchymal to epithelial transition |
| MGMT | O[6]-methylguanine-DNA methyltransferase |
| NF1 | neurofibromatosis 1 |
| PDGFRA | Platelet-derived growth factor receptor A |
| PTEN | Phosphatase and tensin homolog |
| RTK | receptor tyrosine kinases |
| SANTB | South Australian Neurological Tumour Bank |
| scRNAseq | single-cell RNA sequencing |
| NSC medium | StemPro Neural Stem Cell serum-free medium |
| TAM | tumor-associated macrophages |
| hTERT | Human Telomerase reverse transcriptase |
| TERT | Telomerase reverse transcriptase |
| TMZ | temozolomide |
| VEGF | vascular endothelial growth factor |
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15 (Suppl. S2), ii1–ii56. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Li, Q.; Sun, Y.; Liu, B.; Li, J.; Hao, X.; Ge, W.; Zhang, X.; Bao, S.; Gong, J.; Jiang, Z.; et al. ACT001 modulates the NF-κB/MnSOD/ROS axis by targeting IKKβ to inhibit glioblastoma cell growth. J. Mol. Med. (Berl. Ger.) 2020, 98, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Du, X.; Lu, G.; Zhu, J. Survival benefit of glioblastoma patients after FDA approval of temozolomide concomitant with radiation and bevacizumab: A population-based study. Oncotarget 2017, 8, 44015–44031. [Google Scholar] [CrossRef] [PubMed]
- Hottinger, A.F.; Abdullah, K.G.; Stupp, R. Chapter 6—Current Standards of Care in Glioblastoma Therapy; Elsevier Inc: Amsterdam, The Netherlands, 2016; pp. 73–80. [Google Scholar]
- Linde, M.; Brahm, C.; Witt Hamer, P.; Reijneveld, J.; Bruynzeel, A.; Vandertop, W.; Ven, P.; Wagemakers, M.; Weide, H.; Enting, R.; et al. Treatment outcome of patients with recurrent glioblastoma multiforme: A retrospective multicenter analysis. J. Neuro Oncol. 2017, 135, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef]
- Little, S.E.; Popov, S.; Jury, A.; Bax, D.A.; Doey, L.; Al-Sarraj, S.; Jurgensmeier, J.M.; Jones, C. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012, 72, 1614. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Ebert, L.M.; Yu, W.; Gargett, T.; Toubia, J.; Kollis, P.M.; Tea, M.N.; Ebert, B.W.; Bardy, C.; van den Hurk, M.; Bonder, C.S.; et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (FAP) as an excellent target for immunotherapy. Clin. Transl. Immunol. 2020, 9, e1191. [Google Scholar] [CrossRef]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Muller, S.; Liu, S.J.; Di Lullo, E.; Malatesta, M.; Pollen, A.A.; Nowakowski, T.J.; Kohanbash, G.; Aghi, M.; Kriegstein, A.R.; Lim, D.A.; et al. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF- and EGF-driven gliomas. Mol. Syst. Biol. 2016, 12, 889. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Yuan, J.; Levitin, H.M.; Frattini, V.; Bush, E.C.; Boyett, D.M.; Samanamud, J.; Ceccarelli, M.; Dovas, A.; Zanazzi, G.; Canoll, P.; et al. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 2018, 10, 57. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Grot, D.; Stoczynska-Fidelus, E.; Rieske, P. Gaps and doubts in search to recognize glioblastoma cellular origin and tumor initiating cells. J. Oncol. 2020, 2020, 6783627. [Google Scholar] [CrossRef]
- Guelfi, S.; Duffau, H.; Bauchet, L.; Rothhut, B.; Hugnot, J.-P. Vascular Transdifferentiation in the CNS: A Focus on Neural and Glioblastoma Stem-Like Cells. Stem Cells Int. 2016, 2016, 2759403. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95 (Suppl. S1), S2–S7. [Google Scholar] [CrossRef]
- Sadahiro, H.; Yoshikawa, K.; Ideguchi, M.; Kajiwara, K.; Ishii, A.; Ikeda, E.; Owada, Y.; Yasumoto, Y.; Suzuki, M. Pathological features of highly invasive glioma stem cells in a mouse xenograft model. Brain Tumor Pathol. 2014, 31, 77–84. [Google Scholar] [CrossRef]
- Anne, D.; Anna, G.; Thomas, B.; Petr, V.N.; Arnaud, M.; Suresh, P.; Nicolaas, H.C.B.; Sonia, L.; Nicolas, S.; Dzjemma, S.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef]
- Iwadate, Y. Plasticity in glioma stem cell phenotype and its therapeutic implication. Neurol. Med. Chir. 2018, 58, 61–70. [Google Scholar] [CrossRef]
- Australian Institute of Health and Welfare. Brain and Other Central Nervous System Cancers; Australian Institute of Health and Welfare: Canberra, ACT, Australia, 2017.
- Alphandery, E. Glioblastoma treatments: An account of recent industrial developments. Front. Pharm. 2018, 9, 879. [Google Scholar] [CrossRef]
- Weller, M.; van Den Bent, M.; Hopkins, K.; Tonn, J.C.; Stupp, R.; Falini, A.; Cohen-Jonathan-Moyal, E.; Frappaz, D.; Henriksson, R.; Balana, C.; et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014, 15, e395–e403. [Google Scholar] [CrossRef]
- Lowe, S.; Bhat, K.P.; Olar, A. Current clinical management of patients with glioblastoma. Cancer Rep. (Hoboken) 2019, 2, e1216. [Google Scholar] [CrossRef]
- Gomez, G.A.; Oksdath, M.; Brown, M.P.; Ebert, L.M. New approaches to model glioblastoma in vitro using brain organoids: Implications for precision oncology. Transl. Cancer Res. 2019, S606–S611. [Google Scholar] [CrossRef]
- Heffernan, J.M.; Sirianni, R.W. Modeling microenvironmental regulation of glioblastoma stem cells: A biomaterials perspective. Front. Mater. 2018, 5, 7. [Google Scholar] [CrossRef]
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for glioblastoma: Is it working? Drug Discov. Today 2019, 24, 1193–1201. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R. Organoid models of glioblastoma to study brain tumor stem cells. Front. Cell Dev. Biol. 2020, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.N.; Anderson, J.C.; Duarte, C.W.; Shevin, R.S.; Langford, C.P.; Singh, R.; Gillespie, G.Y.; Willey, C.D. Combinatorial drug testing in 3D microtumors derived from GBM patient-derived xenografts reveals cytotoxic synergy in pharmacokinomics-informed pathway interactions. Sci. Rep. 2018, 8, 8412. [Google Scholar] [CrossRef]
- Day, B.W.; Stringer, B.W.; Wilson, J.; Jeffree, R.L.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Inglis, P.; Allan, S.; Winter, C.; et al. Glioma surgical aspirate: A viable source of tumor tissue for experimental research. Cancers 2013, 5, 357–371. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef]
- Stringer, B.W.; Day, B.W.; D’Souza, R.C.J.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgül, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 4902. [Google Scholar] [CrossRef]
- Oksdath, M.; Perrin, S.L.; Bardy, C.; Hilder, E.F.; DeForest, C.A.; Arrua, R.D.; Gomez, G.A. Review: Synthetic scaffolds to control the biochemical, mechanical, and geometrical environment of stem cell-derived brain organoids. APL Bioeng. 2018, 2, 041501. [Google Scholar] [CrossRef]
- Jacob, F.; Ming, G.-L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Tan, S.K.; Jermakowicz, A.; Mookhtiar, A.K.; Nemeroff, C.B.; Schürer, S.C.; Ayad, N.G. Drug repositioning in glioblastoma: A pathway perspective. Front. Pharmacol. 2018, 9, 218. [Google Scholar] [CrossRef]
- Cao, S.; Wang, G.; Ge, F.; Li, X.; Zhu, Q.; Ge, R.S.; Wang, Y. Gossypol inhibits 5α-reductase 1 and 3α-hydroxysteroid dehydrogenase: Its possible use for the treatment of prostate cancer. Fitoterapia 2019, 133, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Davies, B.R.; Han, S.; Zhou, M.; Bai, Y.; Zhang, J.; Xu, Y.; Tang, L.; Wang, H.; Liu, Y.J.; et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J. Transl. Med. 2013, 11, 241. [Google Scholar] [CrossRef]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharm. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Kreisl, T.N.; Kim, L.; Duic, J.P.; Butman, J.A.; Albert, P.S.; Fine, H.A. Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer 2010, 116, 1776–1782. [Google Scholar] [CrossRef]
- Lickliter, J.D.; Cox, J.; McCarron, J.; Martinez, N.R.; Schmidt, C.W.; Lin, H.; Nieda, M.; Nicol, A.J. Small-molecule Bcl-2 inhibitors sensitise tumour cells to immune-mediated destruction. Br. J. Cancer 2007, 96, 600–608. [Google Scholar] [CrossRef]
- Inoue-Yamauchi, A.; Jeng, P.S.; Kim, K.; Chen, H.-C.; Han, S.; Ganesan, Y.T.; Ishizawa, K.; Jebiwott, S.; Dong, Y.; Pietanza, M.C.; et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat. Commun. 2017, 8, 16078. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, A.; Manzione, L. Dasatinib: A new step in molecular target therapy. Ann. Oncol. 2007, 18 (Suppl. S6), vi42–vi46. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Maiorano, B.A.; Pagliara, M.M.; Sammarco, M.G.; Dosa, T.; Martini, M.; Rindi, G.; Bria, E.; Blasi, M.A.; Tortora, G.; et al. Dabrafenib and trametinib in BRAF mutant metastatic conjunctival melanoma. Front. Oncol. 2019, 9, 232. [Google Scholar] [CrossRef]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzym. 2006, 407, 597–612. [Google Scholar]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Palumbo, A.; Lau, G.; Saraceni, M. Abemaciclib: The newest CDK4/6 inhibitor for the treatment of breast cancer. Ann. Pharm. 2019, 53, 178–185. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Celecoxib, a selective cyclooxygenase-2 inhibitor for the treatment of rheumatoid arthritis and osteoarthritis. Clin. Ther. 1999, 21, 1497–1513. [Google Scholar] [CrossRef]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des. Devel. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Rettig, M.P.; Cashen, A.F. Plerixafor, a CXCR4 antagonist for the mobilization of hematopoietic stem cells. Expert Opin Biol. Ther. 2008, 8, 1797–1804. [Google Scholar] [CrossRef]
- Plummer, R.; Lorigan, P.; Steven, N.; Scott, L.; Middleton, M.R.; Wilson, R.H.; Mulligan, E.; Curtin, N.; Wang, D.; Dewji, R.; et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother. Pharm. 2013, 71, 1191–1199. [Google Scholar] [CrossRef]
- You, C.; Dai, X.; Yuan, B.; Wang, Y. Effects of 6-thioguanine and S6-methylthioguanine on transcription in vitro and in human cells. J. Biol. Chem. 2012, 287, 40915–40923. [Google Scholar] [CrossRef]
- Ma, T.; Yamada, S.; Ichwan, S.J.; Iseki, S.; Ohtani, K.; Otsu, M.; Ikeda, M.A. Inability of p53-reactivating compounds Nutlin-3 and RITA to overcome p53 resistance in tumor cells deficient in p53Ser46 phosphorylation. Biochem. Biophys. Res. Commun. 2012, 417, 931–937. [Google Scholar] [CrossRef]
- Huang, L.; Huang, H.; Zhou, X.P.; Liu, J.F.; Li, C.R.; Fang, M.; Wu, J.R. Osimertinib or EGFR-TKIs/chemotherapy in patients with EGFR-mutated advanced nonsmall cell lung cancer: A meta-analysis. Medicine 2019, 98, e17705. [Google Scholar] [CrossRef]
- Wong, S.F. Cetuximab: An epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin. Ther. 2005, 27, 684–694. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Thomas, X.; Elhamri, M. Tipifarnib in the treatment of acute myeloid leukemia. Biologics 2007, 1, 415–424. [Google Scholar]
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Tan, Y.C.; Madan, B.; Amalini, C.; Ong, L.C.; Kheng, B.; Cheong, A.; Zhou, J.; et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011, 1, e44. [Google Scholar] [CrossRef]
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. New Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef]
- Dupuis, N.; Laschet, C.; Franssen, D.; Szpakowska, M.; Gilissen, J.; Geubelle, P.; Soni, A.; Parent, A.S.; Pirotte, B.; Chevigné, A.; et al. Activation of the orphan G protein-coupled receptor GPR27 by surrogate ligands promotes β-Arrestin 2 recruitment. Mol. Pharm. 2017, 91, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, C.; Jung, J.; Yeo, S.G. The histone deacetylase class I, II inhibitor trichostatin A delays peripheral neurodegeneration. J. Mol. Histol. 2019, 50, 167–178. [Google Scholar] [CrossRef]
- Aditya, S.; Rattan, A. Vismodegib: A smoothened inhibitor for the treatment of advanced basal cell carcinoma. Indian Derm. Online J. 2013, 4, 365–368. [Google Scholar] [CrossRef]
- Cherian, M.A.; Ma, C.X. The role of neratinib in HER2-driven breast cancer. Future Oncol. 2017, 13, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91–97. [Google Scholar]
- Ricker, J.L.; Chen, Z.; Yang, X.P.; Pribluda, V.S.; Swartz, G.M.; Van Waes, C. 2-methoxyestradiol inhibits hypoxia-inducible factor 1alpha, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin. Cancer Res. 2004, 10, 8665–86673. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Desano, J.; Qu, Y.; Tang, W.; Meng, Y.; Lawrence, T.S.; Xu, L. Natural IAP inhibitor Embelin enhances therapeutic efficacy of ionizing radiation in prostate cancer. Am. J. Cancer Res. 2011, 1, 128–143. [Google Scholar] [PubMed]
- Dhillon, S. Ivosidenib: First global approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Tallman, M.S.; de Botton, S.; Kantarjian, H.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef]
- Wan, Z.; Sun, J.; Xu, J.; Moharil, P.; Chen, J.; Xu, J.; Zhu, J.; Li, J.; Huang, Y.; Xu, P.; et al. Dual functional immunostimulatory polymeric prodrug carrier with pendent indoximod for enhanced cancer immunochemotherapy. Acta Biomater. 2019, 90, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Günther, J.; Däbritz, J.; Wirthgen, E. Limitations and off-target effects of tryptophan-related IDO inhibitors in cancer treatment. Front. Immunol. 2019, 10, 1801. [Google Scholar] [CrossRef]
- Ransom, J.T. Mechanism of action of mycophenolate mofetil. Ther. Drug Monit. 1995, 17, 681–684. [Google Scholar] [CrossRef]
- Maurer, G.D.; Tritschler, I.; Adams, B.; Tabatabai, G.; Wick, W.; Stupp, R.; Weller, M. Cilengitide modulates attachment and viability of human glioma cells, but not sensitivity to irradiation or temozolomide in vitro. Neuro Oncol. 2009, 11, 747–756. [Google Scholar] [CrossRef]
- Moon, D.-O.; Kim, M.-O.; Kang, C.-H.; Lee, J.-D.; Choi, Y.H.; Kim, G.-Y. JNK inhibitor SP600125 promotes the formation of polymerized tubulin, leading to G2/M phase arrest, endoreduplication, and delayed apoptosis. Exp. Mol. Med. 2009, 41, 665–677. [Google Scholar] [CrossRef]
- Hoffner, B.; Benchich, K. Trametinib: A targeted therapy in metastatic melanoma. J. Adv. Pract. Oncol. 2018, 9, 741–745. [Google Scholar]
- Eagles, J.R.; Jimeno, A. Cobimetinib: Inhibiting MEK1/2 in BRAF V600-mutant melanoma. Drugs Today (Barc. Spain) 2016, 52, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.V.; Logié, A.; Odedra, R.; Heier, A.; Heaton, S.P.; Alferez, D.; Davies, B.R.; Wilkinson, R.W.; Smith, P.D. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br. J. Cancer 2012, 106, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Duncia, J.V.; Santella, J.B., 3rd; Higley, C.A.; Pitts, W.J.; Wityak, J.; Frietze, W.E.; Rankin, F.W.; Sun, J.H.; Earl, R.A.; Tabaka, A.C.; et al. MEK inhibitors: The chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg. Med. Chem. Lett. 1998, 8, 2839–2844. [Google Scholar] [CrossRef]
- Su, Y.; Xu, H.; Xu, Y.; Yu, J.; Xian, Y.; Luo, Q. Azacytidine inhibits the proliferation of human promyelocytic leukemia cells (HL60) by demethylation of MGMT, DAPK and p16 genes. Hematology 2012, 17, 41–46. [Google Scholar] [CrossRef]
- Barnett, C.M. Everolimus: Targeted therapy on the horizon for the treatment of breast cancer. Pharmacotherapy 2012, 32, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Serrato, A.; Lee, J.; Holmes, B.; Landon, K.A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS ONE 2017, 12, e0176599. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Saadane, A.; Masters, S.; DiDonato, J.; Li, J.; Berger, M. Parthenolide inhibits IkappaB kinase, NF-kappaB activation, and inflammatory response in cystic fibrosis cells and mice. Am. J. Respir. Cell Mol. Biol. 2007, 36, 728–736. [Google Scholar] [CrossRef]
- Li, N.; Men, W.; Zheng, Y.; Wang, H.; Meng, X. Oroxin B induces apoptosis by down-regulating MicroRNA-221 resulting in the inactivation of the PTEN/PI3K/AKT pathway in liver cancer. Molecules 2019, 24, 4384. [Google Scholar] [CrossRef]
- Trino, S.; Iacobucci, I.; Erriquez, D.; Laurenzana, I.; De Luca, L.; Ferrari, A.; Ghelli Luserna Di Rorà, A.; Papayannidis, C.; Derenzini, E.; Simonetti, G.; et al. Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: A new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget 2016, 7, 12951–12961. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Moreno, V.; Gupta, A.; Kaye, S.B.; Dean, E.; Middleton, M.R.; Friedlander, M.; Gourley, C.; Plummer, R.; Rustin, G.; et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target. Oncol. 2016, 11, 401–415. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Lukey, P.T.; Harrison, S.A.; Yang, S.; Man, Y.; Holman, B.F.; Rashidnasab, A.; Azzopardi, G.; Grayer, M.; Simpson, J.K.; Bareille, P.; et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 3. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Patel, M.; Kahl, B.S.; Horwitz, S.M.; Foss, F.M.; Porcu, P.; Jones, J.; Burger, J.; Jain, N.; Allen, K.; et al. Duvelisib, an oral dual PI3K-δ,γ inhibitor, shows clinical and pharmacodynamic activity in chronic lymphocytic leukemia and small lymphocytic lymphoma in a phase 1 study. Am. J. Hematol. 2018, 93, 1318–1326. [Google Scholar] [CrossRef]
- Gurbuz, V.; Konac, E.; Varol, N.; Yilmaz, A.; Gurocak, S.; Menevse, S.; Sozen, S. Effects of AG490 and S3I-201 on regulation of the JAK/STAT3 signaling pathway in relation to angiogenesis in TRAIL-resistant prostate cancer cells in vitro. Oncol. Lett. 2014, 7, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Kwon, T.-R.; Jeong, S.-J.; Kim, E.-O.; Sohn, E.J.; Yun, M.; Kim, S.-H. Apoptosis induced by tanshinone IIA and cryptotanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2 signaling in chronic myeloid leukemia K562 Cells. Evid. Based Complementary Altern. Med. 2013, 2013, 805639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ren, Y.; Liu, A.; Han, L.; Zhang, K.; Li, S.; Li, P.; Li, P.; Kang, C.; Wang, X.; et al. STAT3 inhibitor WP1066 attenuates miRNA-21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol. Rep. 2014, 31, 2173–2180. [Google Scholar] [CrossRef]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, e2213. [Google Scholar] [CrossRef] [PubMed]
- Gerson, S.L.; Willson, J.K. O6-alkylguanine-DNA alkyltransferase. A target for the modulation of drug resistance. Hematol. Oncol. Clin. N. Am. 1995, 9, 431–450. [Google Scholar] [CrossRef]
- Lee, A.T.J.; Jones, R.L.; Huang, P.H. Pazopanib in advanced soft tissue sarcomas. Signal Transduct. Target. Ther. 2019, 4, 16. [Google Scholar] [CrossRef]
- Dietrich, J.; Wang, D.; Batchelor, T.T. Cediranib: Profile of a novel anti-angiogenic agent in patients with glioblastoma. Expert Opin Investig. Drugs 2009, 18, 1549–1557. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Akgül, S.; Patch, A.-M.; D’Souza, R.C.J.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.H.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural heterogeneity underlies distinct therapy responses and treatment resistance in glioblastoma. Cancers 2019, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Grundy, T.J.; De Leon, E.; Griffin, K.R.; Stringer, B.W.; Day, B.W.; Fabry, B.; Cooper-White, J.; O’Neill, G.M. Differential response of patient-derived primary glioblastoma cells to environmental stiffness. Sci Rep. 2016, 6, 23353. [Google Scholar] [CrossRef]
- Cornelison, R.C.; Yuan, J.X.; Tate, K.M.; Petrosky, A.; Beeghly, G.F.; Bloomfield, M.; Schwager, S.C.; Berr, A.L.; Cimini, D.; Bafakih, F.F.; et al. A patient-designed tissue-engineered model of the infiltrative glioblastoma microenvironment. bioRxiv 2020. [Google Scholar] [CrossRef]
- Caragher, S.; Chalmers, A.J.; Gomez-Roman, N. Glioblastoma’s next top model: Novel culture systems for brain cancer radiotherapy research. Cancers 2019, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, R.C.J.; Offenhauser, C.; Straube, J.; Baumgartner, U.; Kordowski, A.; Li, Y.; Stringer, B.W.; Alexander, H.; Lwin, Z.; Inglis, P.L.; et al. Q-cell glioblastoma resource: Proteomics analysis reveals unique cell-states are maintained in 3D culture. Cells 2020, 9, 267. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef]
- Kazda, T.; Dziacky, A.; Burkon, P.; Pospisil, P.; Slavik, M.; Rehak, Z.; Jancalek, R.; Slampa, P.; Slaby, O.; Lakomy, R. Radiotherapy of glioblastoma 15 years after the landmark stupp’s trial: More controversies than standards? Radiol. Oncol. 2018, 52, 121–128. [Google Scholar] [CrossRef]
- Kanabur, P.; Guo, S.; Simonds, G.R.; Kelly, D.F.; Gourdie, R.G.; Verbridge, S.S.; Sheng, Z. Patient-derived glioblastoma stem cells respond differentially to targeted therapies. Oncotarget 2016, 7, 86406–86419. [Google Scholar] [CrossRef]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef]
- Kim, D.Y.; Choi, B.Y. Costunolide—A bioactive sesquiterpene lactone with diverse therapeutic potential. Int. J. Mol. Sci. 2019, 20, 2926. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.G.; Jeong, Y.H.; Kim, Y.; Choi, Y.J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Ruda, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.-C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A systematic review of glioblastoma-targeted therapies in phases II, III, IV clinical trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A patient-derived cell atlas informs precision targeting of glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Target | Pathway | Phase II-IV Clinical Trial | FDA-Approved | Future Potential | Ref. |
|---|---|---|---|---|---|---|
| 34 Compounds | 19 Compounds 5 (in Trials) 14 (not in Trials) | 16 Compounds | ||||
| gossypol-acetic acid | 5α-reductase 1 and 3α-hydroxysteroid dehydrogenase | Metabolism | − | − | + | [52] |
| AZD5363 | Akt | PI3K/Akt/mTOR | + | − | − | [53] |
| Disulfiram | ALDH | Metabolism | + | − | − | [54] |
| Talampanel | AMPA | + | − | − | [55] | |
| HA14-1 | Bcl-2 | Apoptosis | − | − | + | [56] |
| ABT-263 | Bcl2/Bcl-XL | Apoptosis | − | − | + | [57] |
| Dasatinib | Bcr-Abl, c-Kit, Src | Angiogenesis | + | − | − | [58] |
| Dabrafenib (GSK2118436) | B-Raf | MAPK | + | − | − | [59] |
| Sorafenib | C/B-Raf | MAPK | + | + | − | [60] |
| Palbociclib (PD-0332991) HCl | CDK | Cell cycle | + | − | − | [61] |
| Abemaciclib | CDK4/6 | Cell cycle | + | − | − | [62] |
| Celecoxib | COX-2 | Neuronal signaling | + | − | − | [63] |
| Pexidartinib (PLX3397) | CSF-1R, c-Kit | Growth factor signaling | + | − | − | [64] |
| Plerixafor (AMD3100) | CXCR4 | GPCR and G Protein | − | + | − | [65] |
| Rucaparib (AG-014699) | PARP | DNA damage | + | − | − | [66] |
| Thioguanine | DNA/RNA synthesis | Epigenetics | − | + | − | [67] |
| RITA (NSC 652287) | E3 Ligase, p53 | Apoptosis | − | − | + | [68] |
| Osimertinib (AZD9291) | EGFR | Growth factor signaling | − | + | − | [69] |
| Cetuximab | EGFR | Growth factor signaling | + | − | − | [70] |
| Tazemetostat (EPZ-6438) | EZH2 | Epigenetics | + | − | − | [71] |
| Tipifarnib | farnesyltransferase | Metabolism | + | − | − | [72] |
| Pacritinib (SB1518) | FLT3, JAK | JAK/STAT | + | − | − | [73] |
| Gilteritinib (ASP2215) | FLT3, TAM Receptor | Growth factor signaling | − | + | − | [74] |
| CID 1375606 | GPR27 | G Protein | − | − | + | [75] |
| Trichostatin A | HDAC I and II | Metabolism | + | − | − | [76] |
| Vismodegib (GDC-0449) | Hedgehog/Smoothened | Stem Cells and Wnt signaling | + | + | − | [77] |
| Neratinib (HKI-272) | HER2 | Growth factor signaling | − | + | − | [78] |
| Crizotinib (PF-02341066) | HGFR, c-Met | Growth factor signaling | − | + | − | [79] |
| 2-Methoxyestradiol (2-MeOE2) | HIF | Angiogenesis | + | − | − | [80] |
| Embelin | IAP | Apoptosis | − | + | − | [81] |
| Ivosidenib (AG-120) | IDH1 | Metabolism | − | + | − | [82] |
| Enasidenib (AG-221) | IDH2 | Metabolism | − | + | − | [83] |
| Indoximod (NLG-8189) | IDO | Metabolism | + | − | − | [84] |
| Epacadostat (INCB024360) | IDO1 | Metabolism | − | + | − | [85] |
| Mycophenolate Mofetil | IMPDH | Metabolism | − | + | − | [86] |
| Cilengitide trifluoroacetate | Integrin | Cytoskeletal Signaling | + | − | − | [87] |
| SP600125 | JNK | MAPK | − | − | + | [88] |
| Trametinib (GSK1120212) | MEK | MAPK | + | − | − | [89] |
| Cobimetinib (GDC-0973, RG7420) | MEK | MAPK | + | − | − | [90] |
| Selumetinib (AZD6244) | MEK1/2 | MAPK | + | − | − | [91] |
| U0126-EtOH | MEK1/2 | MAPK | − | − | + | [92] |
| Azacitidine | MGMT | DNA Damage | − | + | − | [93] |
| Everolimus (RAD001) | mTOR | PI3K/Akt/mTOR | + | + | − | [94] |
| AZD8055 | mTORC1 | PI3K/Akt/mTOR | + | − | − | [95] |
| JR-AB2-011 | mTORC2 | PI3K/Akt/mTOR | − | − | + | [96] |
| Bortezomib (PS-341) | NF-κB | Proteases | + | − | − | [97] |
| Parthenolide | HDAC, IKK-β, NF-κB | NF-κB | − | + | − | [98] |
| Isotretinoin | others | others | + | − | − | |
| Oroxin A | Others | Others | − | − | + | |
| Oroxin B | Others | Cancer | − | − | + | [99] |
| Nutlin-3 | P53, Mdm2 | Apoptosis | + | − | − | [100] |
| Veliparib (ABT-888) | PARP | DNA Damage | + | − | − | [101] |
| Olaparib (AZD2281, Ku-0059436) | PARP | DNA Damage | + | − | − | [102] |
| Imatinib Mesylate (STI571) | PDGFR | Growth factor signaling | + | + | − | [103] |
| Omipalisib (GSK2126458, GSK458) | PI3K | PI3K/Akt/mTOR | − | − | + | [104] |
| Duvelisib (IPI-145, INK1197) | PI3K | Angiogenesis | − | + | − | [105] |
| S3I-201 | STAT | JAK/STAT | − | − | + | [106] |
| Cryptotanshinone | STAT | JAK/STAT | − | − | + | [107] |
| WP1066 | STAT3 | JAK/STAT | − | − | + | [108] |
| Costunolide | TERT | DNA Damage | − | − | + | [109] |
| O6-Benzylguanine | Transferase/ AGT | Metabolism | + | − | − | [110] |
| Pazopanib | Tyrosine kinase | Growth factor signaling | + | + | − | [111] |
| Cediranib (AZD2171) | VEGFR | Growth factor signaling | + | − | − | [112] |
| Yap/TAZ inhibitor-1 | YAP/TAZ | Hippo Pathway | − | − | + | [113] |
| Patient | Age (Years) | Gender | Tumor Type | Tumor Site | Survival (Days) | IDH Status | MGMT Status | TERT Prom. Mutations |
|---|---|---|---|---|---|---|---|---|
| FPW1 [46,118] | 68 | Male | Primary glioblastoma | Right temporal | 242 | WT | unmethylated | none |
| SANTB00442 * | 49 | Male | Primary glioblastoma | Left frontal | 99 | WT. | not available | Not available |
| G18-T Cells | FPW1 Cells | ||
|---|---|---|---|
| Drug Name | Target | Drug Name | Target |
| Pazopanib | c-kit, PDGFR, VEGFR | AZD5363 | Akt |
| Disuliram | ALDH | Pexidartinib | CSF-1R, c-Kit |
| RITA | E3 ligase, p53 | Dafrafenib | Raf |
| Oroxin B | ER | S31-201 | STAT |
| 2-methoxyestradiol | GPR30 | Temozolomide | DNA damage |
| Vismodegib | Hh/GLI | ||
| Costunolide | hTERT | ||
| Trametinib | MEK | ||
| AZD8055 | mTOR | ||
| Partenolide | HDAC, IKK-β, NF-κB | ||
| Imatinib Mesylate | PDGFR | ||
| Omipalisib | PI3K/mTOR | ||
| WP1066 | Stat3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenin, S.; Ponthier, E.; Scheer, K.G.; Yeo, E.C.F.; Tea, M.N.; Ebert, L.M.; Oksdath Mansilla, M.; Poonnoose, S.; Baumgartner, U.; Day, B.W.; et al. A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 4322. https://doi.org/10.3390/ijms22094322
Lenin S, Ponthier E, Scheer KG, Yeo ECF, Tea MN, Ebert LM, Oksdath Mansilla M, Poonnoose S, Baumgartner U, Day BW, et al. A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(9):4322. https://doi.org/10.3390/ijms22094322
Chicago/Turabian StyleLenin, Sakthi, Elise Ponthier, Kaitlin G. Scheer, Erica C. F. Yeo, Melinda N. Tea, Lisa M. Ebert, Mariana Oksdath Mansilla, Santosh Poonnoose, Ulrich Baumgartner, Bryan W. Day, and et al. 2021. "A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma" International Journal of Molecular Sciences 22, no. 9: 4322. https://doi.org/10.3390/ijms22094322
APA StyleLenin, S., Ponthier, E., Scheer, K. G., Yeo, E. C. F., Tea, M. N., Ebert, L. M., Oksdath Mansilla, M., Poonnoose, S., Baumgartner, U., Day, B. W., Ormsby, R. J., Pitson, S. M., & Gomez, G. A. (2021). A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. International Journal of Molecular Sciences, 22(9), 4322. https://doi.org/10.3390/ijms22094322