
Coagulation and Fibrinolysis in Obstructive Sleep Apnoea



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
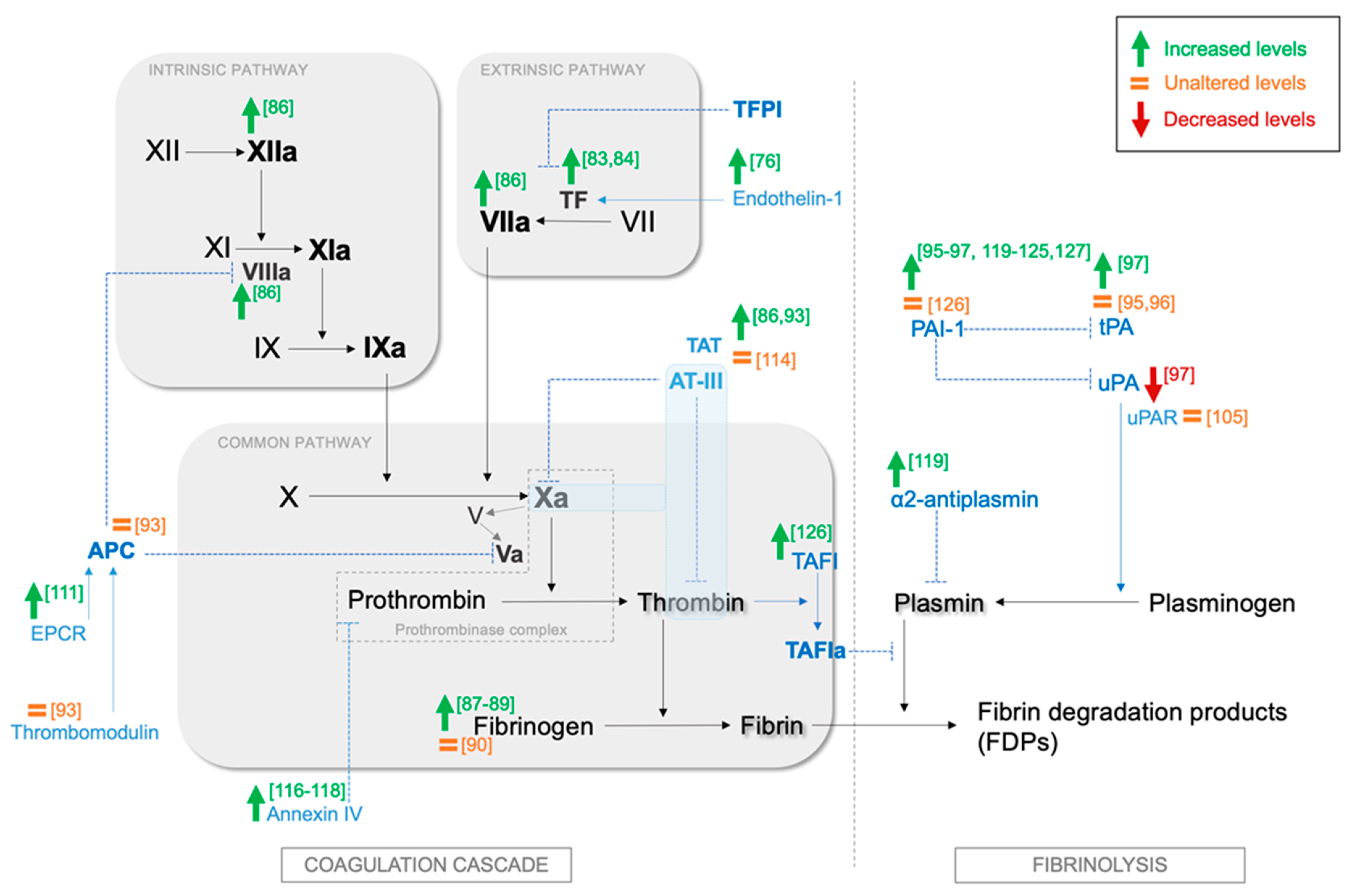
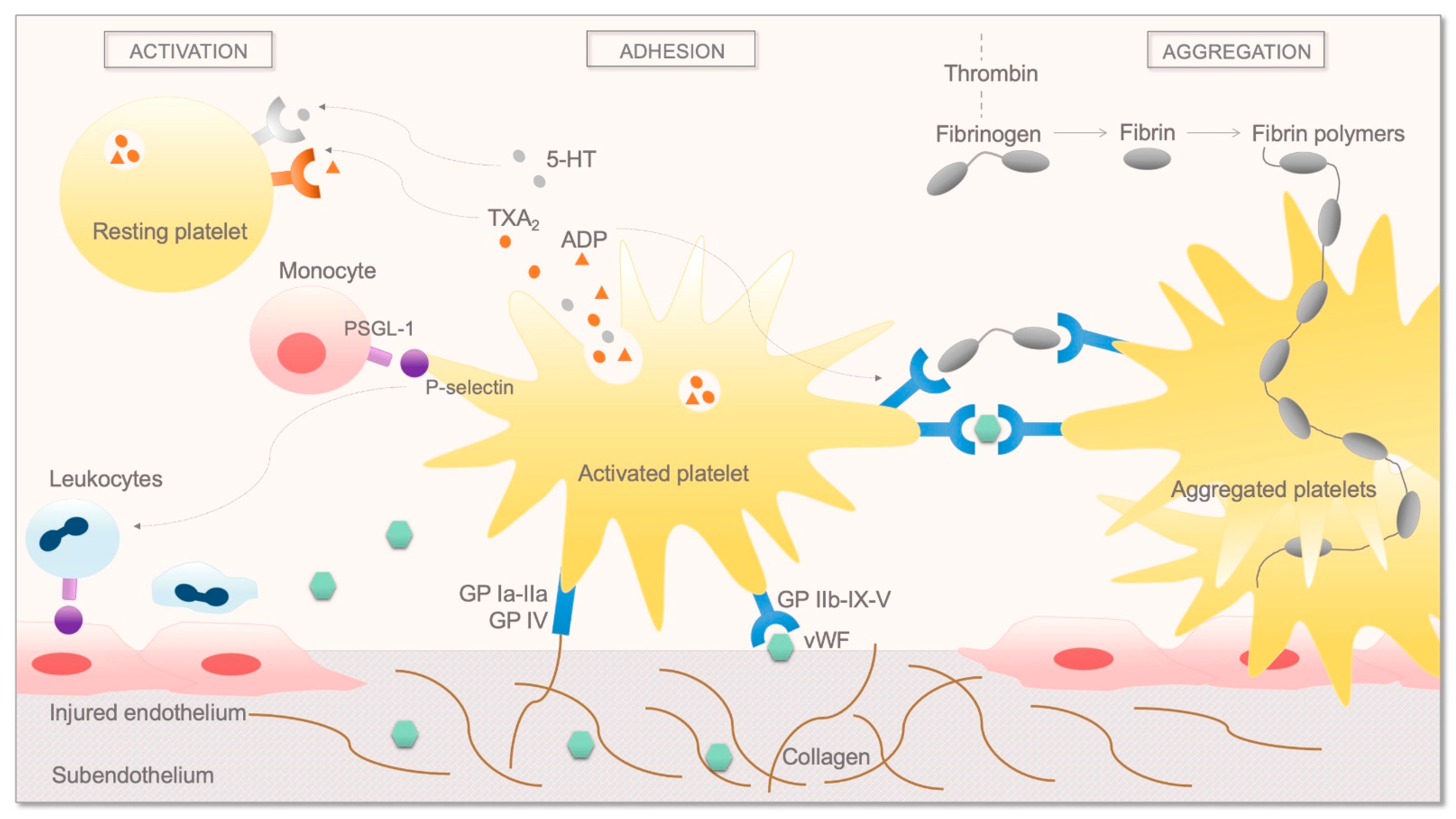
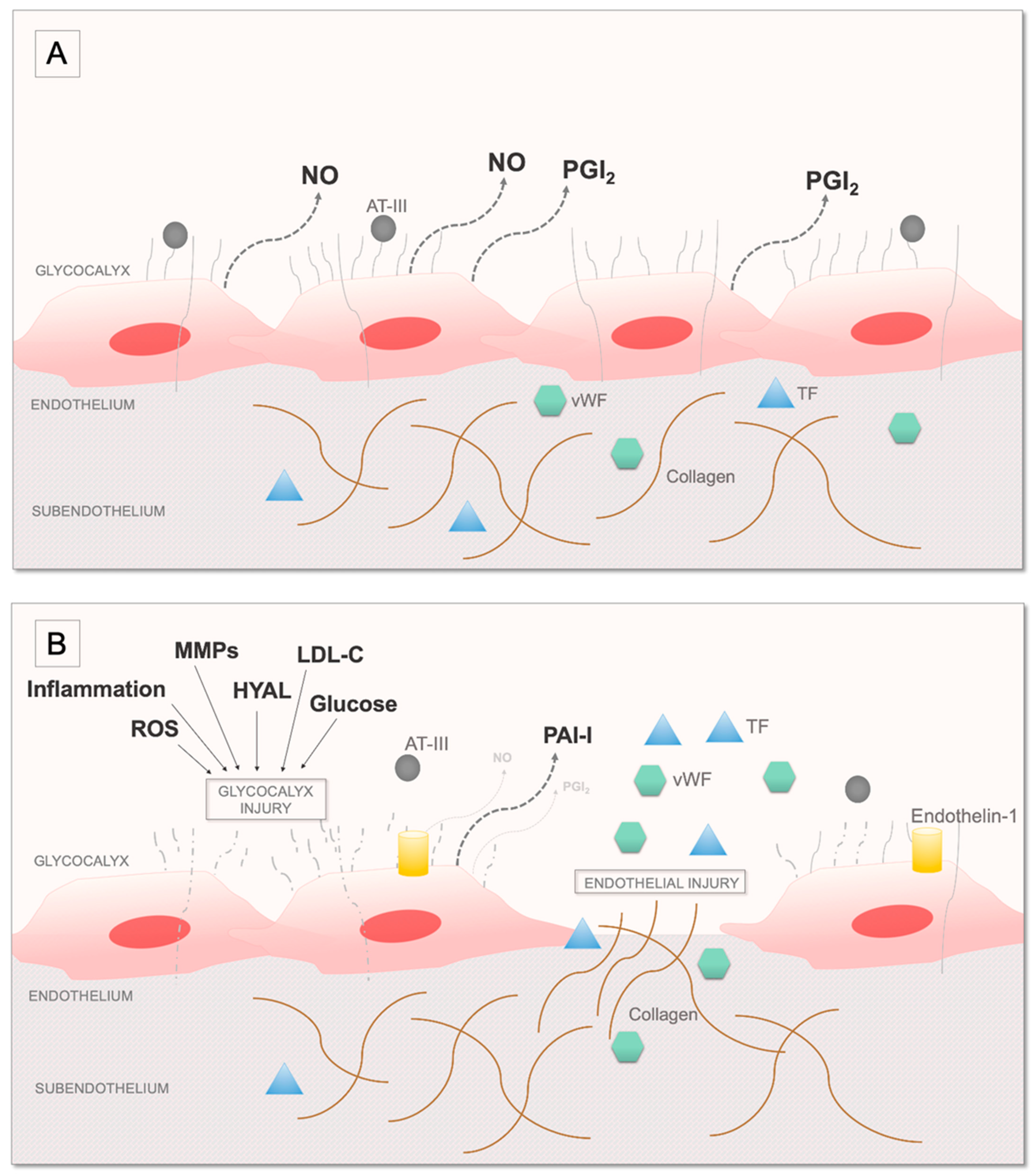
2. Overview of the Coagulation System and Fibrinolysis and the Role of Platelets
3. Current Knowledge on the Effects of OSA on Coagulation, Fibrinolysis, and Platelet Activation
4. The Effect of OSA Treatment on Coagulation, Fibrinolysis, and Platelet Activation
5. Discussion of Major Findings
6. Clinical Implications
7. Implications for Research
8. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohler, M.; Stoewhas, A.C.; Ayers, L.; Senn, O.; Bloch, K.E.; Russi, E.W.; Stradling, J.R. Effects of continuous positive airway pressure therapy withdrawal in patients with obstructive sleep apnea: A randomized controlled trial. Am. J. Respir. Crit. Care Med. 2011, 184, 1192–1199. [Google Scholar] [CrossRef]
- Schwarz, E.I.; Puhan, M.A.; Schlatzer, C.; Stradling, J.R.; Kohler, M. Effect of CPAP therapy on endothelial function in obstructive sleep apnoea: A systematic review and meta-analysis. Respirology 2015, 20, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Dunet, V.; Rey-Bataillard, V.; Allenbach, G.; Beysard, N.; Lovis, A.; Prior, J.O.; Heinzer, R. Effects of continuous positive airway pressure treatment on coronary vasoreactivity measured by (82)Rb cardiac PET/CT in obstructive sleep apnea patients. Sleep Breath. 2016, 20, 673–679. [Google Scholar] [CrossRef]
- Kohler, M.; Stradling, J.R. Mechanisms of vascular damage in obstructive sleep apnea. Nat. Rev. Cardiol. 2010, 7, 677–685. [Google Scholar] [CrossRef]
- Zinchuk, A.V.; Gentry, M.J.; Concato, J.; Yaggi, H.K. Phenotypes in obstructive sleep apnea: A definition, examples and evolution of approaches. Sleep Med. Rev. 2017, 35, 113–123. [Google Scholar] [CrossRef]
- Gabryelska, A.; Białasiewicz, P. Association between excessive daytime sleepiness, REM phenotype and severity of obstructive sleep apnea. Sci. Rep. 2020, 10, 34. [Google Scholar] [CrossRef]
- Sanchez-de-la-Torre, M.; Campos-Rodriguez, F.; Barbe, F. Obstructive sleep apnoea and cardiovascular disease. Lancet Respir. Med. 2013, 1, 61–72. [Google Scholar] [CrossRef]
- von Kanel, R.; Dimsdale, J.E. Hemostatic alterations in patients with obstructive sleep apnea and the implications for cardiovascular disease. Chest 2003, 124, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Li, X.; Zhang, X.; Wang, W.; Chen, J.; Liu, Y.; Fang, X.; Ni, X.; Zhang, J.; Wang, S.; et al. Prothrombotic Factors in Obstructive Sleep Apnea: A Systematic Review with Meta-Analysis. Ear Nose Throat J. 2020, 145561320965208. [Google Scholar] [CrossRef] [PubMed]
- Liak, C.; Fitzpatrick, M. Coagulability in obstructive sleep apnea. Can. Respir. J. 2011, 18, 338–348. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G.; van’t Veer, C.; Cawthern, K.; Butenas, S. The role of the tissue factor pathway in initiation of coagulation. Blood Coagul. Fibrinolysis 1998, 9 (Suppl. S1), S3–S7. [Google Scholar]
- Osterud, B. Tissue factor expression by monocytes: Regulation and pathophysiological roles. Blood Coagul. Fibrinolysis 1998, 9 (Suppl. S1), S9–S14. [Google Scholar] [PubMed]
- Edgington, T.S.; Mackman, N.; Fan, S.T.; Ruf, W. Cellular immune and cytokine pathways resulting in tissue factor expression and relevance to septic shock. Nouv. Rev. Fr. Hematol. 1992, 34, S15–S27. [Google Scholar] [PubMed]
- ten Cate, H.; Bauer, K.A.; Levi, M.; Edgington, T.S.; Sublett, R.D.; Barzegar, S.; Kass, B.L.; Rosenberg, R.D. The activation of factor X and prothrombin by recombinant factor VIIa in vivo is mediated by tissue factor. J. Clin. Investig. 1993, 92, 1207–1212. [Google Scholar] [CrossRef]
- Bryant, J.W.; Shariat-Madar, Z. Human plasma kallikrein-kinin system: Physiological and biochemical parameters. Cardiovasc. Hematol. Agents Med. Chem. 2009, 7, 234–250. [Google Scholar] [CrossRef]
- Doolittle, R.F. Fibrinogen and fibrin. Annu. Rev. Biochem. 1984, 53, 195–229. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.; Alzahrani, S.H.; Mathai, M.; Schroeder, V.; Carter, A.M.; Howell, G.; Koko, T.; Strachan, M.W.; Price, J.F.; Smith, K.A.; et al. A novel mechanism for hypofibrinolysis in diabetes: The role of complement C3. Diabetologia 2012, 55, 1103–1113. [Google Scholar] [CrossRef]
- Whyte, C.S.; Mitchell, J.L.; Mutch, N.J. Platelet-Mediated Modulation of Fibrinolysis. Semin. Thromb. Hemost. 2017, 43, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef]
- Foley, J.H. Plasmin(ogen) at the Nexus of Fibrinolysis, Inflammation, and Complement. Semin. Thromb. Hemost. 2017, 43, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Draxler, D.F.; Sashindranath, M.; Medcalf, R.L. Plasmin: A Modulator of Immune Function. Semin. Thromb. Hemost. 2017, 43, 143–153. [Google Scholar] [CrossRef]
- Miles, L.A.; Greengard, J.S.; Griffin, J.H. A comparison of the abilities of plasma kallikrein, beta-Factor XIIa, Factor XIa and urokinase to activate plasminogen. Thromb. Res. 1983, 29, 407–417. [Google Scholar] [CrossRef]
- Hoylaerts, M.; Rijken, D.C.; Lijnen, H.R.; Collen, D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J. Biol. Chem. 1982, 257, 2912–2919. [Google Scholar] [CrossRef]
- Schneider, M.; Nesheim, M. A study of the protection of plasmin from antiplasmin inhibition within an intact fibrin clot during the course of clot lysis. J. Biol. Chem. 2004, 279, 13333–13339. [Google Scholar] [CrossRef]
- Thorsen, S. The mechanism of plasminogen activation and the variability of the fibrin effector during tissue-type plasminogen activator-mediated fibrinolysis. Ann. N. Y. Acad. Sci. 1992, 667, 52–63. [Google Scholar] [CrossRef]
- Flood, E.C.; Hajjar, K.A. The annexin A2 system and vascular homeostasis. Vasc. Pharmacol. 2011, 54, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Sjowall, C.; Wettero, J. Soluble urokinase plasminogen activator receptor--a valuable biomarker in systemic lupus erythematosus? Clin. Chim. Acta Int. J. Clin. Chem. 2015, 444, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Pedersen, N.; Blasi, F. A soluble, ligand binding mutant of the human urokinase plasminogen activator receptor. J. Biol. Chem. 1991, 266, 8655–8658. [Google Scholar] [CrossRef]
- Takano, K.; Yamaguchi, T.; Uchida, K. Markers of a hypercoagulable state following acute ischemic stroke. Stroke 1992, 23, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Sprengers, E.D.; Kluft, C. Plasminogen activator inhibitors. Blood 1987, 69, 381–387. [Google Scholar] [CrossRef]
- Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Carpenter, S.L.; Mathew, P. Alpha2-antiplasmin and its deficiency: Fibrinolysis out of balance. Haemoph. Off. J. World Fed. Hemoph. 2008, 14, 1250–1254. [Google Scholar] [CrossRef]
- Rijken, D.C.; Uitte de Willige, S. Inhibition of Fibrinolysis by Coagulation Factor XIII. BioMed Res. Int. 2017, 2017, 1209676. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Sussman, I.I.; Hoyer, L. W. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J. Clin. Investig. 1977, 60, 390–404. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Lisman, T.; Weeterings, C.; de Groot, P.G. Platelet aggregation: Involvement of thrombin and fibrin(ogen). Front. Biosci. 2005, 10, 2504–2517. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Hoxie, J.A.; Cunningham, M.; Brass, L.F. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J. Biol. Chem. 1985, 260, 11107–11114. [Google Scholar] [CrossRef]
- Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M.; Stafforini, D.M. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit. Care Med. 2002, 30, S294–S301. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Shao, B.; Miner, J.J.; Yao, L.; Klopocki, A.G.; Maeda, K.; Coggeshall, K.M.; McEver, R.P. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood 2010, 116, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Carlow, D.A.; Gossens, K.; Naus, S.; Veerman, K.M.; Seo, W.; Ziltener, H.J. PSGL-1 function in immunity and steady state homeostasis. Immunol. Rev. 2009, 230, 75–96. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Hartwell, D.; Hrachovinová, I.; Saffaripour, S.; Wagner, D.D. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc. Natl. Acad. Sci. USA 2000, 97, 13835–13840. [Google Scholar] [CrossRef] [PubMed]
- Monković, D.D.; Tracy, P.B. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J. Biol. Chem. 1990, 265, 17132–17140. [Google Scholar] [CrossRef]
- Heemskerk, J.W.; Bevers, E.M.; Lindhout, T. Platelet activation and blood coagulation. Thromb. Haemost. 2002, 88, 186–193. [Google Scholar] [PubMed]
- Michelson, A.D.; Benoit, S.E.; Furman, M.I.; Breckwoldt, W.L.; Rohrer, M.J.; Barnard, M.R.; Loscalzo, J. Effects of nitric oxide/EDRF on platelet surface glycoproteins. Am. J. Physiol. 1996, 270, H1640–H1648. [Google Scholar] [CrossRef] [PubMed]
- McAdam, B.F.; Catella-Lawson, F.; Mardini, I.A.; Kapoor, S.; Lawson, J.A.; FitzGerald, G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: The human pharmacology of a selective inhibitor of COX-2. Proc. Natl. Acad. Sci. USA 1999, 96, 272–277. [Google Scholar] [CrossRef]
- Li, C.; Yang, X.; Feng, J.; Lei, P.; Wang, Y. Proinflammatory and prothrombotic status in emphysematous rats exposed to intermittent hypoxia. Int. J. Clin. Exp. Pathol. 2015, 8, 374–383. [Google Scholar] [PubMed]
- Amitrano, L.; Guardascione, M.A.; Brancaccio, V.; Balzano, A. Coagulation disorders in liver disease. Semin. Liver Dis. 2002, 22, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Gabryelska, A.; Szmyd, B.; Szemraj, J.; Stawski, R.; Sochal, M.; Białasiewicz, P. Patients with obstructive sleep apnea present with chronic upregulation of serum HIF-1α protein. J. Clin. Sleep Med. 2020, 16, 1761–1768. [Google Scholar] [CrossRef]
- Gabryelska, A.; Szmyd, B.; Panek, M.; Szemraj, J.; Kuna, P.; Białasiewicz, P. Serum hypoxia-inducible factor-1α protein level as a diagnostic marker of obstructive sleep apnea. Pol. Arch. Intern. Med. 2020, 130, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Li, N.; Yao, X.; Zhou, L. Potential inflammatory markers in obstructive sleep apnea-hypopnea syndrome. Bosn. J. Basic Med. Sci. 2017, 17, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Gabryelska, A.; Stawski, R.; Sochal, M.; Szmyd, B.; Białasiewicz, P. Influence of one-night CPAP therapy on the changes of HIF-1α protein in OSA patients: A pilot study. J. Sleep Res. 2020, 29, e12995. [Google Scholar] [CrossRef]
- Gabryelska, A.; Sochal, M.; Turkiewicz, S.; Białasiewicz, P. Relationship between HIF-1 and Circadian Clock Proteins in Obstructive Sleep Apnea Patients-Preliminary Study. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Taylor, C.T.; Cummins, E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. N. Y. Acad. Sci. 2009, 1177, 178–184. [Google Scholar] [CrossRef]
- Mackman, N.; Brand, K.; Edgington, T.S. Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. J. Exp. Med. 1991, 174, 1517–1526. [Google Scholar] [CrossRef]
- Begbie, M.; Notley, C.; Tinlin, S.; Sawyer, L.; Lillicrap, D. The Factor VIII acute phase response requires the participation of NFkappaB and C/EBP. Thromb. Haemost. 2000, 84, 216–222. [Google Scholar]
- Liu, W.; Shen, S.-M.; Zhao, X.-Y.; Chen, G.-Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar] [PubMed]
- Ten, V.S.; Pinsky, D.J. Endothelial response to hypoxia: Physiologic adaptation and pathologic dysfunction. Curr. Opin. Crit. Care 2002, 8, 242–250. [Google Scholar] [CrossRef]
- Delaney, C.; Davizon-Castillo, P.; Allawzi, A.; Posey, J.; Gandjeva, A.; Neeves, K.; Tuder, R.M.; Paola, J.D.; Stenmark, K.R.; Nozik, E.S. Platelet activation contributes to hypoxia-induced inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L413–L421. [Google Scholar] [CrossRef]
- Wedzicha, J.A.; Syndercombe-Court, D.; Tan, K.C. Increased platelet aggregate formation in patients with chronic airflow obstruction and hypoxaemia. Thorax 1991, 46, 504–507. [Google Scholar] [CrossRef]
- Rahangdale, S.; Yeh, S.Y.; Novack, V.; Stevenson, K.; Barnard, M.R.; Furman, M.I.; Frelinger, A.L.; Michelson, A.D.; Malhotra, A. The influence of intermittent hypoxemia on platelet activation in obese patients with obstructive sleep apnea. J. Clin. Sleep Med. 2011, 7, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kiouptsi, K.; Gambaryan, S.; Walter, E.; Walter, U.; Jurk, K.; Reinhardt, C. Hypoxia impairs agonist-induced integrin αIIbβ3 activation and platelet aggregation. Sci. Rep. 2017, 7, 7621. [Google Scholar] [CrossRef]
- Ferreira, C.B.; Schoorlemmer, G.H.; Rocha, A.A.; Cravo, S.L. Increased sympathetic responses induced by chronic obstructive sleep apnea are caused by sleep fragmentation. J. Appl. Physiol. (1985) 2020, 129, 163–172. [Google Scholar] [CrossRef]
- Olson, L.J.; Olson, E.J.; Somers, V.K. Obstructive Sleep Apnea and Platelet Activation: Another Potential Link Between Sleep-Disordered Breathing and Cardiovascular Disease. Chest 2004, 126, 339–341. [Google Scholar] [CrossRef]
- von Känel, R.; Dimsdale, J.E. Effects of sympathetic activation by adrenergic infusions on hemostasis in vivo. Eur. J. Haematol. 2000, 65, 357–369. [Google Scholar] [CrossRef]
- Eisensehr, I.; Ehrenberg, B.L.; Noachtar, S.; Korbett, K.; Byrne, A.; McAuley, A.; Palabrica, T. Platelet activation, epinephrine, and blood pressure in obstructive sleep apnea syndrome. Neurology 1998, 51, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; ten Cate, H.; Bauer, K.A.; van der Poll, T.; Edgington, T.S.; Büller, H.R.; van Deventer, S.J.; Hack, C.E.; ten Cate, J.W.; Rosenberg, R.D. Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J. Clin. Investig. 1994, 93, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005, 112, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Gabryelska, A.; Łukasik, Z.M.; Makowska, J.S.; Białasiewicz, P. Obstructive Sleep Apnea: From Intermittent Hypoxia to Cardiovascular Complications via Blood Platelets. Front. Neurol. 2018, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, T.; Ahmad, S.; Gupta, N.; Sahu, A.; Ahmad, Y.; Nair, V.; Chatterjee, T.; Bajaj, N.; Sengupta, S.; Ganju, L.; et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood 2014, 123, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B., Jr. The inflammatory-coagulant axis in the host response to gram-negative sepsis: Regulatory roles of proteins and inhibitors of tissue factor. New Horiz. 1994, 2, 555–565. [Google Scholar] [PubMed]
- Hoyos, C.M.; Melehan, K.L.; Liu, P.Y.; Grunstein, R.R.; Phillips, C.L. Does obstructive sleep apnea cause endothelial dysfunction? A critical review of the literature. Sleep Med. Rev. 2015, 20, 15–26. [Google Scholar] [CrossRef]
- Napoleone, E.; Di Santo, A.; Lorenzet, R. Monocytes upregulate endothelial cell expression of tissue factor: A role for cell-cell contact and cross-talk. Blood 1997, 89, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, R.; Parthasarathy, S.; Quan, S.F. Endothelial dysfunction in obstructive sleep apnea. J. Clin. Sleep Med. 2007, 3, 409–415. [Google Scholar] [CrossRef]
- Phillips, B.G.; Narkiewicz, K.; Pesek, C.A.; Haynes, W.G.; Dyken, M.E.; Somers, V.K. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J. Hypertens. 1999, 17, 61–66. [Google Scholar] [CrossRef]
- Halim, A.; Kanayama, N.; el Maradny, E.; Maehara, K.; Masahiko, H.; Terao, T. Endothelin-1 increased immunoreactive von Willebrand factor in endothelial cells and induced micro thrombosis in rats. Thromb. Res. 1994, 76, 71–78. [Google Scholar] [CrossRef]
- Kambas, K.; Chrysanthopoulou, A.; Kourtzelis, I.; Skordala, M.; Mitroulis, I.; Rafail, S.; Vradelis, S.; Sigalas, I.; Wu, Y.Q.; Speletas, M.; et al. Endothelin-1 signaling promotes fibrosis in vitro in a bronchopulmonary dysplasia model by activating the extrinsic coagulation cascade. J. Immunol. 2011, 186, 6568–6575. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Onisto, M.; Lupo, A.; Gambaro, G. Glycosaminoglycans, proteoglycans and sulodexide and the endothelium: Biological roles and pharmacological effects. Int. Angiol. J. Int. Union Angiol. 2014, 33, 243–254. [Google Scholar]
- Rosenberg, R.D.; Shworak, N.W.; Liu, J.; Schwartz, J.J.; Zhang, L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J. Clin. Investig. 1997, 99, 2062–2070. [Google Scholar] [CrossRef]
- Meszaros, M.; Kis, A.; Kunos, L.; Tarnoki, A.D.; Tarnoki, D.L.; Lazar, Z.; Bikov, A. The role of hyaluronic acid and hyaluronidase-1 in obstructive sleep apnoea. Sci. Rep. 2020, 10, 19484. [Google Scholar] [CrossRef]
- Félétou, M.; Vanhoutte, P.M. Endothelial dysfunction: A multifaceted disorder (The Wiggers Award Lecture). Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H985–H1002. [Google Scholar] [CrossRef]
- El Solh, A.A.; Akinnusi, M.E.; Berim, I.G.; Peter, A.M.; Paasch, L.L.; Szarpa, K.R. Hemostatic implications of endothelial cell apoptosis in obstructive sleep apnea. Sleep Breath. 2008, 12, 331–337. [Google Scholar] [CrossRef]
- Hayashi, M.; Fujimoto, K.; Urushibata, K.; Takamizawa, A.; Kinoshita, O.; Kubo, K. Hypoxia-sensitive molecules may modulate the development of atherosclerosis in sleep apnoea syndrome. Respirology 2006, 11, 24–31. [Google Scholar] [CrossRef]
- von Känel, R.; Loredo, J.S.; Ancoli-Israel, S.; Mills, P.J.; Natarajan, L.; Dimsdale, J.E. Association between polysomnographic measures of disrupted sleep and prothrombotic factors. Chest 2007, 131, 733–739. [Google Scholar] [CrossRef]
- Robinson, G.V.; Pepperell, J.C.; Segal, H.C.; Davies, R.J.; Stradling, J.R. Circulating cardiovascular risk factors in obstructive sleep apnoea: Data from randomised controlled trials. Thorax 2004, 59, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Xu, F.; Babineau, D.C.; Tracy, R.P.; Jenny, N.S.; Patel, S.R.; Redline, S. Sleep-disordered breathing and prothrombotic biomarkers: Cross-sectional results of the Cleveland Family Study. Am. J. Respir. Crit. Care Med. 2010, 182, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Peled, N.; Kassirer, M.; Kramer, M.R.; Rogowski, O.; Shlomi, D.; Fox, B.; Berliner, A.S.; Shitrit, D. Increased erythrocyte adhesiveness and aggregation in obstructive sleep apnea syndrome. Thromb. Res. 2008, 121, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Wessendorf, T.E.; Thilmann, A.F.; Wang, Y.M.; Schreiber, A.; Konietzko, N.; Teschler, H. Fibrinogen levels and obstructive sleep apnea in ischemic stroke. Am. J. Respir. Crit. Care Med. 2000, 162, 2039–2042. [Google Scholar] [CrossRef]
- Akyüz, A.; Akkoyun, D.Ç.; Oran, M.; Değirmenci, H.; Alp, R. Mean platelet volume in patients with obstructive sleep apnea and its relationship with simpler heart rate derivatives. Cardiol. Res. Pract. 2014, 2014, 454701. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, E.K.; Dunoyer-Geindre, S. Human tissue-type plasminogen activator. Thromb. Haemost. 2014, 112, 243–254. [Google Scholar] [CrossRef]
- Kent, B.D.; Ryan, S.; McNicholas, W.T. Obstructive sleep apnea and inflammation: Relationship to cardiovascular co-morbidity. Respir. Physiol. Neurobiol. 2011, 178, 475–481. [Google Scholar] [CrossRef]
- Takagi, T.; Morser, J.; Gabazza, E.C.; Qin, L.; Fujiwara, A.; Naito, M.; Yamaguchi, A.; Kobayashi, T.; D’Alessandro-Gabazza, C.N.; Boveda Ruiz, D.; et al. The coagulation and protein C pathways in patients with sleep apnea. Lung 2009, 187, 209–213. [Google Scholar] [CrossRef]
- Zhang, X.B.; Jiang, X.T.; Cai, F.R.; Zeng, H.Q.; Du, Y.P. Vascular endothelial growth factor levels in patients with obstructive sleep apnea: A meta-analysis. Eur. Arch. Oto-Rhino-Laryngol. 2017, 274, 661–670. [Google Scholar] [CrossRef]
- Bagai, K.; Muldowney, J.A., 3rd; Song, Y.; Wang, L.; Bagai, J.; Artibee, K.J.; Vaughan, D.E.; Malow, B.A. Circadian variability of fibrinolytic markers and endothelial function in patients with obstructive sleep apnea. Sleep 2014, 37, 359–367. [Google Scholar] [CrossRef]
- Rångemark, C.; Hedner, J.A.; Carlson, J.T.; Gleerup, G.; Winther, K. Platelet function and fibrinolytic activity in hypertensive and normotensive sleep apnea patients. Sleep 1995, 18, 188–194. [Google Scholar] [CrossRef]
- Steffanina, A.; Proietti, L.; Antonaglia, C.; Palange, P.; Angelici, E.; Canipari, R. The Plasminogen System and Transforming Growth Factor-beta in Subjects with Obstructive Sleep Apnea Syndrome: Effects of CPAP Treatment. Respir. Care 2015, 60, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Lyngbaek, S.; Sehestedt, T.; Marott, J.L.; Hansen, T.W.; Olsen, M.H.; Andersen, O.; Linneberg, A.; Madsbad, S.; Haugaard, S.B.; Eugen-Olsen, J.; et al. CRP and suPAR are differently related to anthropometry and subclinical organ damage. Int. J. Cardiol. 2013, 167, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Baran, M.; Mollers, L.N.; Andersson, S.; Jonsson, I.M.; Ekwall, A.K.; Bjersing, J.; Tarkowski, A.; Bokarewa, M. Survivin is an essential mediator of arthritis interacting with urokinase signalling. J. Cell. Mol. Med. 2009, 13, 3797–3808. [Google Scholar] [CrossRef] [PubMed]
- Kunos, L.; Horvath, P.; Kis, A.; Tarnoki, D.L.; Tarnoki, A.D.; Lazar, Z.; Bikov, A. Circulating Survivin Levels in Obstructive Sleep Apnoea. Lung 2018, 196, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Stavaras, C.; Pastaka, C.; Papala, M.; Gravas, S.; Tzortzis, V.; Melekos, M.; Seitanidis, G.; Gourgoulianis, K.I. Sexual function in pre- and post-menopausal women with obstructive sleep apnea syndrome. Int. J. Impot. Res. 2012, 24, 228–233. [Google Scholar] [CrossRef]
- Kadir, R.R.A.; Bayraktutan, U. Urokinase Plasminogen Activator: A Potential Thrombolytic Agent for Ischaemic Stroke. Cell. Mol. Neurobiol. 2020, 40, 347–355. [Google Scholar] [CrossRef]
- Gozal, D.; Jortani, S.; Snow, A.B.; Kheirandish-Gozal, L.; Bhattacharjee, R.; Kim, J.; Capdevila, O.S. Two-dimensional differential in-gel electrophoresis proteomic approaches reveal urine candidate biomarkers in pediatric obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2009, 180, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- von Känel, R.; Malan, N.T.; Hamer, M.; Lambert, G.W.; Schlaich, M.; Reimann, M.; Malan, L. Three-year changes of prothrombotic factors in a cohort of South Africans with a high clinical suspicion of obstructive sleep apnea. Thromb. Haemost. 2016, 115, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Bocskei, R.M.; Meszaros, M.; Tarnoki, A.D.; Tarnoki, D.L.; Kunos, L.; Lazar, Z.; Bikov, A. Circulating Soluble Urokinase-Type Plasminogen Activator Receptor in Obstructive Sleep Apnoea. Medicina (Kaunas, Lithuania) 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Moller, B.; Todd, R.F., 3rd; Christensen, T.; Andreasen, P.A.; Gliemann, J.; Petersen, C.M. Urokinase receptor. An activation antigen in human T lymphocytes. J. Immunol. 1994, 152, 505–516. [Google Scholar]
- Chavakis, T.; Willuweit, A.K.; Lupu, F.; Preissner, K.T.; Kanse, S.M. Release of soluble urokinase receptor from vascular cells. Thromb. Haemost. 2001, 86, 686–693. [Google Scholar] [CrossRef]
- Thuno, M.; Macho, B.; Eugen-Olsen, J. suPAR: The molecular crystal ball. Disease Markers 2009, 27, 157–172. [Google Scholar] [CrossRef]
- Lund, L.R.; Ellis, V.; Ronne, E.; Pyke, C.; Dano, K. Transcriptional and post-transcriptional regulation of the receptor for urokinase-type plasminogen activator by cytokines and tumour promoters in the human lung carcinoma cell line A549. Biochem. J. 1995, 310, 345–352. [Google Scholar] [CrossRef]
- Shetty, S.; Idell, S. A urokinase receptor mRNA binding protein from rabbit lung fibroblasts and mesothelial cells. Am. J. Physiol. 1998, 274, L871–L882. [Google Scholar] [CrossRef]
- Kohli, M.; Sharma, S.K.; Upadhyay, V.; Varshney, S.; Sengupta, S.; Basak, T.; Sreenivas, V. Urinary EPCR and dermcidin as potential novel biomarkers for severe adult OSA patients. Sleep Med. 2019, 64, 92–100. [Google Scholar] [CrossRef]
- Keeling, D.M.; Wilson, A.J.; Mackie, I.J.; Isenberg, D.A.; Machin, S.J. Role of beta 2-glycoprotein I and anti-phospholipid antibodies in activation of protein C in vitro. J. Clin. Pathol. 1993, 46, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhou, L.; Cao, Y.; Chen, P.; Chen, Y.; Zong, D.; Ouyang, R. Relation between serum leptin levels, lipid profiles and neurocognitive deficits in Chinese OSAHS patients. Int. J. Neurosci. 2017, 127, 981–987. [Google Scholar] [CrossRef]
- García Suquia, A.; Alonso-Fernández, A.; de la Peña, M.; Romero, D.; Piérola, J.; Carrera, M.; Barceló, A.; Soriano, J.B.; Arque, M.; Fernández-Capitán, C.; et al. High D-dimer levels after stopping anticoagulants in pulmonary embolism with sleep apnoea. Eur. Respir. J. 2015, 46, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Russo-Marie, F. Annexin V and phospholipid metabolism. Clin. Chem. Lab. Med. 1999, 37, 287–291. [Google Scholar] [CrossRef]
- El Solh, A.A.; Akinnusi, M.E.; Baddoura, F.H.; Mankowski, C.R. Endothelial cell apoptosis in obstructive sleep apnea: A link to endothelial dysfunction. Am. J. Respir. Crit. Care Med. 2007, 175, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Bikov, A.; Kunos, L.; Pállinger, É.; Lázár, Z.; Kis, A.; Horváth, G.; Losonczy, G.; Komlósi, Z.I. Diurnal variation of circulating microvesicles is associated with the severity of obstructive sleep apnoea. Sleep Breath. 2017, 21, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Jung, K.H.; Chu, K.; Kim, S.H.; Ji, K.H.; Park, H.K.; Kim, H.C.; Lee, S.T.; Lee, S.K.; Roh, J.K. Increased circulating endothelial microparticles and carotid atherosclerosis in obstructive sleep apnea. J. Clin. Neurol. (Seoul, Korea) 2010, 6, 89–98. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Zakrzewska, E.; Kicinski, P.; Przybylska-Kuc, S.; Dybala, A.; Myslinski, W.; Pastryk, J.; Tomaszewski, T.; Mosiewicz, J. Evaluation of Fibrinolytic Inhibitors: Alpha-2-Antiplasmin and Plasminogen Activator Inhibitor 1 in Patients with Obstructive Sleep Apnoea. PLoS ONE 2016, 11, e0166725. [Google Scholar] [CrossRef]
- Martin, R.A.; Strosnider, C.; Giersch, G.; Womack, C.J.; Hargens, T.A. The effect of acute aerobic exercise on hemostasis in obstructive sleep apnea. Sleep Breath. 2017, 21, 623–629. [Google Scholar] [CrossRef]
- von Känel, R.; Natarajan, L.; Ancoli-Israel, S.; Mills, P.J.; Wolfson, T.; Gamst, A.C.; Loredo, J.S.; Dimsdale, J.E. Effect of continuous positive airway pressure on day/night rhythm of prothrombotic markers in obstructive sleep apnea. Sleep Med. 2013, 14, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, M.; Karttunen, V.; Wartiovaara-Kautto, U.; Riutta, A.; Uchiyama, S.; Hillbom, M. Fibrinolytic activity and platelet function in subjects with obstructive sleep apnoea and a patent foramen ovale: Is there an option for prevention of ischaemic stroke? Stroke Res. Treat. 2012, 2012, 945849. [Google Scholar] [CrossRef][Green Version]
- Ifergane, G.; Ovanyan, A.; Toledano, R.; Goldbart, A.; Abu-Salame, I.; Tal, A.; Stavsky, M.; Novack, V. Obstructive Sleep Apnea in Acute Stroke: A Role for Systemic Inflammation. Stroke 2016, 47, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- von Känel, R.; Natarajan, L.; Ancoli-Israel, S.; Mills, P.J.; Loredo, J.S.; Dimsdale, J.E. Day/Night rhythm of hemostatic factors in obstructive sleep apnea. Sleep 2010, 33, 371–377. [Google Scholar] [CrossRef]
- Gileles-Hillel, A.; Alonso-Álvarez, M.L.; Kheirandish-Gozal, L.; Peris, E.; Cordero-Guevara, J.A.; Terán-Santos, J.; Martinez, M.G.; Jurado-Luque, M.J.; Corral-Peñafiel, J.; Duran-Cantolla, J.; et al. Inflammatory Markers and Obstructive Sleep Apnea in Obese Children: The NANOS Study. Mediat. Inflamm. 2014, 2014, 605280. [Google Scholar] [CrossRef]
- Niżankowska-Jędrzejczyk, A.; Almeida, F.R.; Lowe, A.A.; Kania, A.; Nastałek, P.; Mejza, F.; Foley, J.H.; Niżankowska-Mogilnicka, E.; Undas, A. Modulation of inflammatory and hemostatic markers in obstructive sleep apnea patients treated with mandibular advancement splints: A parallel, controlled trial. J. Clin. Sleep Med. 2014, 10, 255–262. [Google Scholar] [CrossRef]
- von Känel, R.; Loredo, J.S.; Ancoli-Israel, S.; Mills, P.J.; Dimsdale, J.E. Elevated plasminogen activator inhibitor 1 in sleep apnea and its relation to the metabolic syndrome: An investigation in 2 different study samples. Metabolism 2007, 56, 969–976. [Google Scholar] [CrossRef]
- Liao, H.; Hyman, M.C.; Lawrence, D.A.; Pinsky, D.J. Molecular regulation of the PAI-1 gene by hypoxia: Contributions of Egr-1, HIF-1alpha, and C/EBPalpha. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen Activator Inhibitor Type-1 as a Regulator of Fibrosis. J. Cell. Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Pákó, J.; Kunos, L.; Mészáros, M.; Tárnoki, D.L.; Tárnoki, Á.D.; Horváth, I.; Bikov, A. Decreased Levels of Anti-Aging Klotho in Obstructive Sleep Apnea. Rejuvenation Res. 2020, 23, 256–261. [Google Scholar] [CrossRef]
- Takeshita, K.; Yamamoto, K.; Ito, M.; Kondo, T.; Matsushita, T.; Hirai, M.; Kojima, T.; Nishimura, M.; Nabeshima, Y.; Loskutoff, D.J.; et al. Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “Klotho” mouse. Semin. Thromb. Hemost. 2002, 28, 545–554. [Google Scholar] [CrossRef]
- Inoue, K.; Takano, H.; Yanagisawa, R.; Sakurai, M.; Shimada, A.; Sato, M.; Yoshino, S.; Yoshikawa, T. Role of interleukin-6 in fibrinolytic changes induced by lipopolysaccharide in mice. Blood Coagul. Fibrinolysis 2006, 17, 307–309. [Google Scholar] [CrossRef]
- Miljić, P.; Heylen, E.; Willemse, J.; Djordjević, V.; Radojković, D.; Colović, M.; Elezović, I.; Hendriks, D. Thrombin activatable fibrinolysis inhibitor (TAFI): A molecular link between coagulation and fibrinolysis. Srp. Arh. Celok. Lek. 2010, 138 (Suppl. S1), 74–78. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Tarnoki, D.L.; Tarnoki, A.D.; Karlinger, K.; Lazar, Z.; Losonczy, G.; Kunos, L.; Bikov, A. Complement system activation in obstructive sleep apnea. J. Sleep Res. 2018, 27, e12674. [Google Scholar] [CrossRef]
- Hui, D.S.; Ko, F.W.; Fok, J.P.; Chan, M.C.; Li, T.S.; Tomlinson, B.; Cheng, G. The effects of nasal continuous positive airway pressure on platelet activation in obstructive sleep apnea syndrome. Chest 2004, 125, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Akinnusi, M.E.; Paasch, L.L.; Szarpa, K.R.; Wallace, P.K.; El Solh, A.A. Impact of Nasal Continuous Positive Airway Pressure Therapy on Markers of Platelet Activation in Patients with Obstructive Sleep Apnea. Respiration 2009, 77, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bokinsky, G.; Miller, M.; Ault, K.; Husband, P.; Mitchell, J. Spontaneous platelet activation and aggregation during obstructive sleep apnea and its response to therapy with nasal continuous positive airway pressure. A preliminary investigation. Chest 1995, 108, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Geiser, T.; Buck, F.; Meyer, B.J.; Bassetti, C.; Haeberli, A.; Gugger, M. In vivo platelet activation is increased during sleep in patients with obstructive sleep apnea syndrome. Respiration 2002, 69, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, W.H.; Oswald, J.; Walter, R.; Kuhn, M. Blood viscosity and platelet function in patients with obstructive sleep apnea syndrome treated with nasal continuous positive airway pressure. Clin. Hemorheol. Microcirc. 2002, 27, 201–207. [Google Scholar] [PubMed]
- Michelson, A.D.; Ellis, P.A.; Barnard, M.R.; Matic, G.B.; Viles, A.F.; Kestin, A.S. Downregulation of the platelet surface glycoprotein Ib-IX complex in whole blood stimulated by thrombin, adenosine diphosphate, or an in vivo wound. Blood 1991, 77, 770–779. [Google Scholar] [CrossRef]
- Horváth, P.; Lázár, Z.; Gálffy, G.; Puskás, R.; Kunos, L.; Losonczy, G.; Mészáros, M.; Tárnoki, Á.D.; Tárnoki, D.L.; Bikov, A. Circulating P-Selectin Glycoprotein Ligand 1 and P-Selectin Levels in Obstructive Sleep Apnea Patients. Lung 2020, 198, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, H.M.; Cofta, S.; Bielawska, L.; Płóciniczak, A.; Piorunek, T.; Wysocka, E. Circulating P-Selectin and Its Glycoprotein Ligand in Nondiabetic Obstructive Sleep Apnea Patients. Adv. Exp. Med. Biol. 2020, 1279, 61–69. [Google Scholar] [CrossRef]
- Bravo Mde, L.; Serpero, L.D.; Barceló, A.; Barbé, F.; Agustí, A.; Gozal, D. Inflammatory proteins in patients with obstructive sleep apnea with and without daytime sleepiness. Sleep Breath. 2007, 11, 177–185. [Google Scholar] [CrossRef]
- Zamarrón-Sanz, C.; Ricoy-Galbaldon, J.; Gude-Sampedro, F.; Riveiro-Riveiro, A. Plasma levels of vascular endothelial markers in obstructive sleep apnea. Arch. Med. Res. 2006, 37, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar]
- Ayers, L.; Ferry, B.; Craig, S.; Nicoll, D.; Stradling, J.R.; Kohler, M. Circulating cell-derived microparticles in patients with minimally symptomatic obstructive sleep apnoea. Eur. Respir. J. 2009, 33, 574–580. [Google Scholar] [CrossRef]
- Maruyama, K.; Morishita, E.; Sekiya, A.; Omote, M.; Kadono, T.; Asakura, H.; Hashimoto, M.; Kobayashi, M.; Nakatsumi, Y.; Takada, S.; et al. Plasma levels of platelet-derived microparticles in patients with obstructive sleep apnea syndrome. J. Atheroscler. Thromb. 2012, 19, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.N.; Yun, H.C.; Yoo, J.H.; Lee, S.H. Association Between Hypercoagulability and Severe Obstructive Sleep Apnea. JAMA Otolaryngol. Head Neck Surg. 2017, 143, 996–1002. [Google Scholar] [CrossRef]
- Barceló, A.; Morell-Garcia, D.; Sanchís, P.; Peña-Zarza, J.A.; Bauça, J.M.; Piérola, J.; Peña, M.; Toledo-Pons, N.; Giménez, P.; Ribot, C.; et al. Prothrombotic state in children with obstructive sleep apnea. Sleep Med. 2019, 53, 101–105. [Google Scholar] [CrossRef]
- Zicari, A.M.; Occasi, F.; Di Mauro, F.; Lollobrigida, V.; Di Fraia, M.; Savastano, V.; Loffredo, L.; Nicita, F.; Spalice, A.; Duse, M. Mean Platelet Volume, Vitamin D and C Reactive Protein Levels in Normal Weight Children with Primary Snoring and Obstructive Sleep Apnea Syndrome. PLoS ONE 2016, 11, e0152497. [Google Scholar] [CrossRef] [PubMed]
- Saygin, M.; Ozturk, O.; Ozguner, M.F.; Akkaya, A.; Varol, E. Hematological Parameters as Predictors of Cardiovascular Disease in Obstructive Sleep Apnea Syndrome Patients. Angiology 2016, 67, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Dahlbäck, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef]
- Phillips, C.L.; McEwen, B.J.; Morel-Kopp, M.C.; Yee, B.J.; Sullivan, D.R.; Ward, C.M.; Tofler, G.H.; Grunstein, R.R. Effects of continuous positive airway pressure on coagulability in obstructive sleep apnoea: A randomised, placebo-controlled crossover study. Thorax 2012, 67, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; Kita, H.; Noguchi, T.; Otsuka, N.; Tsuboi, T.; Nakamura, T.; Shimizu, K.; Mishima, M.; Ohi, M. Improvement of factor VII clotting activity following long-term NCPAP treatment in obstructive sleep apnoea syndrome. QJM 1998, 91, 627–633. [Google Scholar] [CrossRef][Green Version]
- Dorkova, Z.; Petrasova, D.; Molcanyiova, A.; Popovnakova, M.; Tkacova, R. Effects of Continuous Positive Airway Pressure on Cardiovascular Risk Profile in Patients with Severe Obstructive Sleep Apnea and Metabolic Syndrome. Chest 2008, 134, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; Ohi, M.; Kita, H.; Noguchi, T.; Otsuka, N.; Tsuboi, T.; Mishima, M.; Kuno, K. Effects of NCPAP therapy on fibrinogen levels in obstructive sleep apnea syndrome. Am. J. Respir. Crit. Care Med. 1996, 153, 1972–1976. [Google Scholar] [CrossRef] [PubMed]
- von Kanel, R.; Loredo, J.S.; Ancoli-Israel, S.; Dimsdale, J.E. Association between sleep apnea severity and blood coagulability: Treatment effects of nasal continuous positive airway pressure. Sleep Breath. 2006, 10, 139–146. [Google Scholar] [CrossRef]
- Kheirandish-Gozal, L.; Gileles-Hillel, A.; Alonso-Alvarez, M.L.; Peris, E.; Bhattacharjee, R.; Teran-Santos, J.; Duran-Cantolla, J.; Gozal, D. Effects of adenotonsillectomy on plasma inflammatory biomarkers in obese children with obstructive sleep apnea: A community-based study. Int. J. Obes. (Lond) 2015, 39, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Gumbau, V.; Bruna, M.; Canelles, E.; Guaita, M.; Mulas, C.; Bases, C.; Celma, I.; Puche, J.; Marcaida, G.; Oviedo, M.; et al. A prospective study on inflammatory parameters in obese patients after sleeve gastrectomy. Obes. Surg. 2014, 24, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Kamio, K.; Haida, M.; Ono, Y.; Miyachi, H.; Yamamoto, M.; Shinohara, Y.; Ando, Y. Platelet activation in patients with obstructive sleep apnea syndrome and effects of nasal-continuous positive airway pressure. Tokai J. Exp. Clin. Med. 2002, 27, 107–112. [Google Scholar] [PubMed]
- Zhang, X.; Yin, K.; Wang, H.; Su, M.; Yang, Y. Effect of continuous positive airway pressure treatment on elderly Chinese patients with obstructive sleep apnea in the prethrombotic state. Chin. Med. J. (Engl.) 2003, 116, 1426–1428. [Google Scholar]
- Oga, T.; Chin, K.; Tabuchi, A.; Kawato, M.; Morimoto, T.; Takahashi, K.; Handa, T.; Takahashi, K.; Taniguchi, R.; Kondo, H.; et al. Effects of Obstructive Sleep Apnea with Intermittent Hypoxia on Platelet Aggregability. J. Atheroscler. Thromb. 2009, 16, 862–869. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Y.; Xu, H.; Qian, Y.; Zou, J.; Yi, H.; Guan, J.; Yin, S. Association between Upper-airway Surgery and Ameliorative Risk Markers of Endothelial Function in Obstructive Sleep Apnea. Sci. Rep. 2019, 9, 20157. [Google Scholar] [CrossRef] [PubMed]
- Ayers, L.; Turnbull, C.; Petousi, N.; Ferry, B.; Kohler, M.; Stradling, J. Withdrawal of Continuous Positive Airway Pressure Therapy for 2 Weeks in Obstructive Sleep Apnoea Patients Results in Increased Circulating Platelet and Leucocyte-Derived Microvesicles. Respiration 2016, 91, 412–413. [Google Scholar] [CrossRef] [PubMed]
- Ayers, L.; Stoewhas, A.C.; Ferry, B.; Stradling, J.; Kohler, M. Elevated levels of endothelial cell-derived microparticles following short-term withdrawal of continuous positive airway pressure in patients with obstructive sleep apnea: Data from a randomized controlled trial. Respiration 2013, 85, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, L.; Wang, N.; Ge, X.; Pinto, J.M.; Wu, X.; Wei, Y. Can upper airway surgery for OSA protect against cardiovascular sequelae via effects on coagulation? Acta Otolaryngol. 2016, 136, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Kohler, H.P.; Grant, P.J. Plasminogen-activator inhibitor type 1 and coronary artery disease. N. Engl. J. Med. 2000, 342, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Meade, T.W.; Ruddock, V.; Stirling, Y.; Chakrabarti, R.; Miller, G.J. Fibrinolytic activity, clotting factors, and long-term incidence of ischaemic heart disease in the Northwick Park Heart Study. Lancet 1993, 342, 1076–1079. [Google Scholar] [CrossRef]
- Lévy, P.; Kohler, M.; McNicholas, W.T.; Barbé, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pépin, J.L. Obstructive sleep apnoea syndrome. Nat. Rev. Dis. Primers 2015, 1, 15015. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Jiang, T.; Wang, W.; Hu, S.; Shi, Y.; Lin, Y. Circulating fibrinogen levels are elevated in patients with obstructive sleep apnea: A systemic review and meta-analysis. Sleep Med. 2020, 68, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Fernández, A.; Toledo-Pons, N.; García-Río, F. Obstructive sleep apnea and venous thromboembolism: Overview of an emerging relationship. Sleep Med. Rev. 2020, 50, 101233. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bikov, A.; Meszaros, M.; Schwarz, E.I. Coagulation and Fibrinolysis in Obstructive Sleep Apnoea. Int. J. Mol. Sci. 2021, 22, 2834. https://doi.org/10.3390/ijms22062834
Bikov A, Meszaros M, Schwarz EI. Coagulation and Fibrinolysis in Obstructive Sleep Apnoea. International Journal of Molecular Sciences. 2021; 22(6):2834. https://doi.org/10.3390/ijms22062834
Chicago/Turabian StyleBikov, Andras, Martina Meszaros, and Esther Irene Schwarz. 2021. "Coagulation and Fibrinolysis in Obstructive Sleep Apnoea" International Journal of Molecular Sciences 22, no. 6: 2834. https://doi.org/10.3390/ijms22062834
APA StyleBikov, A., Meszaros, M., & Schwarz, E. I. (2021). Coagulation and Fibrinolysis in Obstructive Sleep Apnoea. International Journal of Molecular Sciences, 22(6), 2834. https://doi.org/10.3390/ijms22062834