
Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L

 ,
,  , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Structural Characterization of Mpro in Apo and MG-132 Bound Forms
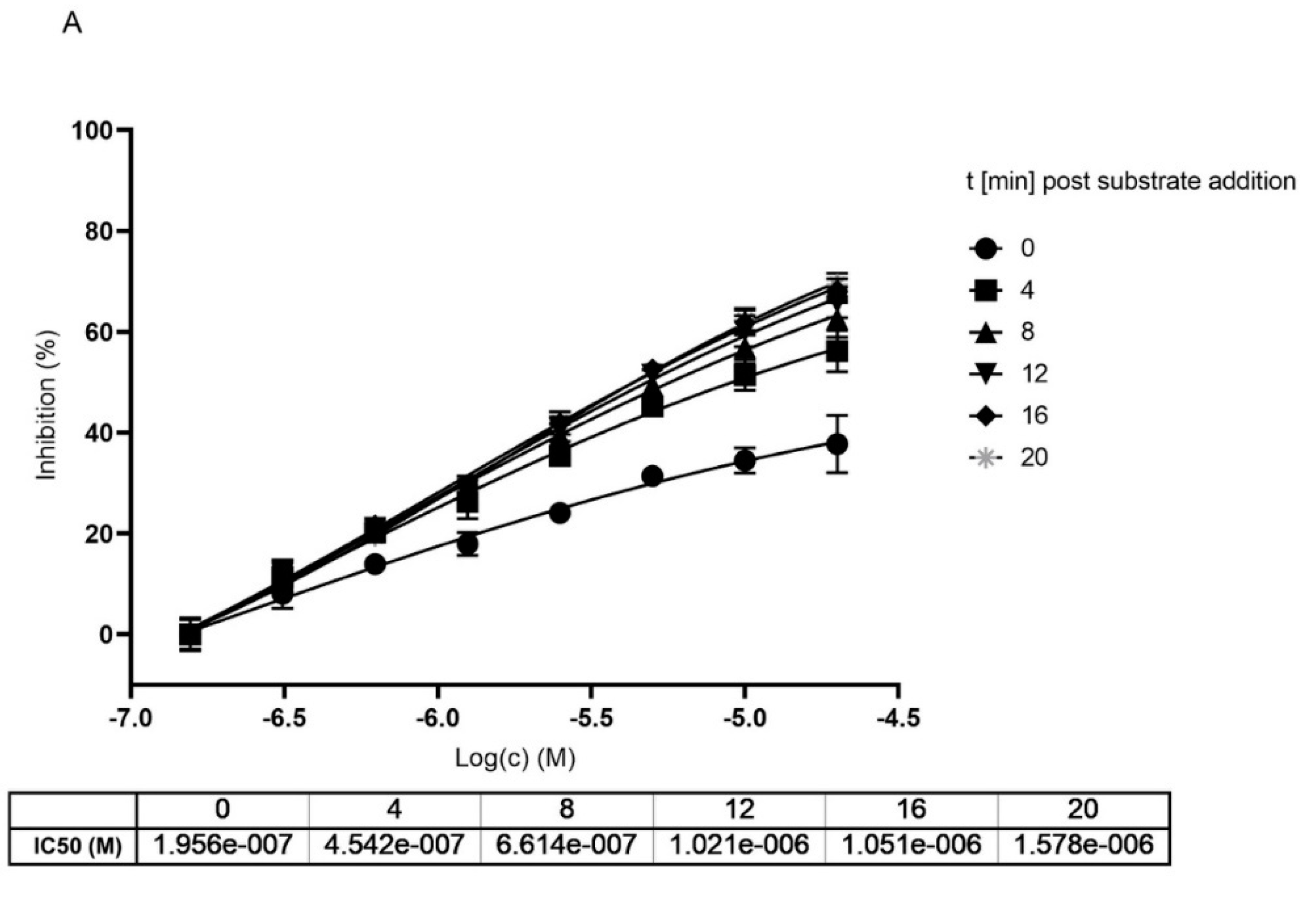
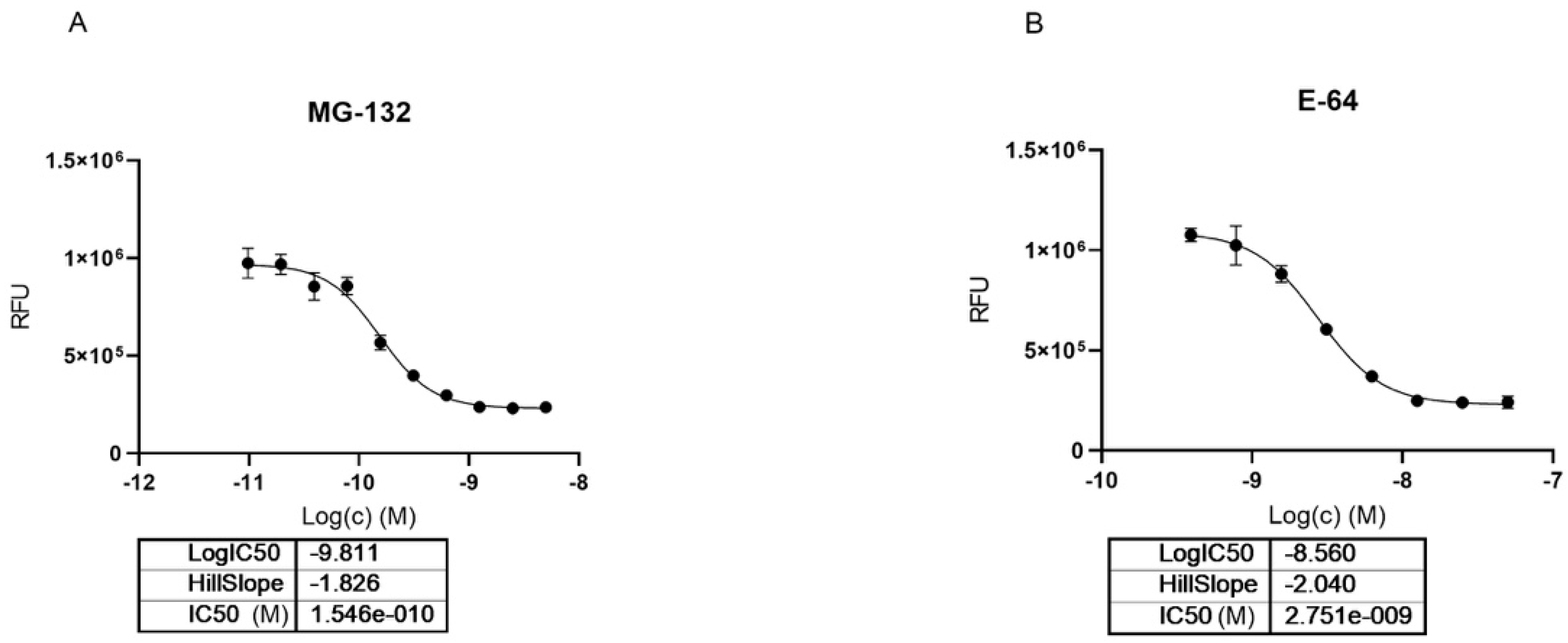
2.2. Mpro Inhibition Mechanism by MG-132
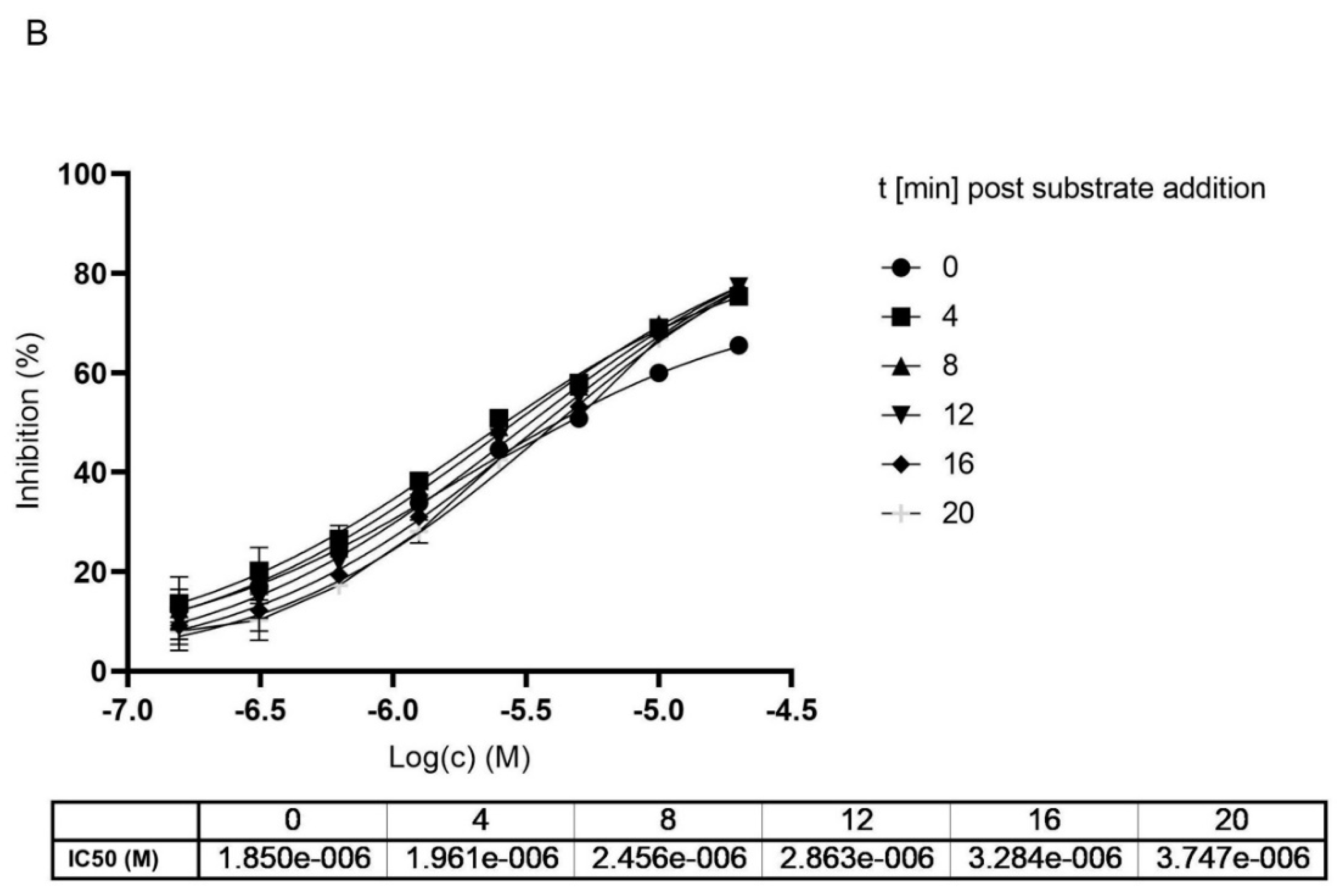
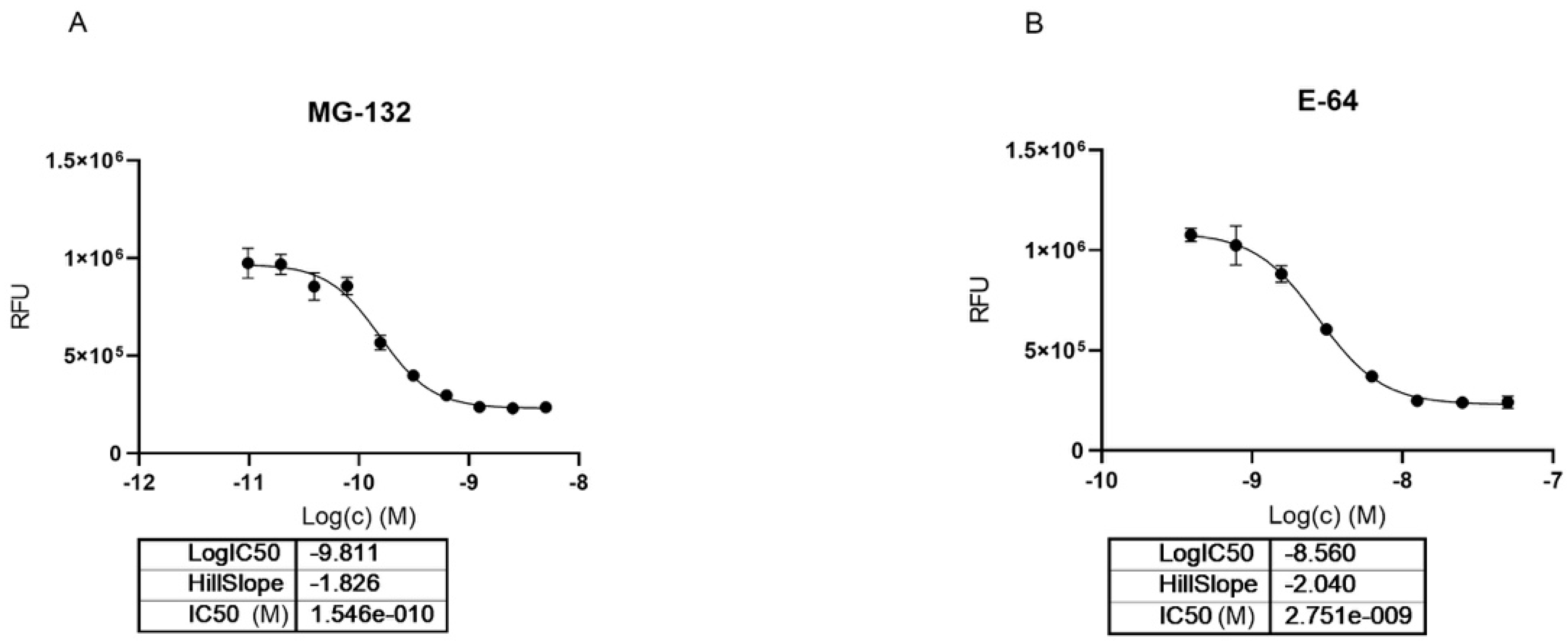
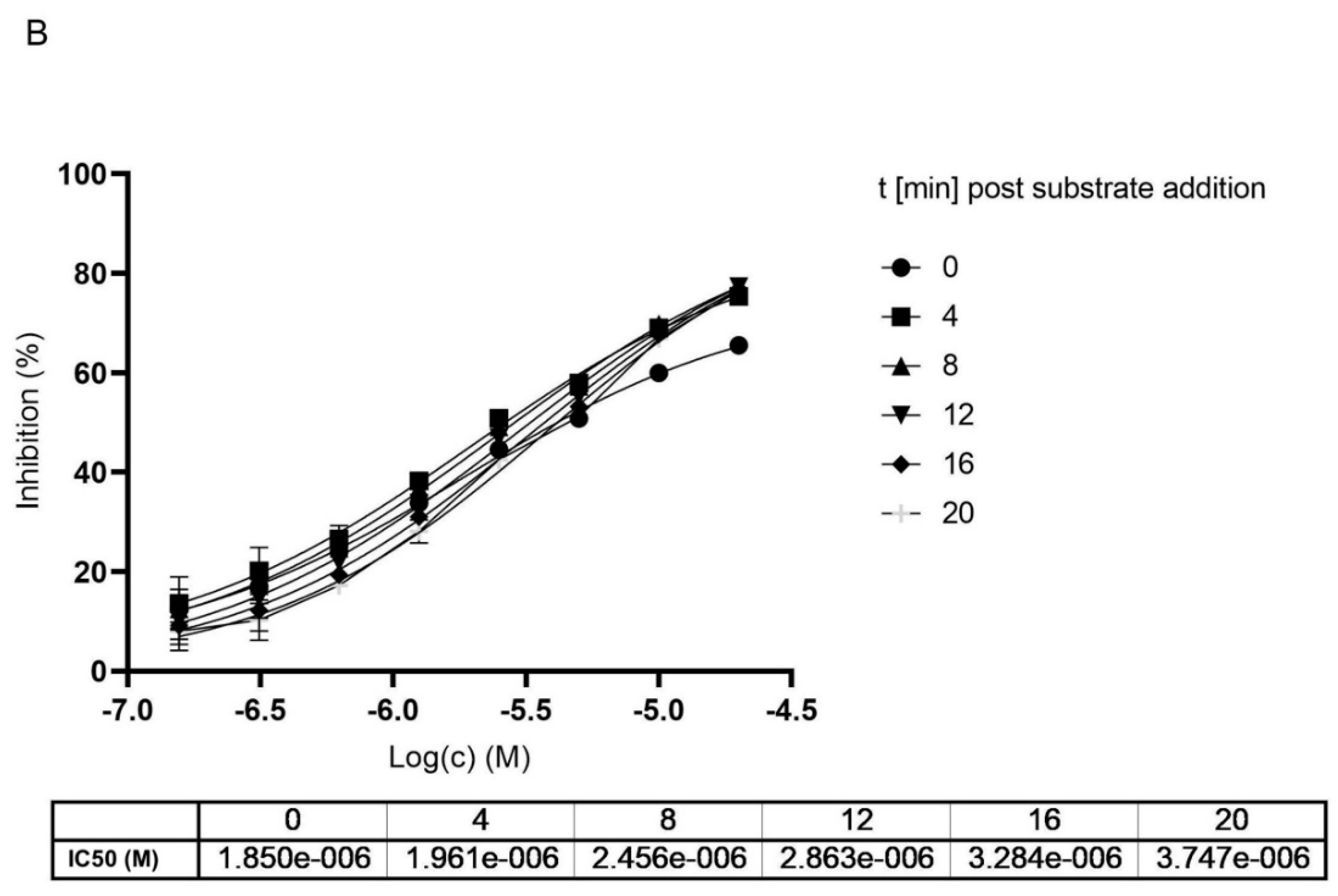
2.3. MG-132 and Cathepsin-L Inhibition
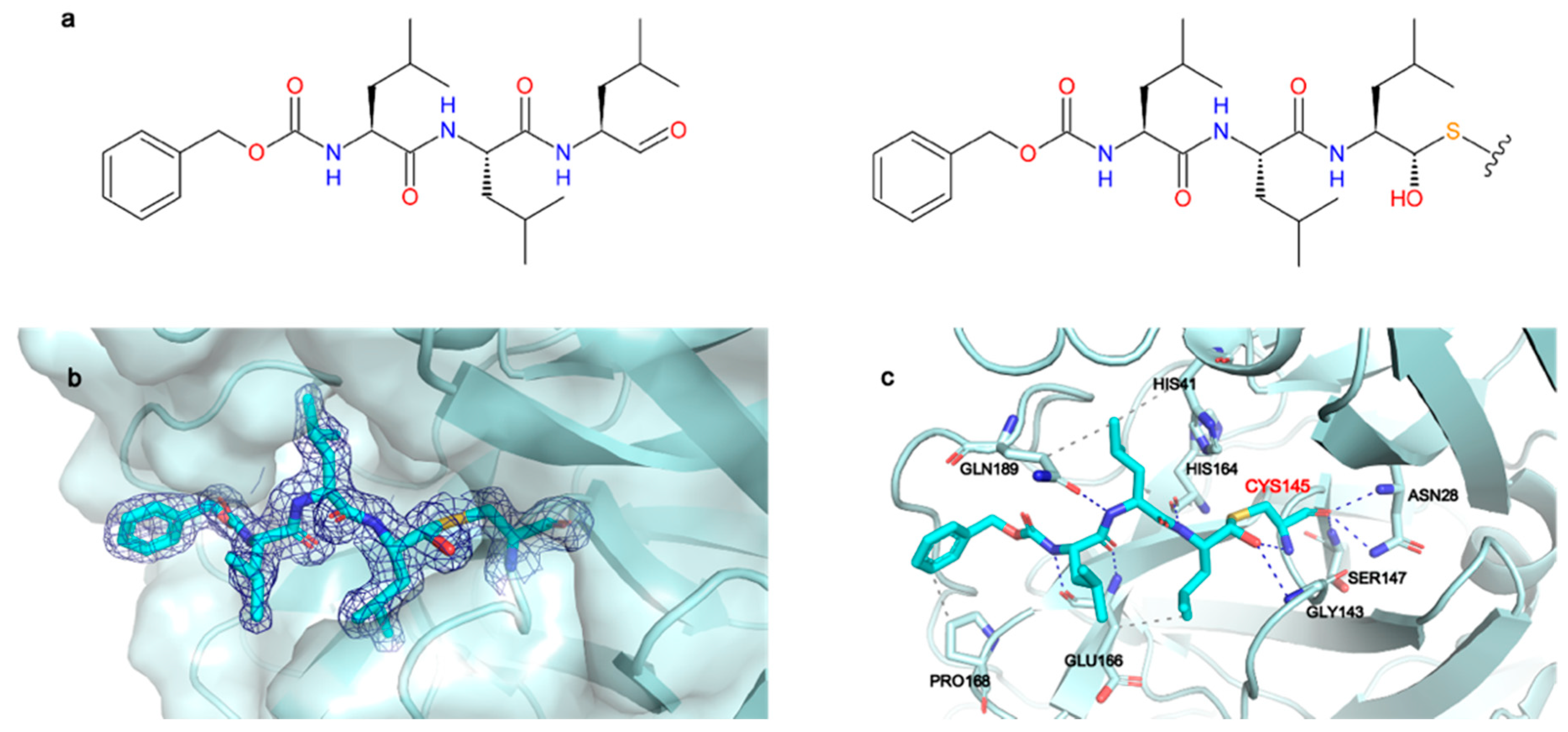
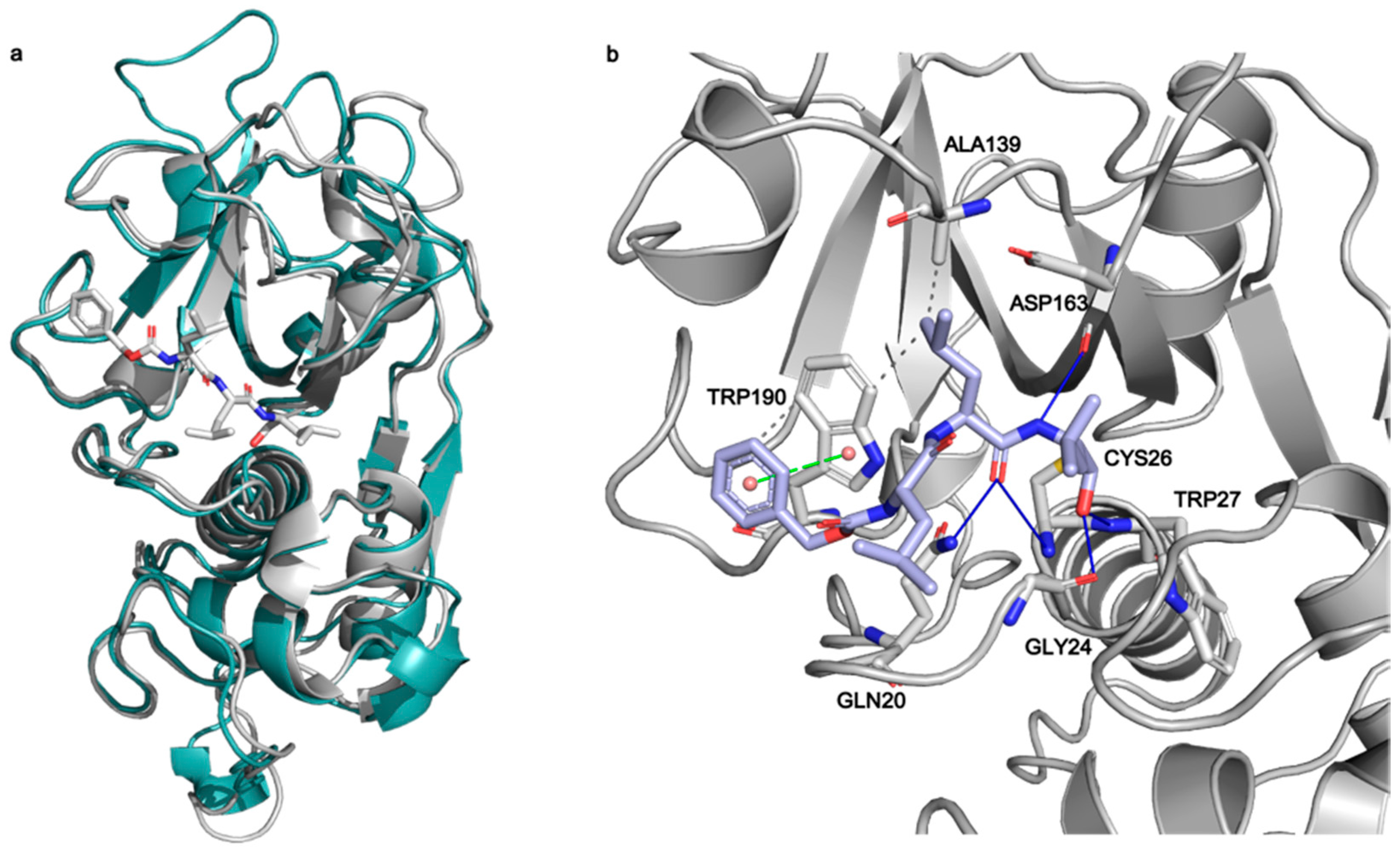
2.4. Docking of MG-132 in Cathepsin-L Binding Site
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Mpro Enzymatic Activity
4.3. Cathepsin-L Inhibition Assay
4.4. Thermostability Assay
4.5. Crystallization
4.6. Data Collection, Data Reduction, Structure Determination, Refinement and Final Model Analysis
4.7. Molecular Docking
4.8. Sequence Alignment
4.9. Docking Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laboratory Testing for 2019 Novel Coronavirus (2019-nCoV) in Suspected Human Cases. Available online: https://www.who.int/publications-detail-redirect/10665-331501 (accessed on 16 June 2021).
- WHO Coronavirus (COVID-19) Dashboard|WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/ (accessed on 16 June 2021).
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Vandyck, K.; Deval, J. Considerations for the Discovery and Development of 3-Chymotrypsin-like Cysteine Protease Inhibitors Targeting SARS-CoV-2 Infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease as a Clinical Candidate for the Potential Treatment of COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pfizer. A Phase 1B, 2-Part, Double-Blind, Placebo-Controlled, Sponsor-Open Study, to Evaluate the Safety, Tolerability and Pharmacokinetics of Single Ascending (24-Hour, Part 1) and Multiple Ascending (120-Hour, Part 2) Intravenous Infusions of pf-07304814 In Hospitalized Participants with COVID-19; Pfizer: New York, NY, USA, 2021. [Google Scholar]
- University of Dundee. A Randomised Double-Blind. Placebo-Controlled Trial of Brensocatib (INS1007) in Patients with Severe COVID-19; University of Dundee: Dundee, UK, 2021. [Google Scholar]
- RedHill Biopharma Limited. Phase 2/3 Study of Upamostat, a Serine Protease Inhibitor, or Placebo for Treatment of COVID-19 Disease; RedHill Biopharma Limited: Raleigh, NC, USA, 2021. [Google Scholar]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-NCoV. Chembiochem 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021. [Google Scholar] [CrossRef]
- Guo, N.; Peng, Z. MG132, a Proteasome Inhibitor, Induces Apoptosis in Tumor Cells. Asia Pac. J. Clin. Oncol. 2013, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goldberg, A.L. Proteasome Inhibitors: Valuable New Tools for Cell Biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe Acute Respiratory Syndrome Coronavirus Replication Is Severely Impaired by MG132 Due to Proteasome-Independent Inhibition of M-Calpain. J. Virol. 2012, 86, 10112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimaru, H.; Hosokawa, K.; Sugimoto, A.; Tanaka, R.; Watanabe, T.; Fujimuro, M. MG132 Exerts Anti-Viral Activity against HSV-1 by Overcoming Virus-Mediated Suppression of the ERK Signaling Pathway. Sci. Rep. 2020, 10, 6671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zhou, X.; Peppelenbosch, M.P.; Pan, Q. Inhibition of Hepatitis E Virus Replication by Proteasome Inhibitor Is Nonspecific. Arch. Virol. 2015, 160, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Kaspari, M.; Tavalai, N.; Stamminger, T.; Zimmermann, A.; Schilf, R.; Bogner, E. Proteasome Inhibitor MG132 Blocks Viral DNA Replication and Assembly of Human Cytomegalovirus. FEBS Lett. 2008, 582, 666–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Zhang, J.; Cheung, C.; Suarez, A.; McManus, B.M.; Yang, D. Proteasome Inhibition Reduces Coxsackievirus B3 Replication in Murine Cardiomyocytes. Am. J. Pathol. 2003, 163, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-M.; Li, Y.-C.; Chen, P.; Ye, S.; Xie, S.-H.; Xia, W.-J.; Yang, J.-H. MG-132 Attenuates Cardiac Deterioration of Viral Myocarditis via AMPK Pathway. Biomed. Pharmacother. 2020, 126, 110091. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Dutch, R.E. Cathepsin L Is Involved in Proteolytic Processing of the Hendra Virus Fusion Protein. J. Virol. 2005, 79, 12714–12720. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.-P. Cysteine Protease Cathepsins in Cardiovascular Disease: From Basic Research to Clinical Trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C.; et al. Cathepsin L Plays a Key Role in SARS-CoV-2 Infection in Humans and Humanized Mice and Is a Promising Target for New Drug Development. Signal. Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J.M. Cathepsin L Functionally Cleaves the Severe Acute Respiratory Syndrome Coronavirus Class I Fusion Protein Upstream of Rather than Adjacent to the Fusion Peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Cho, C.-C.D.; Geng, Z.Z.; Ma, X.R.; Allen, R.; Shaabani, N.; Vatansever, E.C.; Alugubelli, Y.R.; Ma, Y.; Ellenburg, W.H.; et al. Cellular Activities of SARS-CoV-2 Main Protease Inhibitors Reveal Their Unique Characteristics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Gossen, J.; Albani, S.; Hanke, A.; Joseph, B.P.; Bergh, C.; Kuzikov, M.; Costanzi, E.; Manelfi, C.; Storici, P.; Gribbon, P.; et al. A Blueprint for High Affinity SARS-CoV-2 Mpro Inhibitors from Activity-Based Compound Library Screening Guided by Analysis of Protein Dynamics. ACS Pharmacol. Transl. Sci. 2021, 4, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.; Riley, B.T.; Kumaran, D.; Andi, B.; Fuchs, M.R.; McSweeney, S.; Keedy, D.A. The Temperature-Dependent Conformational Ensemble of SARS-CoV-2 Main Protease (Mpro). bioRxiv 2021. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural Plasticity of SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-ray Crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Wang, Q.; Xing, Y.; Feng, L.; Kong, J.; Peng, C.; Zhang, L.; Yang, H.; Lu, M. Identification of Proteasome and Caspase Inhibitors Targeting SARS-CoV-2 Mpro. Sig. Transduct. Target. Ther. 2021, 6, 214. [Google Scholar] [CrossRef]
- Galasiti Kankanamalage, A.C.; Kim, Y.; Damalanka, V.C.; Rathnayake, A.D.; Fehr, A.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Lushington, G.H.; Perlman, S.; et al. Structure-Guided Design of Potent and Permeable Inhibitors of MERS Coronavirus 3CL Protease That Utilize a Piperidine Moiety as a Novel Design Element. Eur. J. Med. Chem. 2018, 150, 334–346. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against Mpro and Cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- Hung, H.-C.; Ke, Y.-Y.; Huang, S.Y.; Huang, P.-N.; Kung, Y.-A.; Chang, T.-Y.; Yen, K.-J.; Peng, T.-T.; Chang, S.-E.; Huang, C.-T.; et al. Discovery of M Protease Inhibitors Encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00872-20. [Google Scholar] [CrossRef]
- Krippendorff, B.-F.; Neuhaus, R.; Lienau, P.; Reichel, A.; Huisinga, W. Mechanism-Based Inhibition: Deriving KI and Kinact Directly from Time-Dependent IC50 Values. J. Biomol. Screen. 2009, 14, 913–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Rafferty, B.; Caldwell, G.W.; Masucci, J.A. Rapidly Distinguishing Reversible and Irreversible CYP450 Inhibitors by Using Fluorometric Kinetic Analyses. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, M.; Lee, J.S.; Whig, K.; Segrist, E.; Li, M.; Jurado, K.; Samby, K.; Ramage, H.; Schultz, D.; Cherry, S. Drug Repurposing Screens Reveal FDA Approved Drugs Active against SARS-Cov-2. 2020. Available online: https://doi.org/10.1101/2020.06.19.161042 (accessed on 16 June 2021).
- Ito, H.; Watanabe, M.; Kim, Y.-T.; Takahashi, K. Inhibition of Rat Liver Cathepsins B and L by the Peptide Aldehyde Benzyloxycarbonyl-Leucyl-Leucyl-Leucinal and Its Analogues. J. Enzym. Inhib. Med. Chem. 2009, 24, 279–286. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, J.M.; Zhao, B.; Smith, W.W.; Janson, C.A.; DesJarlais, R.L.; Tomaszek, T.A.; Carr, T.J.; Thompson, S.K.; Oh, H.J.; Yamashita, D.S.; et al. Use of Papain as a Model for the Structure-Based Design of Cathepsin K Inhibitors: Crystal Structures of Two Papain-Inhibitor Complexes Demonstrate Binding to S’-Subsites. J. Med. Chem. 1998, 41, 4567–4576. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why Is Tanimoto Index an Appropriate Choice for Fingerprint-Based Similarity Calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, R.T.; Sivaraman, J. Structural Basis for Reversible and Irreversible Inhibition of Human Cathepsin L by Their Respective Dipeptidyl Glyoxal and Diazomethylketone Inhibitors. J. Struct. Biol. 2011, 173, 14–19. [Google Scholar] [CrossRef]
- Pfizer Initiates Phase 1 Study of Novel Oral Antiviral Therapeutic Agent Against SARS-CoV-2 Pfpfizeruscom. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-initiates-phase-1-study-novel-oral-antiviral (accessed on 17 August 2021).
- PF-07321332. Available online: https://go.drugbank.com/drugs/DB16691 (accessed on 17 August 2021).
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What Coronavirus 3C-like Protease Tells Us: From Structure, Substrate Selectivity, to Inhibitor Design. Med. Res. Rev. 2021, 41, 1965–1998. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R.J. Cysteine Proteases as Therapeutic Targets: Does Selectivity Matter? A Systematic Review of Calpain and Cathepsin Inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.-P. Cathepsin L-Selective Inhibitors: A Potentially Promising Treatment for COVID-19 Patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti-COVID-19 Drug Design. Sci. Adv. 2020. [Google Scholar] [CrossRef] [PubMed]
- Biniossek, M.L.; Nägler, D.K.; Becker-Pauly, C.; Schilling, O. Proteomic Identification of Protease Cleavage Sites Characterizes Prime and Non-Prime Specificity of Cysteine Cathepsins B, L, and S. J. Proteome Res. 2011, 10, 5363–5373. [Google Scholar] [CrossRef]
- Choe, Y.; Leonetti, F.; Greenbaum, D.C.; Lecaille, F.; Bogyo, M.; Brömme, D.; Ellman, J.A.; Craik, C.S. Substrate Profiling of Cysteine Proteases Using a Combinatorial Peptide Library Identifies Functionally Unique Specificities. J. Biol. Chem. 2006, 281, 12824–12832. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.R.; Alugubelli, Y.R.; Ma, Y.; Vantasever, E.C.; Scott, D.A.; Qiao, Y.; Yu, G.; Xu, S.; Liu, W.R. MPI8 Is Potent Against SARS-CoV-2 by Inhibiting Dually and Selectively the SARS-CoV-2 Main Protease and the Host Cathepsin L. ChemMedChem 2021. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Belovodskiy, A.; Hena, M.; Kandadai, A.S.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Khan, M.B.; Arutyunova, E.; Lu, J.; et al. Peptidomimetic α-Acyloxymethylketone Warheads with Six-Membered Lactam P1 Glutamine Mimic: SARS-CoV-2 3CL Protease Inhibition, Coronavirus Antiviral Activity, and in Vitro Biological Stability. J. Med. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.-O.; Pedersen, N.C. Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016, 12, e1005531. [Google Scholar] [CrossRef]
- Lausi, A.; Polentarutti, M.; Onesti, S.; Plaisier, J.R.; Busetto, E.; Bais, G.; Barba, L.; Cassetta, A.; Campi, G.; Lamba, D.; et al. Status of the Crystallography Beamlines at Elettra. Eur. Phys. J. Plus 2015, 130, 43. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How Good Are My Data and What Is the Resolution? Acta Cryst. D 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie AG, W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 235–242. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75 Pt 10, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PKaprediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Altschul, S. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schaffer, A.A.; Yu, Y.-K. Protein Database Searches Using Compositionally Adjusted Substitution Matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Asaad, N.; Bethel, P.A.; Coulson, M.D.; Dawson, J.E.; Ford, S.J.; Gerhardt, S.; Grist, M.; Hamlin, G.A.; James, M.J.; Jones, E.V.; et al. Dipeptidyl Nitrile Inhibitors of Cathepsin L. Bioorganic. Med. Chem. Lett. 2009, 19, 4280–4283. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costanzi, E.; Kuzikov, M.; Esposito, F.; Albani, S.; Demitri, N.; Giabbai, B.; Camasta, M.; Tramontano, E.; Rossetti, G.; Zaliani, A.; et al. Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L. Int. J. Mol. Sci. 2021, 22, 11779. https://doi.org/10.3390/ijms222111779
Costanzi E, Kuzikov M, Esposito F, Albani S, Demitri N, Giabbai B, Camasta M, Tramontano E, Rossetti G, Zaliani A, et al. Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L. International Journal of Molecular Sciences. 2021; 22(21):11779. https://doi.org/10.3390/ijms222111779
Chicago/Turabian StyleCostanzi, Elisa, Maria Kuzikov, Francesca Esposito, Simone Albani, Nicola Demitri, Barbara Giabbai, Marianna Camasta, Enzo Tramontano, Giulia Rossetti, Andrea Zaliani, and et al. 2021. "Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L" International Journal of Molecular Sciences 22, no. 21: 11779. https://doi.org/10.3390/ijms222111779
APA StyleCostanzi, E., Kuzikov, M., Esposito, F., Albani, S., Demitri, N., Giabbai, B., Camasta, M., Tramontano, E., Rossetti, G., Zaliani, A., & Storici, P. (2021). Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L. International Journal of Molecular Sciences, 22(21), 11779. https://doi.org/10.3390/ijms222111779