
Partial Agonistic Actions of Sex Hormone Steroids on TRPM3 Function

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
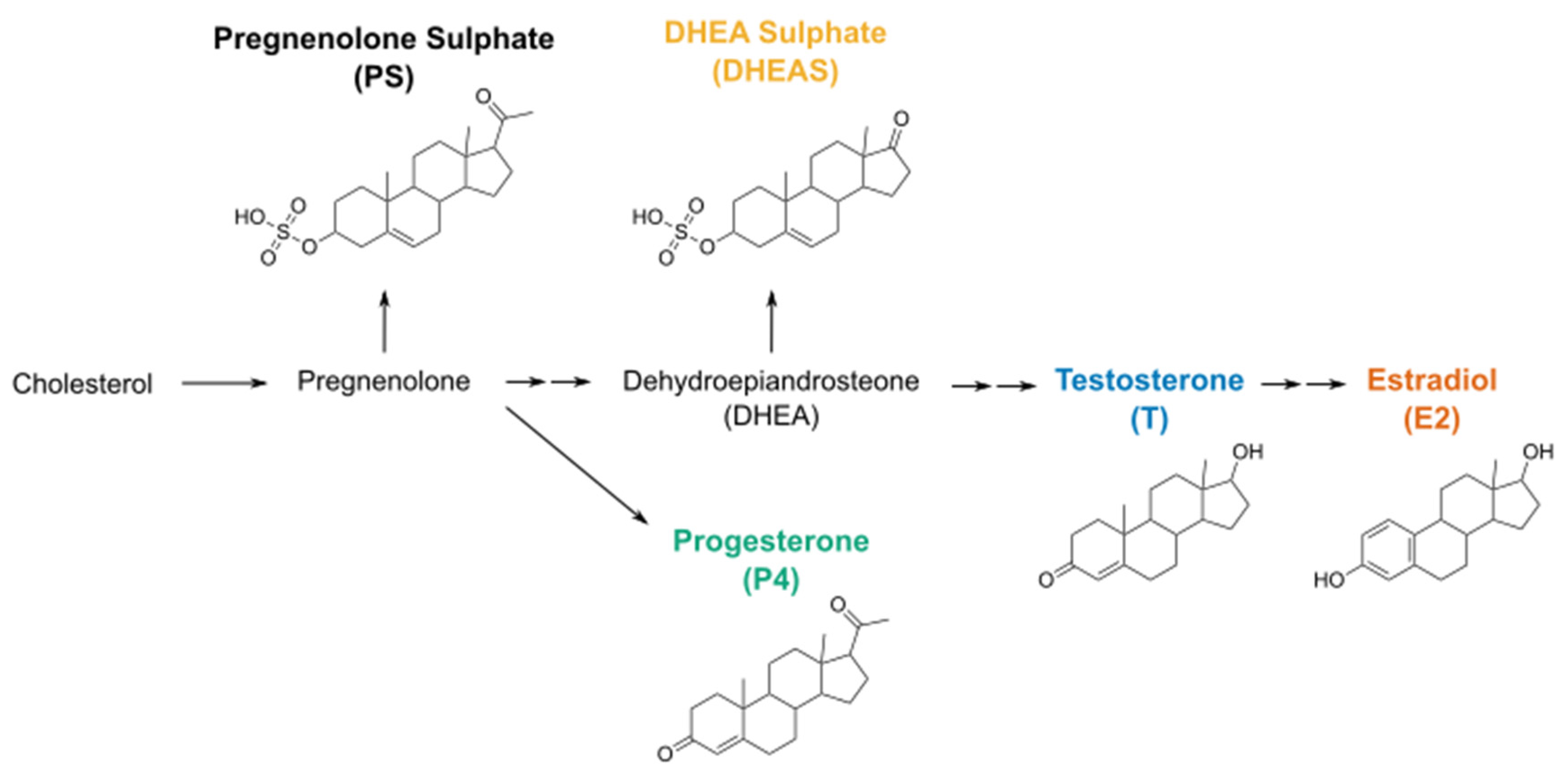
:1. Introduction
2. Results
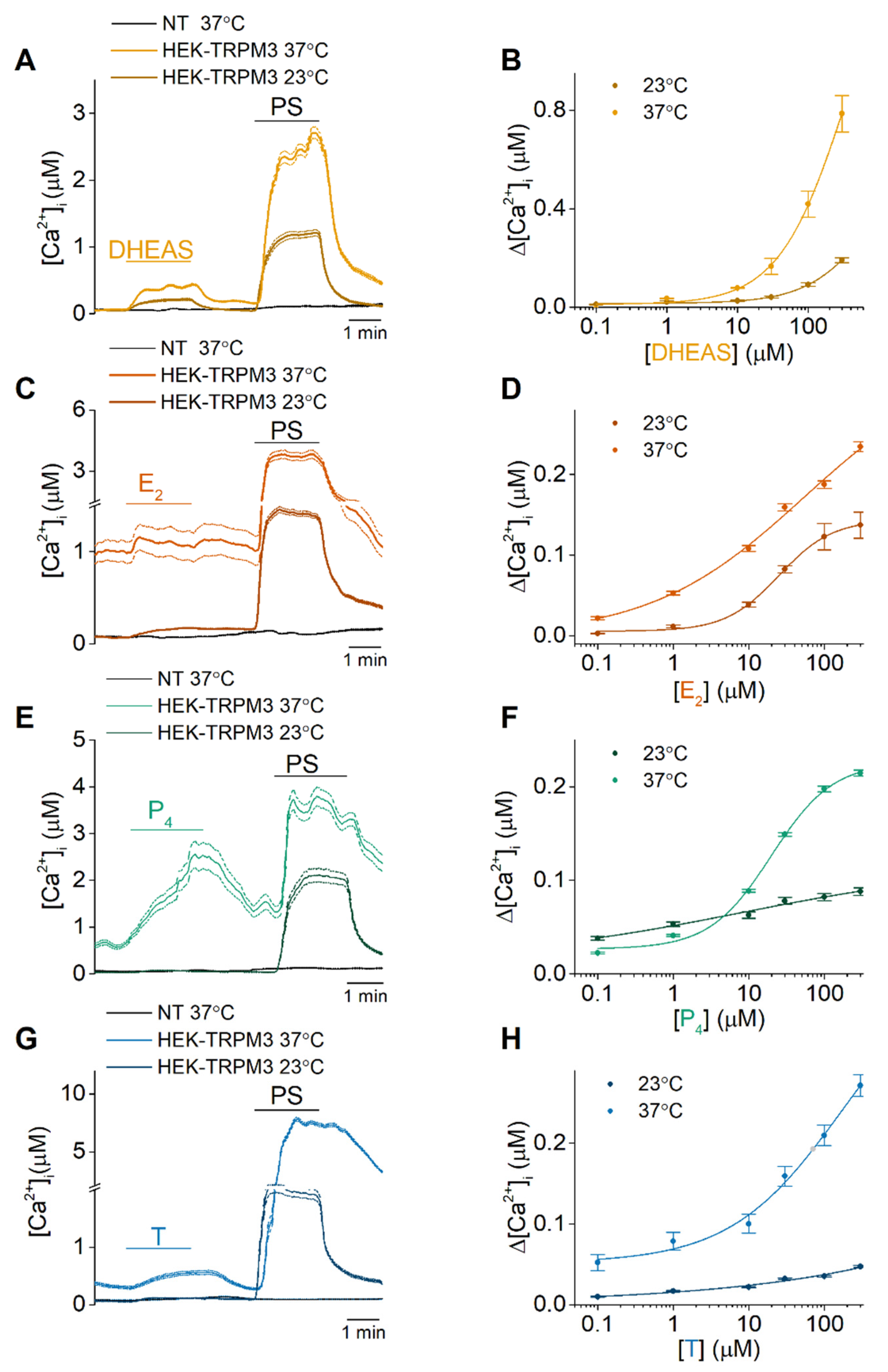
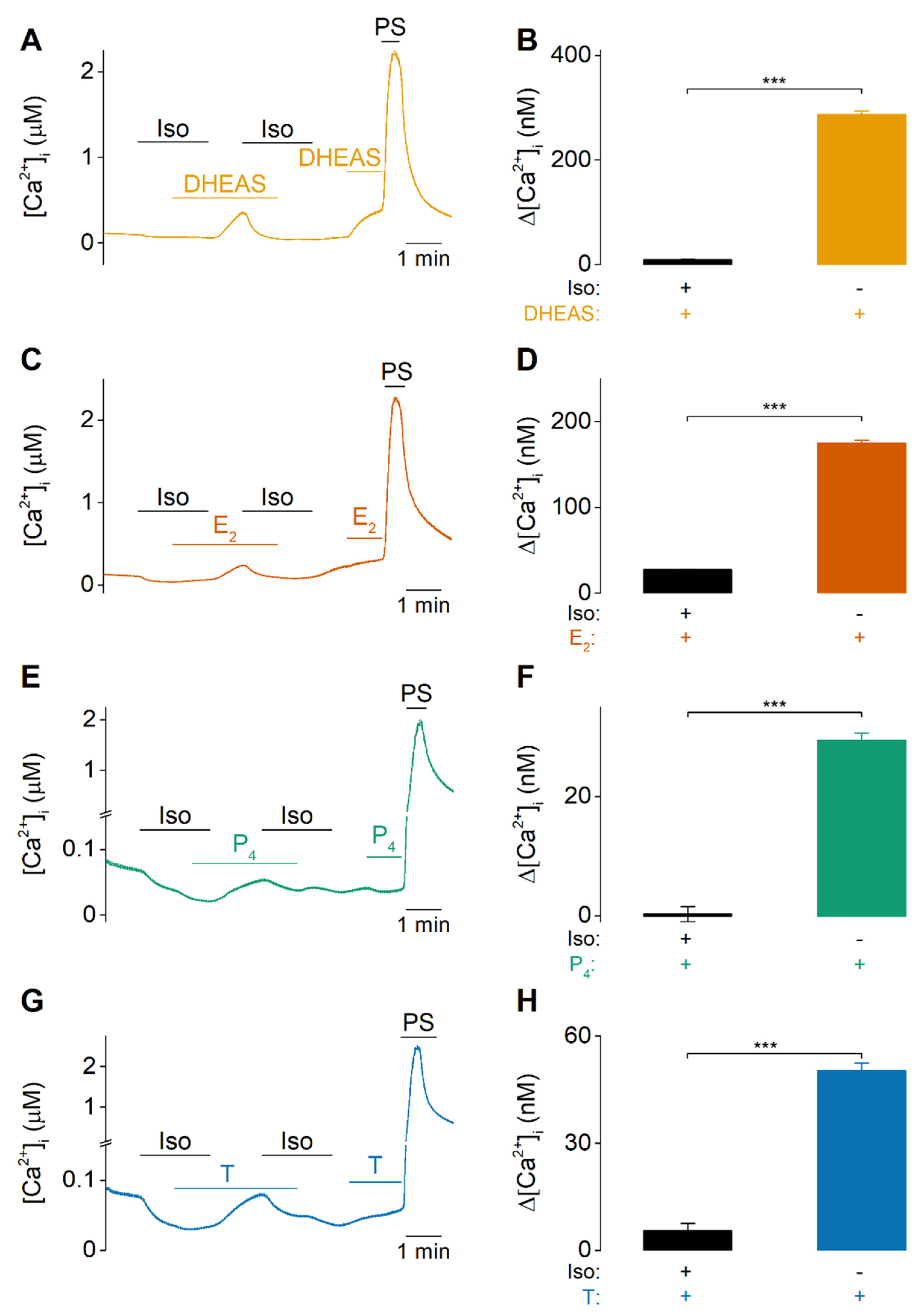
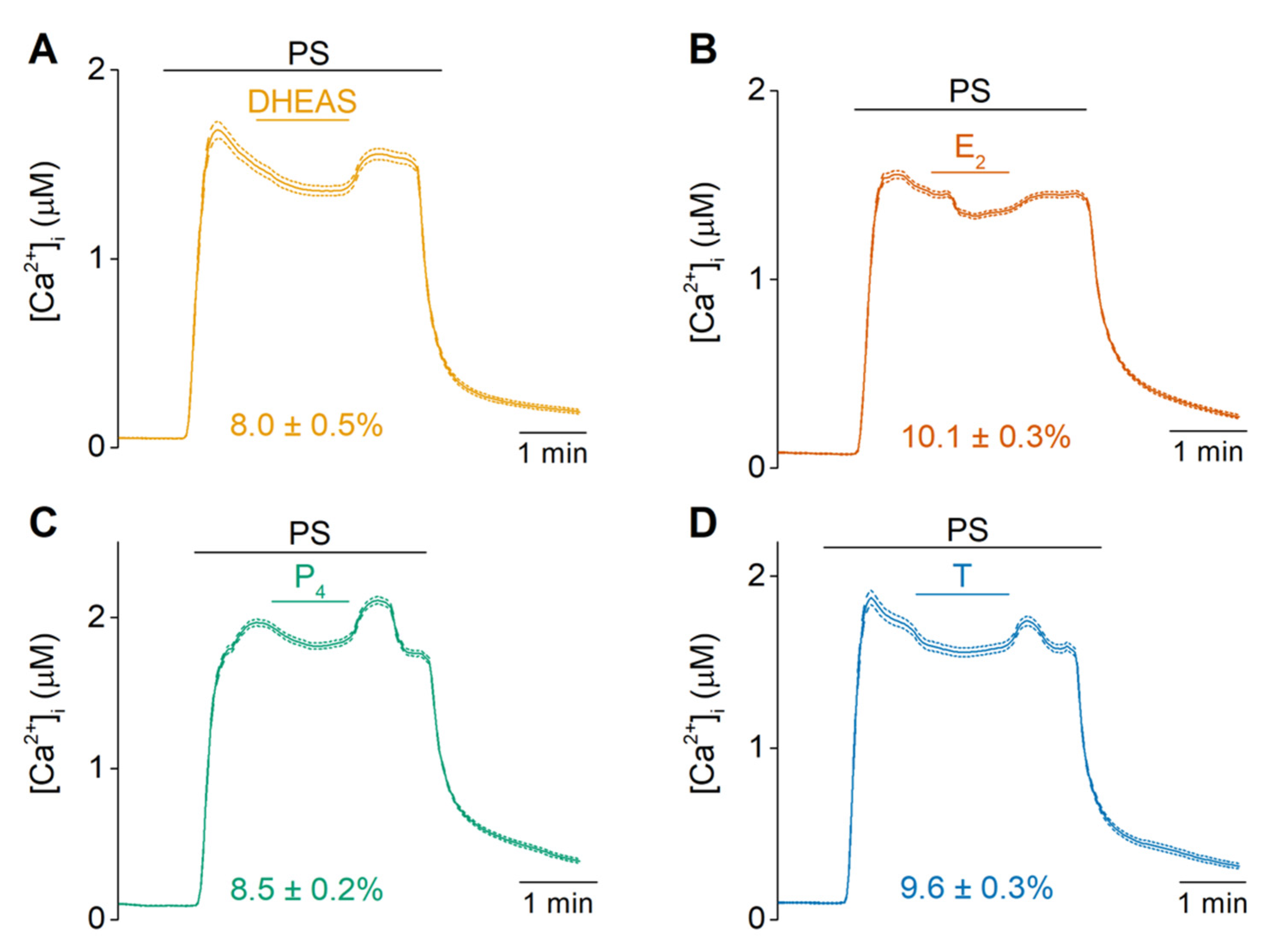
2.1. DHEAS, E2, P4 and Testosterone Act as Weak Agonists of TRPM3 at Body Temperature
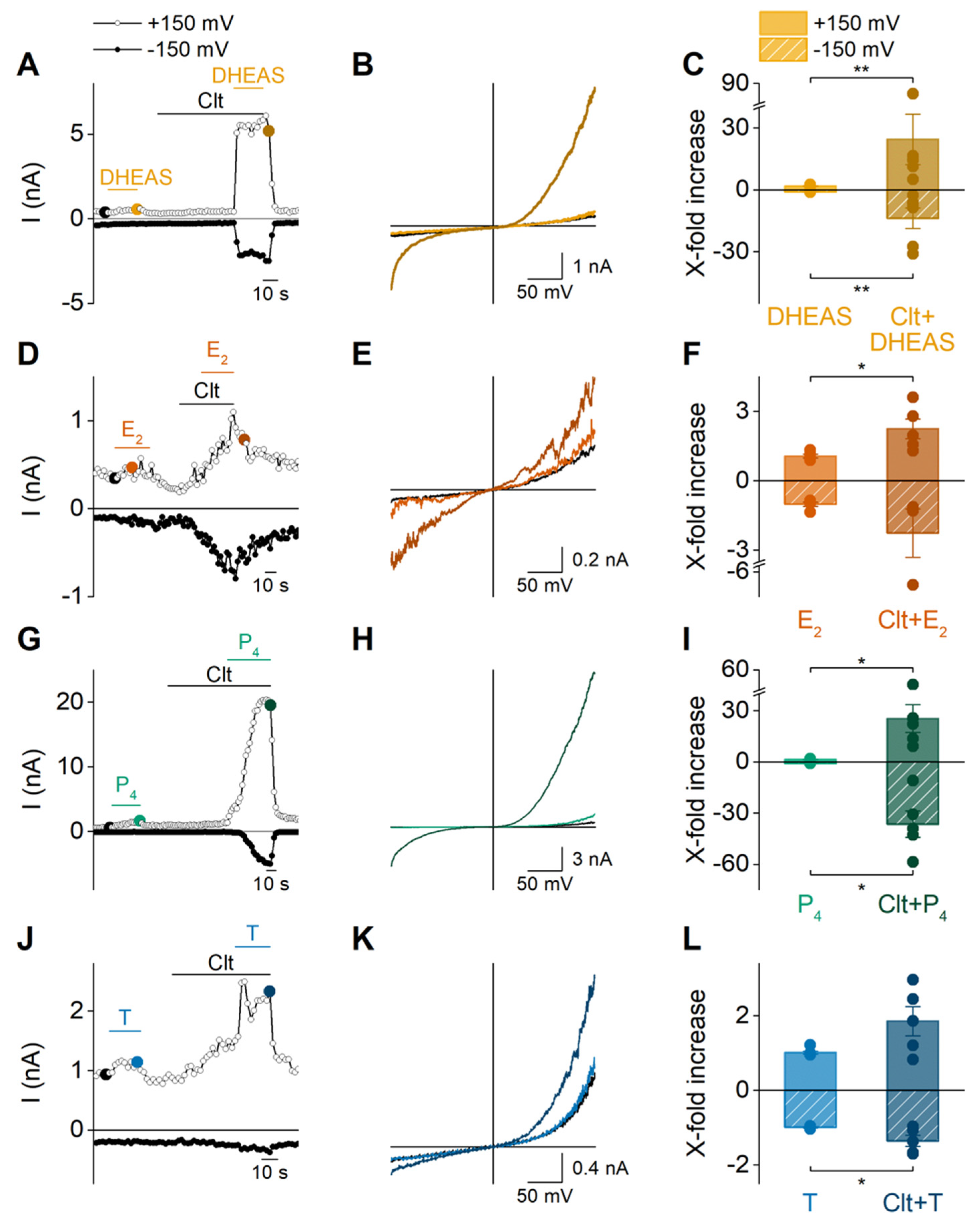
2.2. Sex Steroid Hormones Share a Similar Interaction Site and Gating Features
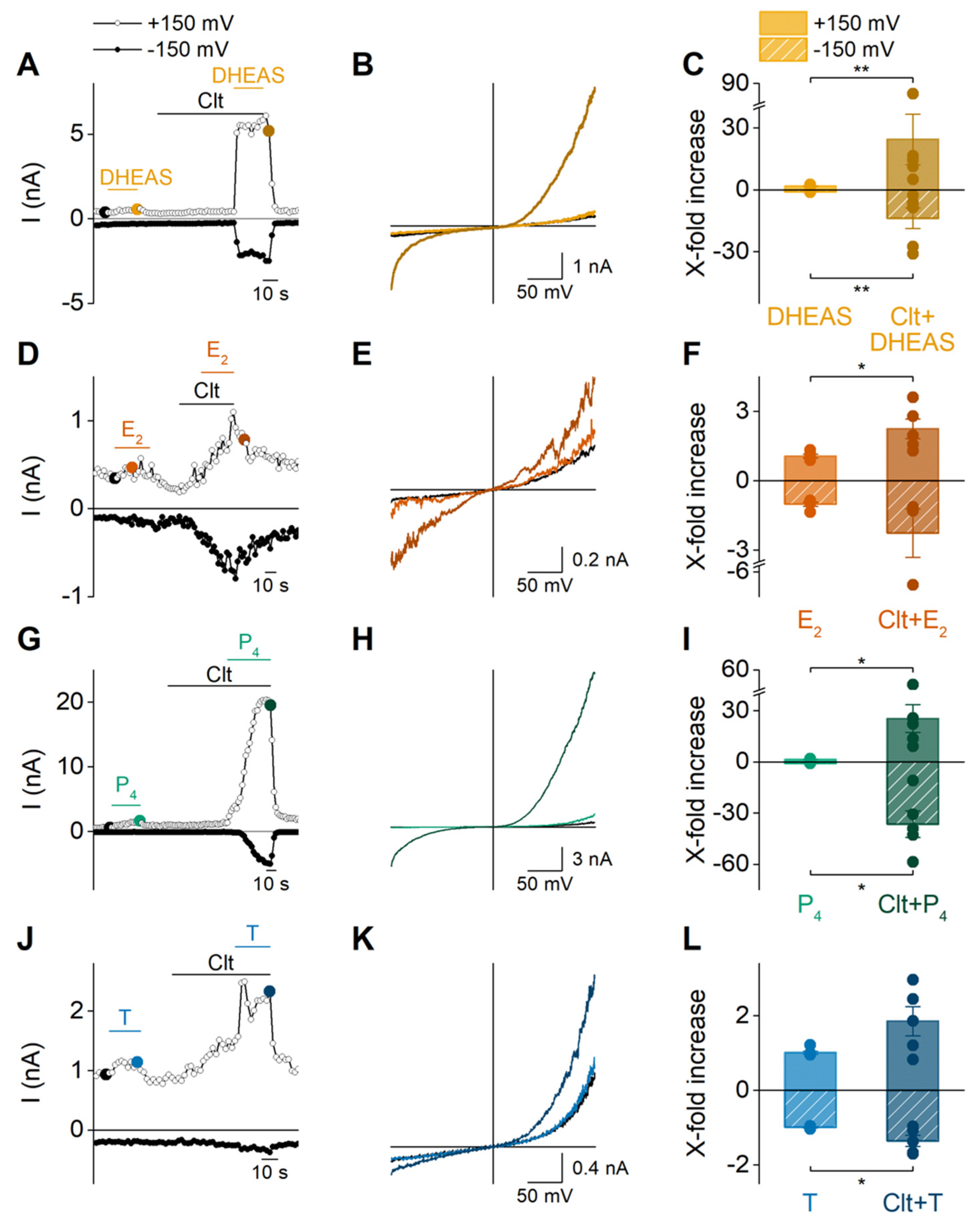
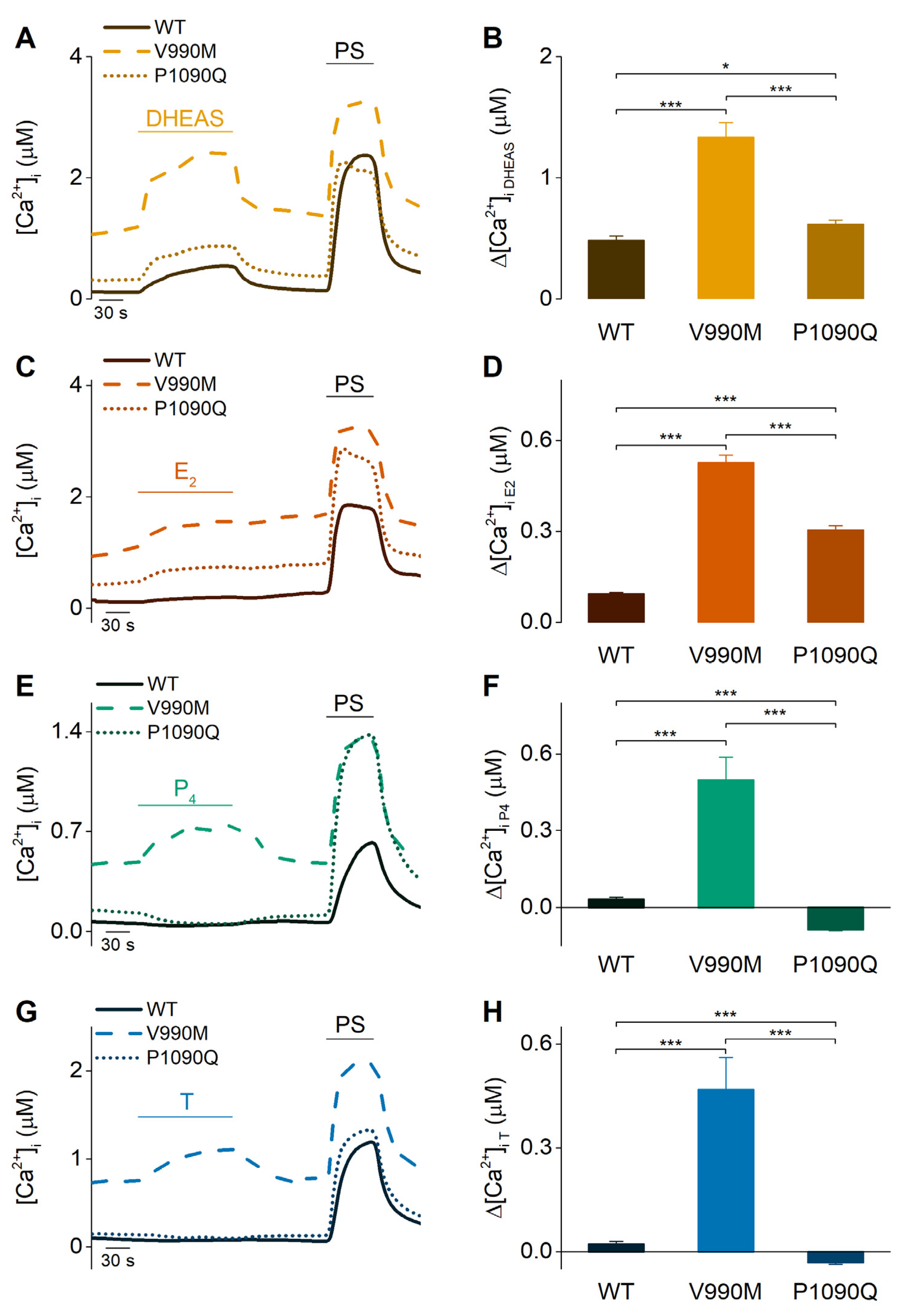
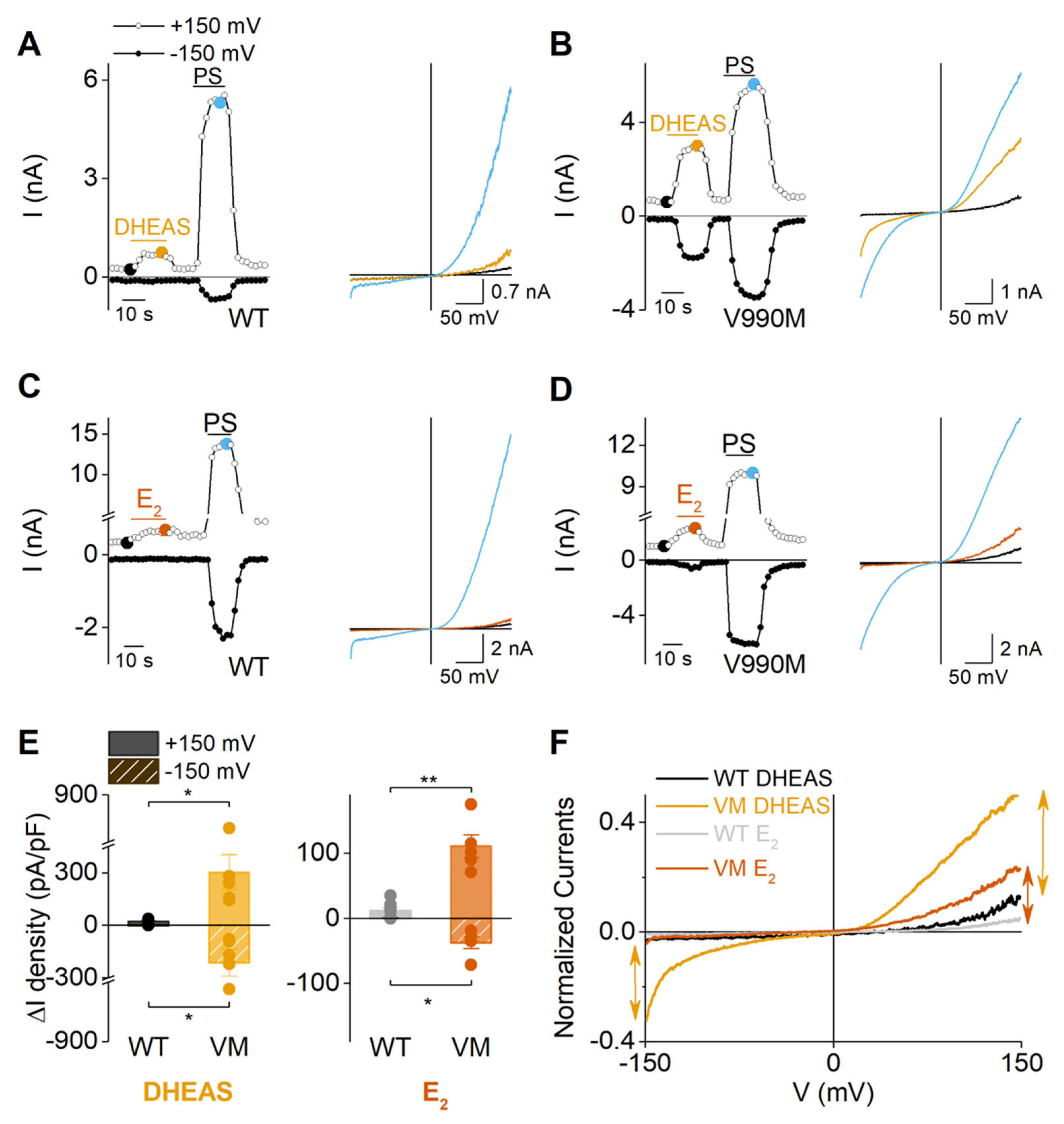
2.3. Sex Steroid Hormones May Contribute to the Disease Phenotype of Patients Carrying Trpm3 Gene Alterations
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Calcium Imaging
4.3. Patch Clamp Recordings
4.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grimm, S.L.; Hartig, S.M.; Edwards, D.P. Progesterone Receptor Signaling Mechanisms. J. Mol. Biol. 2016, 428, 3831–3849. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2016, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.; Verma, A.; Bivens, C.B.; Schwartz, Z.; Boyan, B.D. Rapid steroid hormone actions via membrane receptors. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y. Membrane Progesterone Receptors: Evidence for Neuroprotective, Neurosteroid Signaling and Neuroendocrine Functions in Neuronal Cells. Neuroendocrinology 2012, 96, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asuthkar, S.; Demirkhanyan, L.; Sun, X.; Elustondo, P.A.; Krishnan, V.; Baskaran, P.; Velpula, K.K.; Thyagarajan, B.; Pavlov, E.V.; Zakharian, E. The TRPM8 Protein Is a Testosterone Receptor. J. Biol. Chem. 2015, 290, 2670–2688. [Google Scholar] [CrossRef] [Green Version]
- De Logu, F.; Tonello, R.; Materazzi, S.; Nassini, R.; Fusi, C.; Coppi, E.; Puma, S.L.; Marone, I.M.; Sadofsky, L.R.; Morice, A.H.; et al. TRPA1 Mediates Aromatase Inhibitor–Evoked Pain by the Aromatase Substrate Androstenedione. Cancer Res. 2016, 76, 7024–7035. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Reséndiz, K.A.; Enciso-Pablo, O.; González-Ramírez, R.; Juárez-Contreras, R.; Rosenbaum, T.; Morales-Lázaro, S.L. Steroids and TRP Channels: A Close Relationship. Int. J. Mol. Sci. 2020, 21, 3819. [Google Scholar] [CrossRef]
- Wagner, T.F.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Düfer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic β cells. Nature 2008, 10, 1421–1430. [Google Scholar] [CrossRef]
- Naylor, J.; Li, J.; Milligan, C.J.; Zeng, F.; Sukumar, P.; Hou, B.; Sedo, A.; Yuldasheva, N.Y.; Majeed, Y.; Beri, D.; et al. Pregnenolone Sulphate- and Cholesterol-Regulated TRPM3 Channels Coupled to Vascular Smooth Muscle Secretion and Contraction. Circ. Res. 2010, 106, 1507–1515. [Google Scholar] [CrossRef]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 Is a Nociceptor Channel Involved in the Detection of Noxious Heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.; Mannebach, S.; Mathar, I.; Weissgerber, P.; Freichel, M.; Loodin, A.P.; Fecher-Trost, C.; Belkacemi, A.; Beck, A.; Philipp, S.E. Control of Insulin Release by Transient Receptor Potential Melastatin 3 (TRPM3) Ion Channels. Cell. Physiol. Biochem. 2020, 54, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Carbajo, L.; Alpizar, Y.A.; Startek, J.B.; López-López, J.R.; Pérez-García, M.T.; Talavera, K. Activation of the cation channel TRPM3 in perivascular nerves induces vasodilation of resistance arteries. J. Mol. Cell. Cardiol. 2019, 129, 219–230. [Google Scholar] [CrossRef]
- Vandewauw, I.; De Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; Van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018, 555, 662–666. [Google Scholar] [CrossRef]
- Held, K.; Kichko, T.; De Clercq, K.; Klaassen, H.; Van Bree, R.; Vanherck, J.-C.; Marchand, A.; Reeh, P.; Chaltin, P.; Voets, T.; et al. Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc. Natl. Acad. Sci. USA 2015, 112, E1363–E1372. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Yudin, Y.; Kim, N.; Tao, Y.-X.; Rohacs, T. TRPM3 Channels Play Roles in Heat Hypersensitivity and Spontaneous Pain after Nerve Injury. J. Neurosci. 2021, 41, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Kraft, R.; Sauerbruch, S.; Schultz, G.; Harteneck, C. Molecular and Functional Characterization of the Melastatin-related Cation Channel TRPM3. J. Biol. Chem. 2003, 278, 21493–21501. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Chen, J.; Sun, L.; Wu, S.; Gray, K.R.; Rich, A.; Huang, M.; Lin, J.-H.; Feder, J.N.; Janovitz, E.B.; et al. Expression and Characterization of Human Transient Receptor Potential Melastatin 3 (hTRPM3). J. Biol. Chem. 2003, 278, 20890–20897. [Google Scholar] [CrossRef] [Green Version]
- Majeed, Y.; Tumova, S.; Green, B.L.; Seymour, V.A.; Woods, D.M.; Agarwal, A.K.; Naylor, J.; Jiang, S.; Picton, H.M.; Porter, K.E.; et al. Pregnenolone sulphate-independent inhibition of TRPM3 channels by progesterone. Cell Calcium 2012, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Held, K.; Voets, T.; Vriens, J. TRPM3 in temperature sensing and beyond. Temperature 2015, 2, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Vriens, J.; Held, K.; Janssens, A.; Tóth, B.I.; Kerselaers, S.; Nilius, B.; Vennekens, R.; Voets, T. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat. Chem. Biol. 2014, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Voets, T.; Vriens, J. Signature and Pathophysiology of Non-canonical Pores in Voltage-Dependent Cation Channels. Rev. Physiol. Biochem. Pharm. 2016, 170, 67–99. [Google Scholar] [CrossRef]
- Held, K.; Gruss, F.; Aloi, V.D.; Janssens, A.; Ulens, C.; Voets, T.; Vriens, J. Mutations in the voltage-sensing domain affect the alternative ion permeation pathway in the TRPM3 channel. J. Physiol. 2018, 596, 2413–2432. [Google Scholar] [CrossRef] [PubMed]
- Dyment, D.A.; Terhal, P.A.; Rustad, C.F.; Tveten, K.; Griffith, C.; Jayakar, P.; Shinawi, M.; Ellingwood, S.; Smith, R.; Van Gassen, K.; et al. De novo substitutions of TRPM3 cause intellectual disability and epilepsy. Eur. J. Hum. Genet. 2019, 27, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Agathe, J.-M.D.S.; Van-Gils, J.; Lasseaux, E.; Arveiler, B.; Lacombe, D.; Pfirrmann, C.; Raclet, V.; Gaston, L.; Plaisant, C.; Aupy, J.; et al. Confirmation and Expansion of the Phenotype Associated with the Recurrent p.Val837Met Variant in TRPM3. Eur. J. Med. Genet. 2020, 63, 103942. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, L.; Liao, H.; Yang, S.; Kuang, X.; Ning, Z.; Liao, C.; Chen, B. A Chinese patient with developmental and epileptic encephalopathies (DEE) carrying a TRPM3 gene mutation: A paediatric case report. BMC Pediatr. 2021, 21, 256. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.W.; Chatron, N.; Cabet, S.; Labalme, A.; Carneiro, M.; Poirot, I.; Delvert, C.; Gleizal, A.; Lesca, G.; Putoux, A. Description of a novel patient with the TRPM3 recurrent p.Val837Met variant. Eur. J. Med. Genet. 2021, 64, 104320. [Google Scholar] [CrossRef] [PubMed]
- Van Hoeymissen, E.; Held, K.; Freitas, A.C.N.; Janssens, A.; Voets, T.; Vriens, J. Gain of channel function and modified gating properties in TRPM3 mutants causing intellectual disability and epilepsy. eLife 2020, 9, e57190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Yudin, Y.; Rohacs, T. Disease-associated mutations in the human TRPM3 render the channel overactive via two distinct mechanisms. eLife 2020, 9, e55634. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, M.F.; Cambiasso, M.; Holschbach, M.A.; Cabrera, R. Oestrogens and Progestagens: Synthesis and Action in the Brain. J. Neuroendocr. 2016, 28. [Google Scholar] [CrossRef]
- Straub, I.; Krügel, U.; Mohr, F.; Teichert, J.; Rizun, O.; Konrad, M.; Oberwinkler, J.; Schaefer, M. Flavanones That Selectively Inhibit TRPM3 Attenuate Thermal Nociception In Vivo. Mol. Pharmacol. 2013, 84, 736–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeed, Y.; Agarwal, A.; Naylor, J.; Seymour, V.; Jiang, S.; Muraki, K.; Fishwick, C.; Beech, D. Cis-isomerism and other chemical requirements of steroidal agonists and partial agonists acting at TRPM3 channels. Br. J. Pharmacol. 2010, 161, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Drews, A.; Mohr, F.; Rizun, O.; Wagner, T.F.J.; Dembla, S.; Rudolph, S.; Lambert, S.; Konrad, M.; Philipp, S.E.; Behrendt, M.; et al. Structural requirements of steroidal agonists of transient receptor potential melastatin 3 (TRPM 3) cation channels. Br. J. Pharmacol. 2014, 171, 1019–1032. [Google Scholar] [CrossRef]
- Riley, J.L.; Robinson, M.E.; Wise, E.A.; Price, D. A meta-analytic review of pain perception across the menstrual cycle. Pain 1999, 81, 225–235. [Google Scholar] [CrossRef]
- Frye, C.; Cuevas, C.; Kanarek, R. Diet and estrous cycle influence pain sensitivity in rats. Pharmacol. Biochem. Behav. 1993, 45, 255–260. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Roh, D.-H.; Seo, H.-S.; Kang, S.-Y.; Han, H.J.; Beitz, A.; Lee, J.-H. Intrathecal injection of the neurosteroid, DHEAS, produces mechanical allodynia in mice: Involvement of spinal sigma-1 and GABAA receptors. Br. J. Pharmacol. 2009, 157, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Kibaly, C.; Meyer, L.; Patte-Mensah, C.; Mensah-Nyagan, A.G. Biochemical and functional evidence for the control of pain mechanisms by dehydroepiandrosterone endogenously synthesized in the spinal cord. FASEB J. 2007, 22, 93–104. [Google Scholar] [CrossRef]
- Bartley, E.J.; Palit, S.; Kuhn, B.L.; Kerr, K.L.; Terry, E.L.; DelVentura, J.L.; Rhudy, J.L. Natural Variation in Testosterone is Associated with Hypoalgesia in Healthy Women. Clin. J. Pain 2015, 31, 730–739. [Google Scholar] [CrossRef]
- Edinger, K.L.; Frye, C.A. Testosterone’s Analgesic, Anxiolytic, and Cognitive-Enhancing Effects May Be Due in Part to Actions of Its 5α-Reduced Metabolites in the Hippocampus. Behav. Neurosci. 2004, 118, 1352–1364. [Google Scholar] [CrossRef]
- Becker, M.; Hesse, V. Minipuberty: Why Does it Happen? Horm. Res. Paediatr. 2020, 93, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, O. Steroid analysis for medical diagnosis. J. Chromatogr. A 2001, 935, 267–278. [Google Scholar] [CrossRef]
- White, B.; Harrison, J.R.; Mehlmann, L. Endocrine and Reproductive Physiology, 5th ed.; Elsevier: St. Louis, MO, USA, 2019; p. 288. [Google Scholar]
- Jang, Y.; Lee, Y.; Kim, S.M.; Yang, Y.D.; Jung, J.; Oh, U. Quantitative analysis of TRP channel genes in mouse organs. Arch. Pharmacal Res. 2012, 35, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Oberwinkler, J.; Philipp, S.E. TRPM3. Handb. Exp. Pharmacol. 2014, 222, 427–459. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Held, K.; Aloi, V.D.; Freitas, A.C.N.; Janssens, A.; Segal, A.; Przibilla, J.; Philipp, S.E.; Wang, Y.T.; Voets, T.; Vriens, J. Pharmacological properties of TRPM3 isoforms are determined by the length of the pore loop. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persoons, E.; Kerselaers, S.; Voets, T.; Vriens, J.; Held, K. Partial Agonistic Actions of Sex Hormone Steroids on TRPM3 Function. Int. J. Mol. Sci. 2021, 22, 13652. https://doi.org/10.3390/ijms222413652
Persoons E, Kerselaers S, Voets T, Vriens J, Held K. Partial Agonistic Actions of Sex Hormone Steroids on TRPM3 Function. International Journal of Molecular Sciences. 2021; 22(24):13652. https://doi.org/10.3390/ijms222413652
Chicago/Turabian StylePersoons, Eleonora, Sara Kerselaers, Thomas Voets, Joris Vriens, and Katharina Held. 2021. "Partial Agonistic Actions of Sex Hormone Steroids on TRPM3 Function" International Journal of Molecular Sciences 22, no. 24: 13652. https://doi.org/10.3390/ijms222413652
APA StylePersoons, E., Kerselaers, S., Voets, T., Vriens, J., & Held, K. (2021). Partial Agonistic Actions of Sex Hormone Steroids on TRPM3 Function. International Journal of Molecular Sciences, 22(24), 13652. https://doi.org/10.3390/ijms222413652