Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
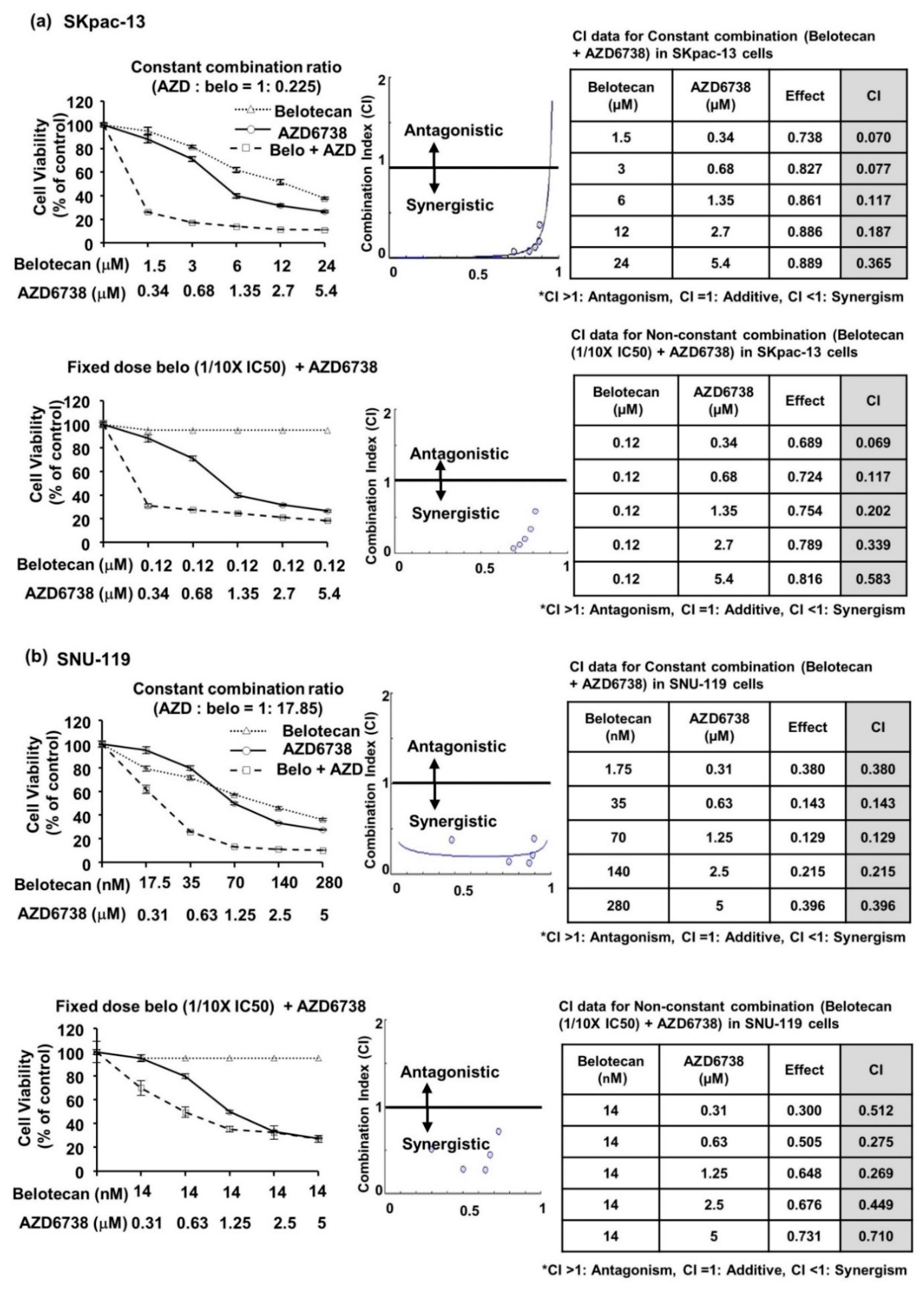
2.1. AZD6738 Synergizes with Belotecan for Cytotoxicity in Ovarian Cancer Cell Line
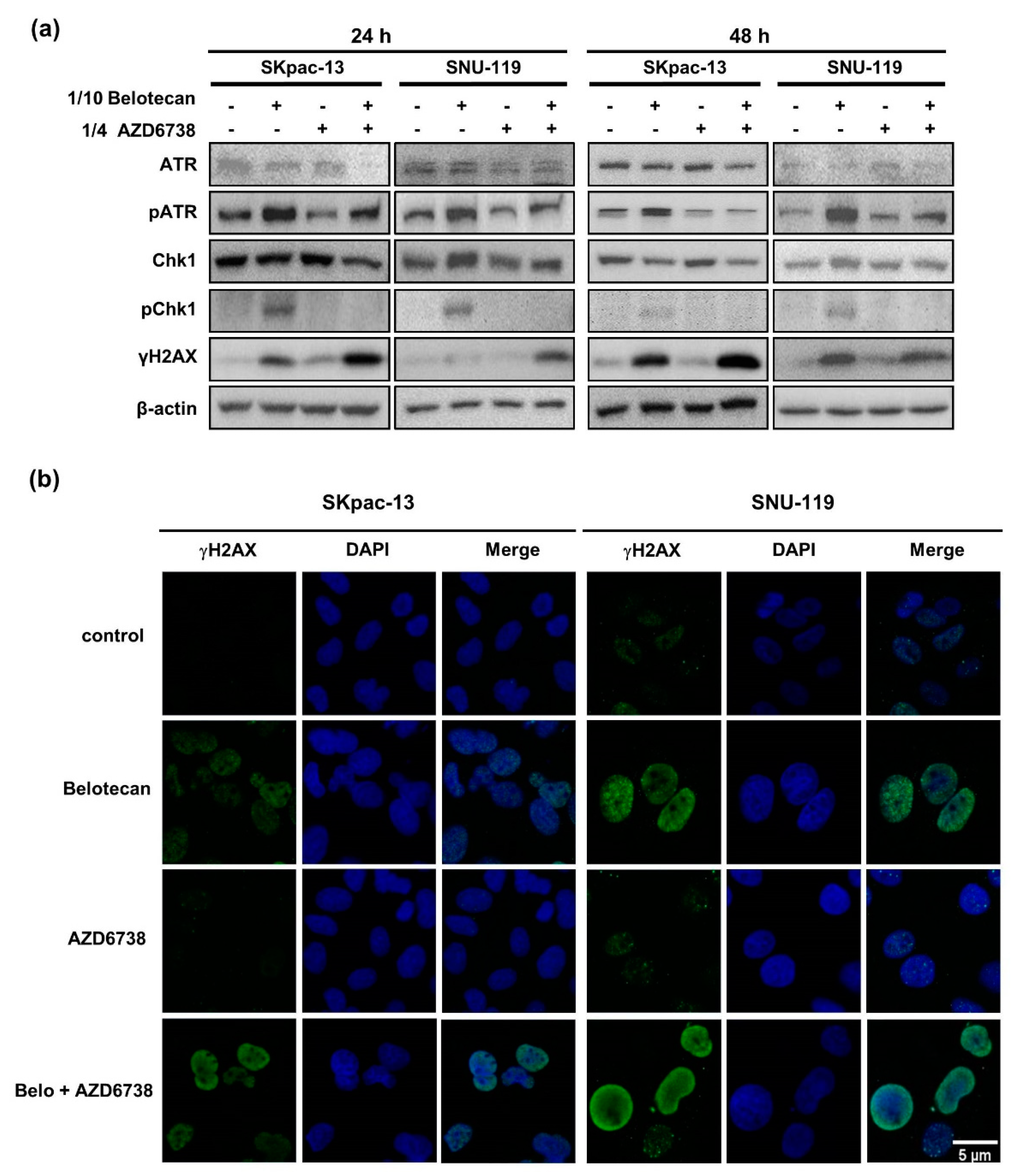
2.2. AZD6738 Induces DNA DSB by Suppressing DNA Damage Response Caused by Belotecan
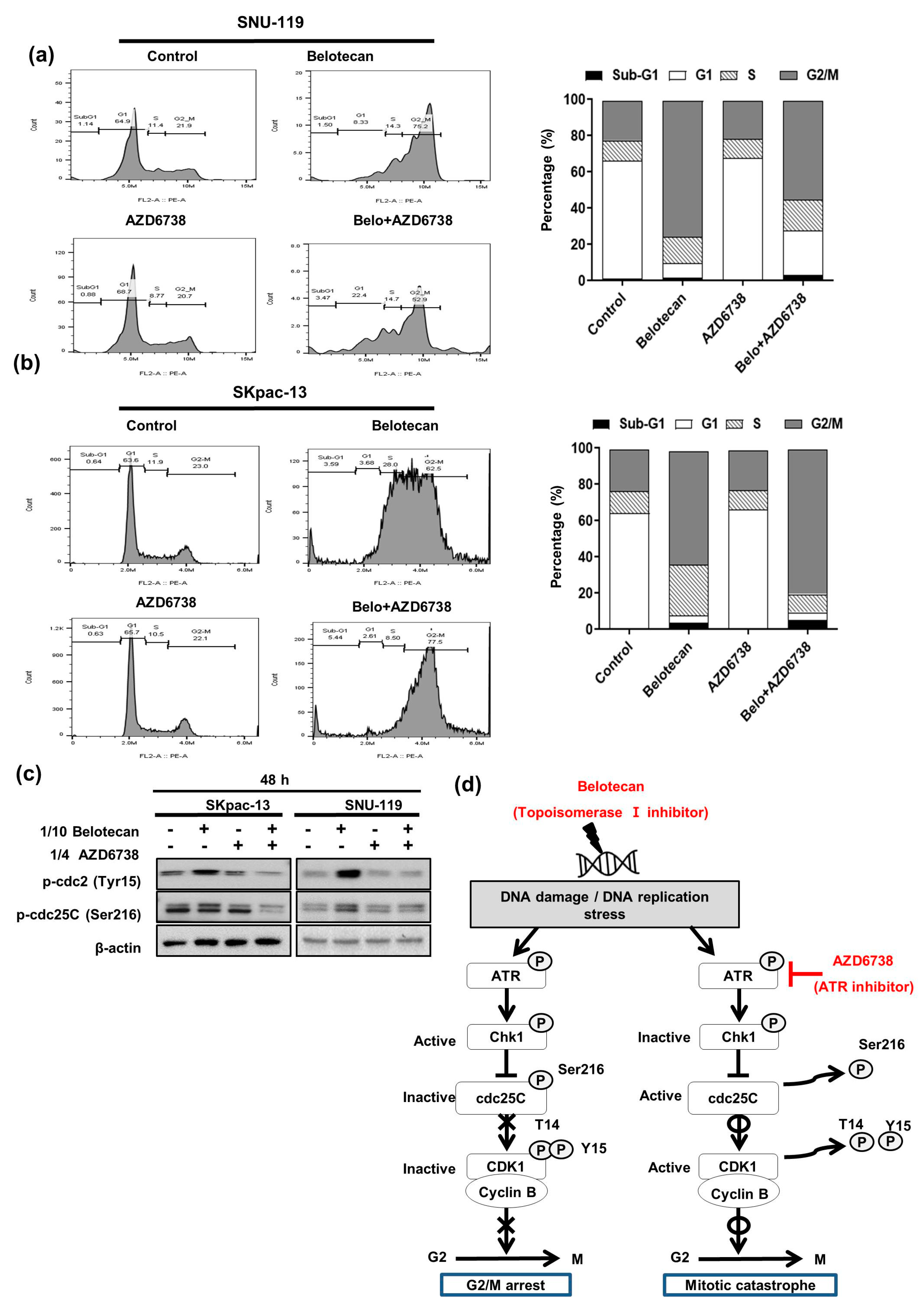
2.3. Mitotic Catastrophe Caused by AZD6738 in Combination with Belotecan May Account for Synergistic Mechanisms
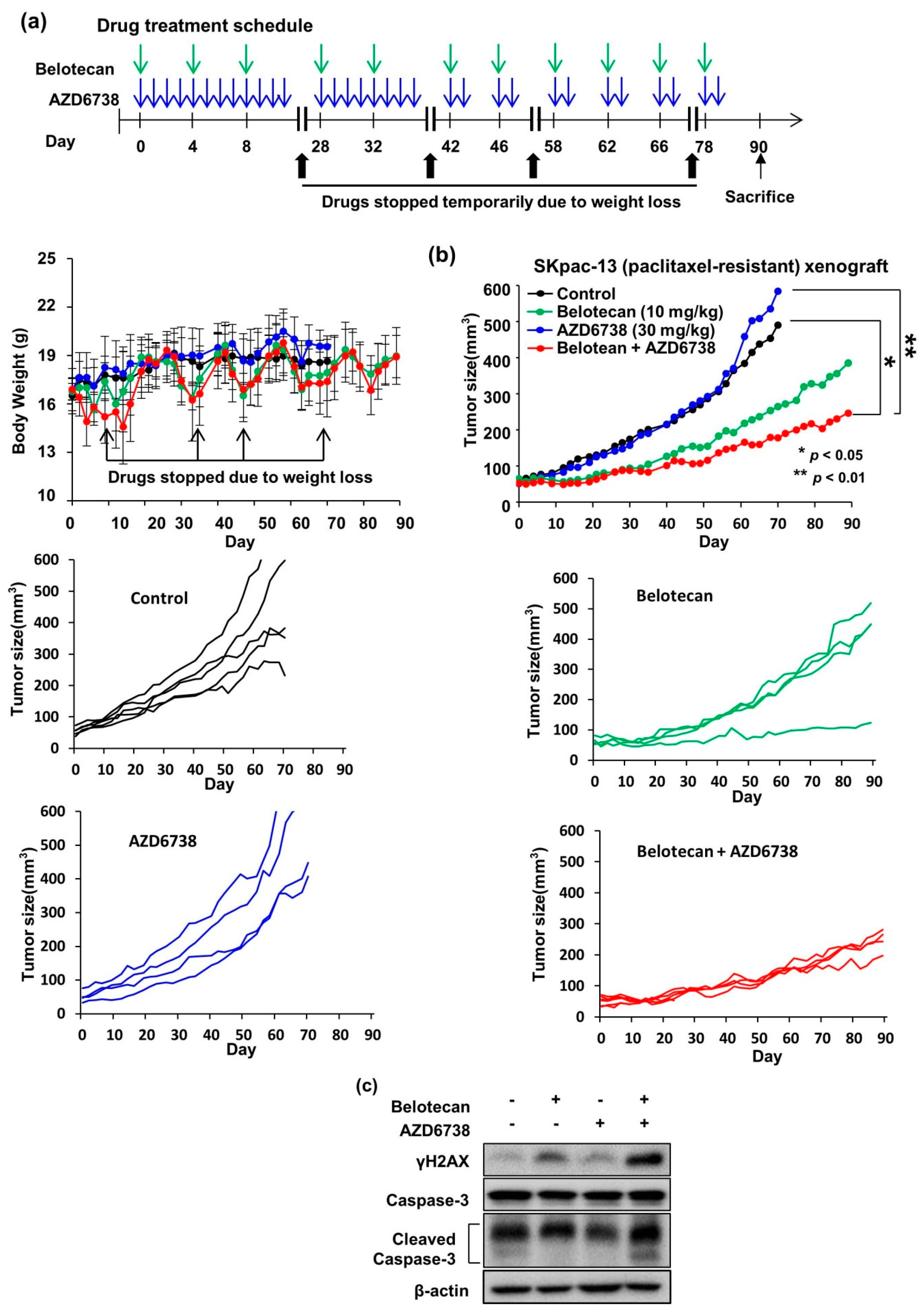
2.4. AZD6738 and Belotecan Combination Effectively Suppresses Tumor Growth in a Xenograft Model
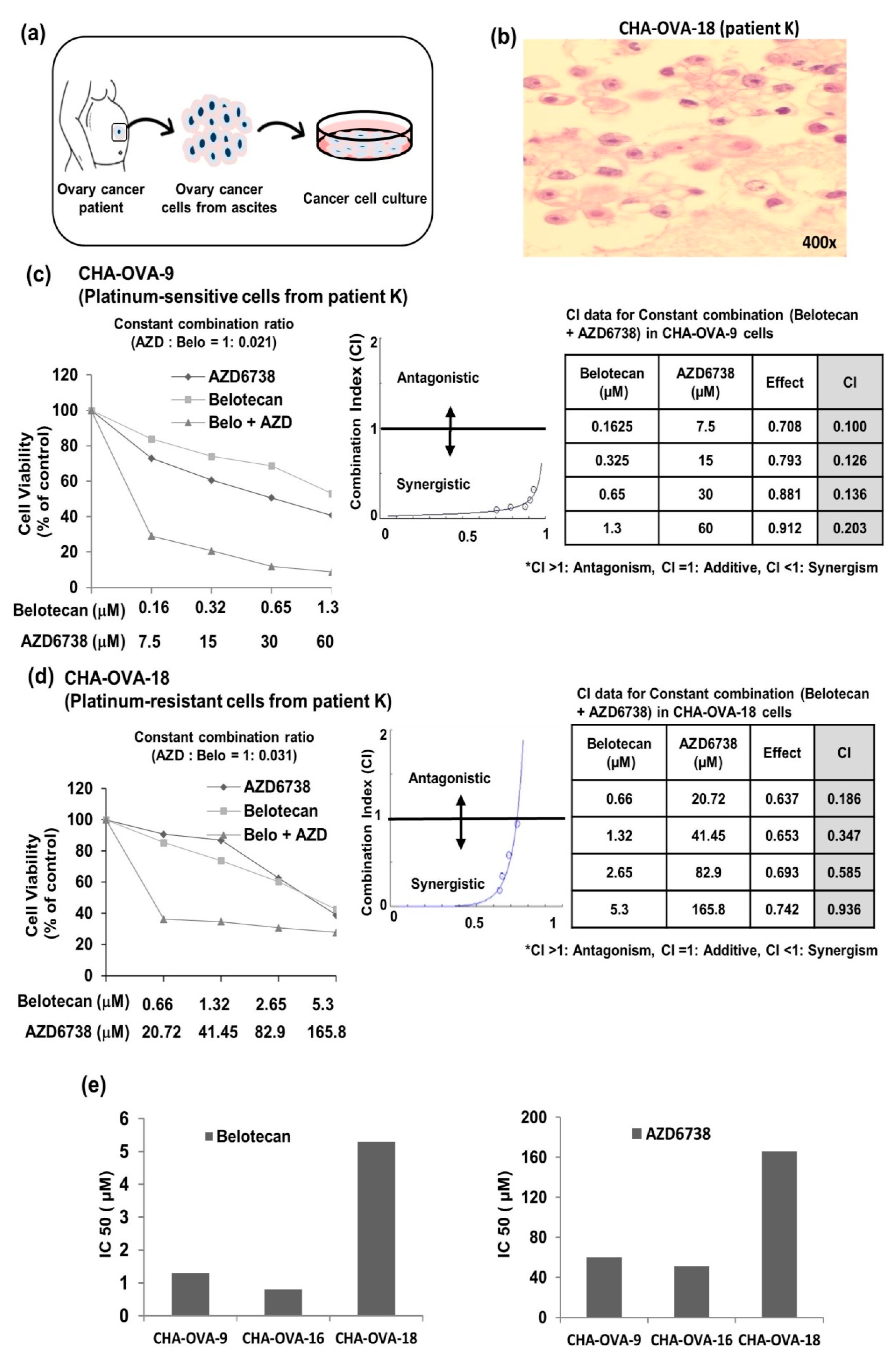
2.5. Analysis of Patient Fluid Samples Supports Synergism of AZD6738, ATR Inhibitor, and Belotecan in Ovarian Cancer
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Line and Cell Culture
4.3. Cell Viability Assay
4.4. Apoptosis Assay
4.5. Western Blot Analysis
4.6. Immunocytochemistry
4.7. Cell Cycle Analysis
4.8. In Vivo Study
4.9. Clinical Sample
4.10. H&E Staining of Cell Block
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Marsden, D.E.; Friedlander, M.; Hacker, N.F. Current Management of Epithelial Ovarian Carcinoma: A Review. In Seminars in Surgical Oncology; John Wiley & Sons, Inc.: New York, NY, USA, 2000; pp. 11–19. [Google Scholar]
- Tummala, M.; McGuire, W. Recurrent ovarian cancer. Clin. Adv. Hematol. Oncol. 2005, 3, 723–736. [Google Scholar] [PubMed]
- Alberts, D.S. Treatment of Refractory and Recurrent Ovarian Cancer. Semin Oncol. 1999, 26, 8–14. [Google Scholar] [PubMed]
- Lorusso, D.; Pietragalla, A.; Mainenti, S.; Masciullo, V.; Di Vagno, G.; Scambia, G. Review role of topotecan in gynaecological cancers: Current indications and perspectives. Crit. Rev. Oncol. Hematol. 2010, 74, 163–174. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W., III; Markman, M. Primary ovarian cancer chemotherapy: Current standards of care. Br. J. Cancer 2003, 89, S3. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, J.; Vergote, I.; Du Bois, A.; Eisenhauer, E. Combination therapy with gemcitabine and carboplatin in recurrent ovarian cancer. Int. J. Gynecol. Cancer 2005, 15, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Gore, M. Review of the use of topotecan in ovarian carcinoma. Expert Opin. Pharmacother. 2004, 5, 2333–2340. [Google Scholar] [CrossRef]
- Washington, C.R.; Richardson, D.L.; Moore, K.N. Olaparib in the treatment of ovarian cancer. Future Oncol. 2019, 15, 3435–3449. [Google Scholar] [CrossRef]
- Heo, Y.-A.; Duggan, S.T. Niraparib: A review in ovarian cancer. Target. Oncol. 2018, 13, 533–539. [Google Scholar] [CrossRef]
- Colomba, E.; Pautier, P.; Pommeret, F.; Leary, A. Rucaparib in the landscape of PARP inhibition in ovarian cancer. Expert Rev. Anticancer. Ther. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Rafii, S.; Gourley, C.; Kumar, R.; Geuna, E.; Ang, J.E.; Rye, T.; Chen, L.-M.; Shapira-Frommer, R.; Friedlander, M.; Matulonis, U. Baseline clinical predictors of antitumor response to the PARP inhibitor olaparib in germline BRCA1/2 mutated patients with advanced ovarian cancer. Oncotarget 2017, 8, 47154. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. Obstet. Gynecol. Surv. 2014, 69, 402–404. [Google Scholar] [CrossRef]
- Downs, L.S., Jr.; Judson, P.L.; Argenta, P.A.; Ghebre, R.; Geller, M.A.; Bliss, R.L.; Boente, M.P.; Nahhas, W.A.; Abu-Ghazaleh, S.Z.; Chen, M.D. A prospective randomized trial of thalidomide with topotecan compared with topotecan alone in women with recurrent epithelial ovarian carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2008, 112, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, J.-M.; Kim, J.-K.; Ahn, S.-K.; Lee, S.-J.; Kim, M.-Y.; Jew, S.-S.; Park, J.-G.; Hong, C.I. Antitumor activity of 7-[2-(N-isopropylamino) ethyl]-(20S)-camptothecin, CKD602, as a potent DNA topoisomerase I inhibitor. Arch. Pharmacal Res. 1998, 21, 581–590. [Google Scholar] [CrossRef]
- Kim, H.S.; Park, N.H.; Kang, S.; Seo, S.S.; Chung, H.H.; Kim, J.W.; Song, Y.S.; Kang, S.B. Comparison of the efficacy between topotecan-and belotecan-, a new camptothecin analog, based chemotherapies for recurrent epithelial ovarian cancer: A single institutional experience. J. Obstet. Gynaecol. Res. 2010, 36, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-P.; Seo, S.-S.; Ryu, S.-Y.; Kim, J.-H.; Bang, Y.-J.; Park, S.-Y.; Nam, J.-H.; Kang, S.-B.; Lee, K.-H.; Song, Y.S. Phase II evaluation of CKD-602, a camptothecin analog, administered on a 5-day schedule to patients with platinum-sensitive or-resistant ovarian cancer. Gynecol. Oncol. 2008, 109, 359–363. [Google Scholar] [CrossRef]
- Nam, E.J.; Kim, J.W.; Kim, J.H.; Kim, S.; Kim, S.W.; Jang, S.Y.; Lee, D.W.; Jung, Y.W.; Kim, Y.T. Efficacy and toxicity of belotecan with and without cisplatin in patients with recurrent ovarian cancer. Am. J. Clin. Oncol. 2010, 33, 233–237. [Google Scholar]
- Kim, H.S.; Kang, S.B.; Seo, S.S.; Han, S.S.; Kim, J.W.; Park, N.H.; Kang, S.B.; Lee, H.P.; Song, Y.S. Phase I/IIa study of combination chemotherapy with CKD-602 and cisplatin in patients with recurrent epithelial ovarian cancer. Ann. N. Y. Acad. Sci. 2009, 1171, 627–634. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Huen, M.S.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res. 2008, 18, 8. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Liang, X.; Yang, Q.; Wang, W.; Liu, T.; Hu, J. VE-822 mediated inhibition of ATR signaling sensitizes chondrosarcoma to cisplatin via reversion of the DNA damage response. Onco. Targets Ther. 2019, 12, 6083. [Google Scholar] [CrossRef] [PubMed]
- Moeglin, E.; Desplancq, D.; Conic, S.; Oulad-Abdelghani, M.; Stoessel, A.; Chiper, M.; Vigneron, M.; Didier, P.; Tora, L.; Weiss, E. Uniform Widespread Nuclear Phosphorylation of Histone H2AX Is an Indicator of Lethal DNA Replication Stress. Cancers 2019, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- DiPaola, R.S. To arrest or not to G2-M Cell-cycle arrest: Commentary re: AK Tyagi et al., Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G2-M arrest, and apoptosis. Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar] [PubMed]
- Krebs, M.G.; Lopez, J.; El-Khoueiry, A.; Bang, Y.; Postel-Vinay, S.; Abida, W.; Carter, L.; Xu, W.; Im, S.; Pierce, A. Phase I study of AZD6738, an inhibitor of ataxia telangiectasia Rad3-related (ATR), in combination with olaparib or durvalumab in patients (pts) with advanced solid cancers. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Huntoon, C.J.; Flatten, K.S.; Hendrickson, A.E.W.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef]
- Nam, A.-R.; Jin, M.H.; Park, J.E.; Bang, J.-H.; Oh, D.-Y.; Bang, Y.-J. Therapeutic targeting of the DNA damage response using an ATR inhibitor in biliary tract cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2019, 51, 1167. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Farolfi, A.; Gurioli, G.; Fugazzola, P.; Burgio, S.L.; Casanova, C.; Ravaglia, G.; Altavilla, A.; Costantini, M.; Amadori, A.; Framarini, M. Immune System and DNA Repair Defects in Ovarian Cancer: Implications for Locoregional Approaches. Int. J. Mol. Sci. 2019, 20, 2569. [Google Scholar] [CrossRef]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019, 153, 135–148. [Google Scholar] [CrossRef]
- George, E.; Kim, H.; Krepler, C.; Wenz, B.; Makvandi, M.; Tanyi, J.L.; Brown, E.; Zhang, R.; Brafford, P.; Jean, S. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight 2017, 2, e89760. [Google Scholar] [CrossRef]
- Checkley, S.; MacCallum, L.; Yates, J.; Jasper, P.; Luo, H.; Tolsma, J.; Bendtsen, C. Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci. Rep. 2015, 5, 13545. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, J.K.; Kim, B.; DeWitt, J.P.; Lee, J.E.; Han, J.H.; Kim, S.K.; Oh, C.W.; Kim, C.Y. Combination therapy of cilengitide with belotecan against experimental glioblastoma. Int. J. Cancer 2013, 133, 749–756. [Google Scholar] [CrossRef]
- Yu, N.Y.; Conway, C.; Pena, R.L.; Chen, J.Y. STEALTH® liposomal CKD-602, a topoisomerase I inhibitor, improves the therapeutic index in human tumor xenograft models. Anticancer. Res. 2007, 27, 2541–2545. [Google Scholar] [PubMed]
- Lee, J.-M.; Lee, J.-H.; Kim, J.-K.; Shin, H.-J.; Lee, H.-K.; Lee, S.-J.; Hong, C.-I. Pharmacokinetic Study of CKD-602, A New Camptothecin Derivative: Distribution, Metabolism and Excretion. Yakhak Hoeji 1998, 42, 437–446. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, J.; Ghosh, M.; Kim, T.H.; Park, N.; Pandey, K.; Cho, Y.B.; Hong, S.D.; Katuwal, N.B.; Kang, M.; An, H.J.; et al. Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 1223. https://doi.org/10.3390/ijms22031223
Hur J, Ghosh M, Kim TH, Park N, Pandey K, Cho YB, Hong SD, Katuwal NB, Kang M, An HJ, et al. Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer. International Journal of Molecular Sciences. 2021; 22(3):1223. https://doi.org/10.3390/ijms22031223
Chicago/Turabian StyleHur, Jin, Mithun Ghosh, Tae Heon Kim, Nahee Park, Kamal Pandey, Young Bin Cho, Sa Deok Hong, Nar Bahadur Katuwal, Minsil Kang, Hee Jung An, and et al. 2021. "Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer" International Journal of Molecular Sciences 22, no. 3: 1223. https://doi.org/10.3390/ijms22031223
APA StyleHur, J., Ghosh, M., Kim, T. H., Park, N., Pandey, K., Cho, Y. B., Hong, S. D., Katuwal, N. B., Kang, M., An, H. J., & Moon, Y. W. (2021). Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer. International Journal of Molecular Sciences, 22(3), 1223. https://doi.org/10.3390/ijms22031223