
Hepatocyte-Specific Deficiency of BAP31 Amplified Acetaminophen-Induced Hepatotoxicity via Attenuating Nrf2 Signaling Activation in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
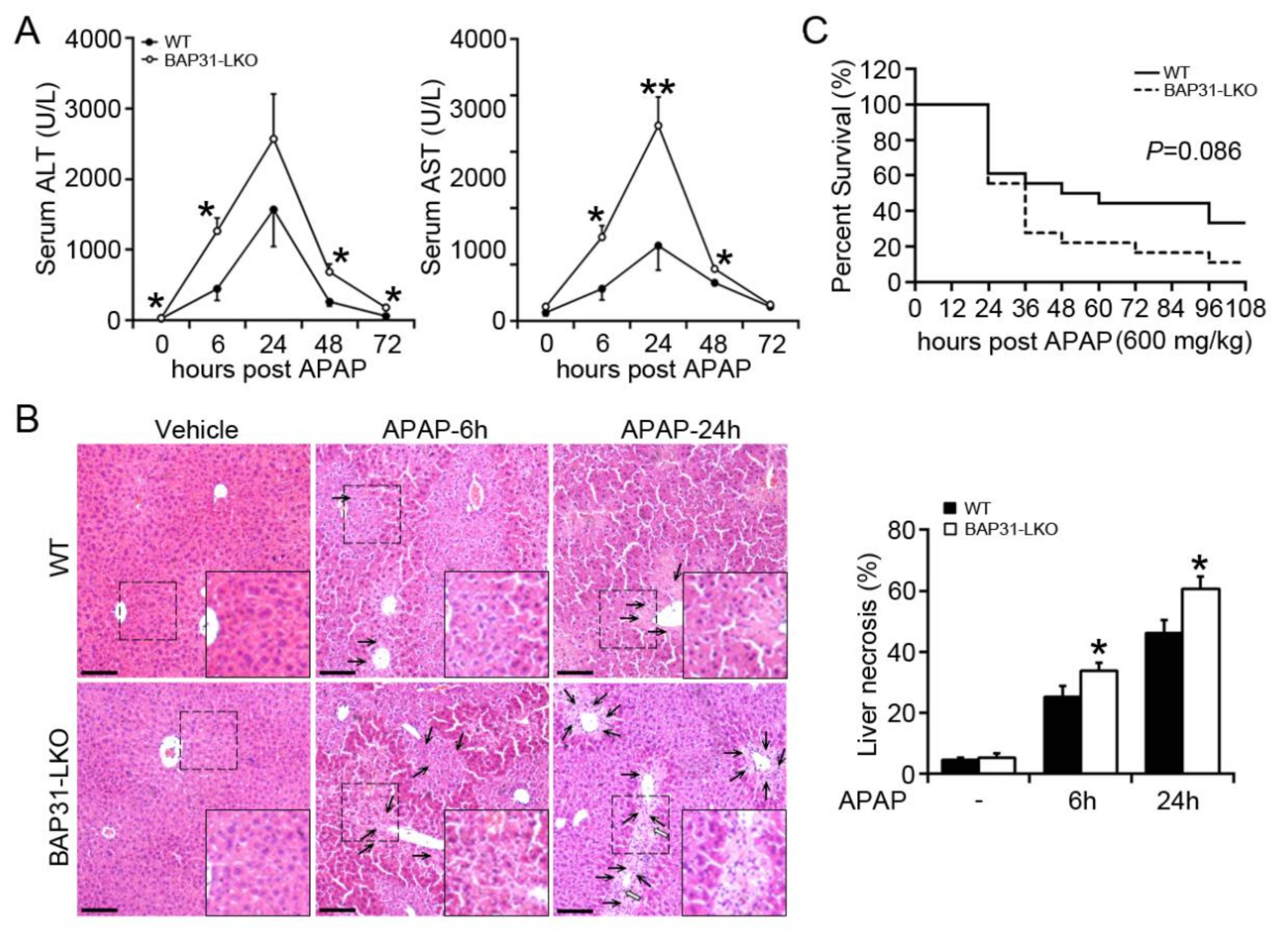
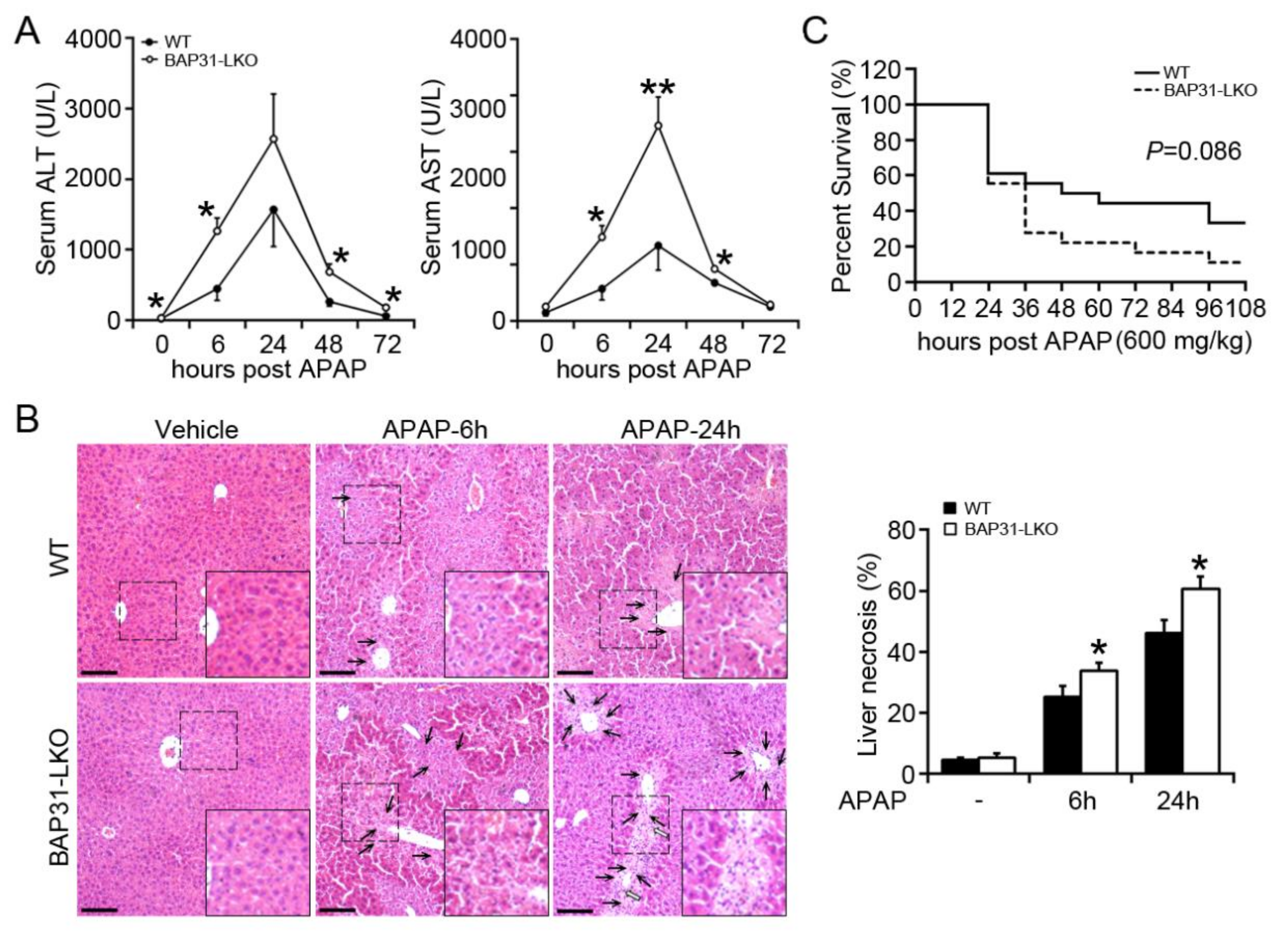
2.1. BAP31-Deficiency Increased APAP-Induced Liver Injury
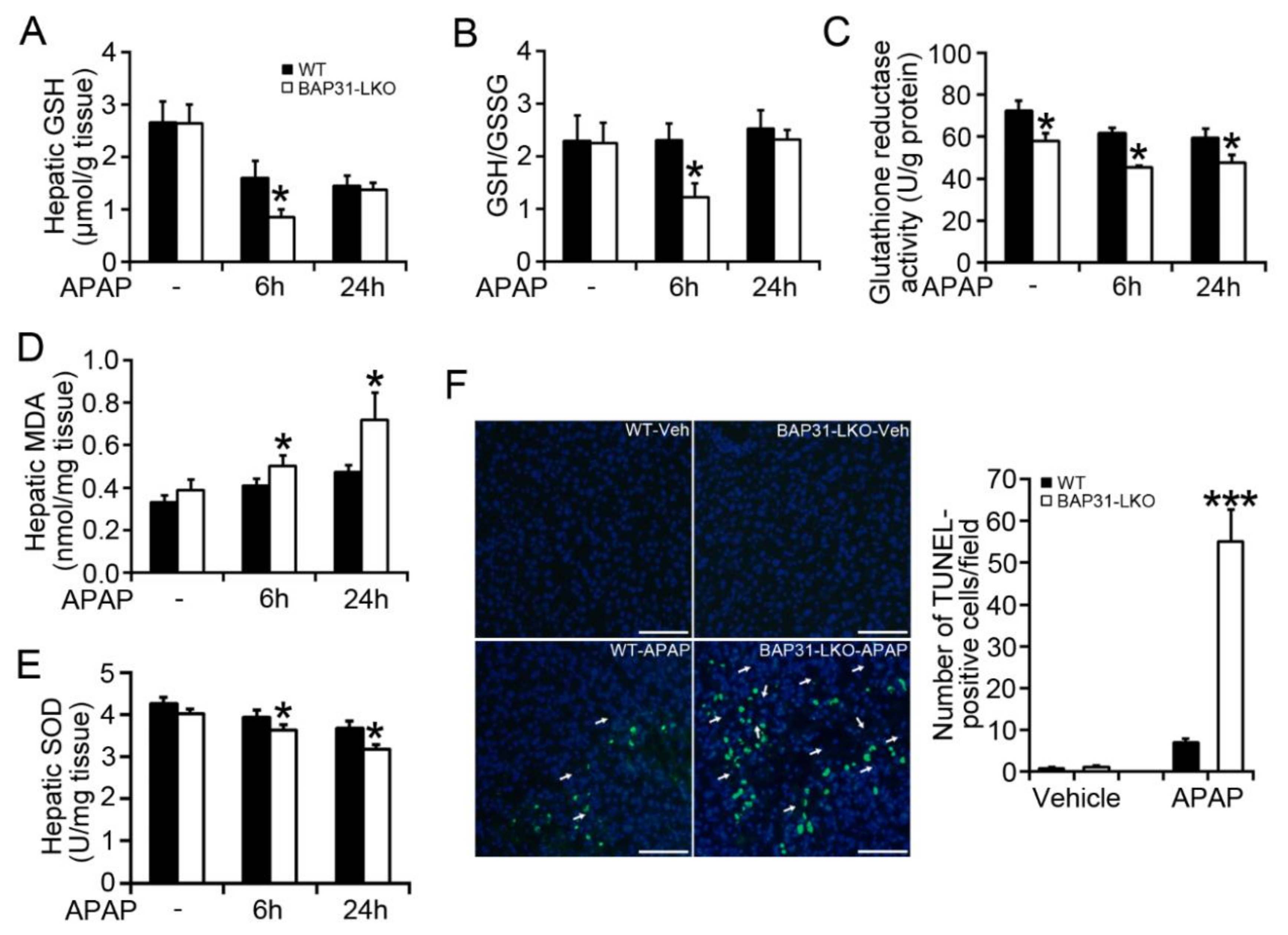
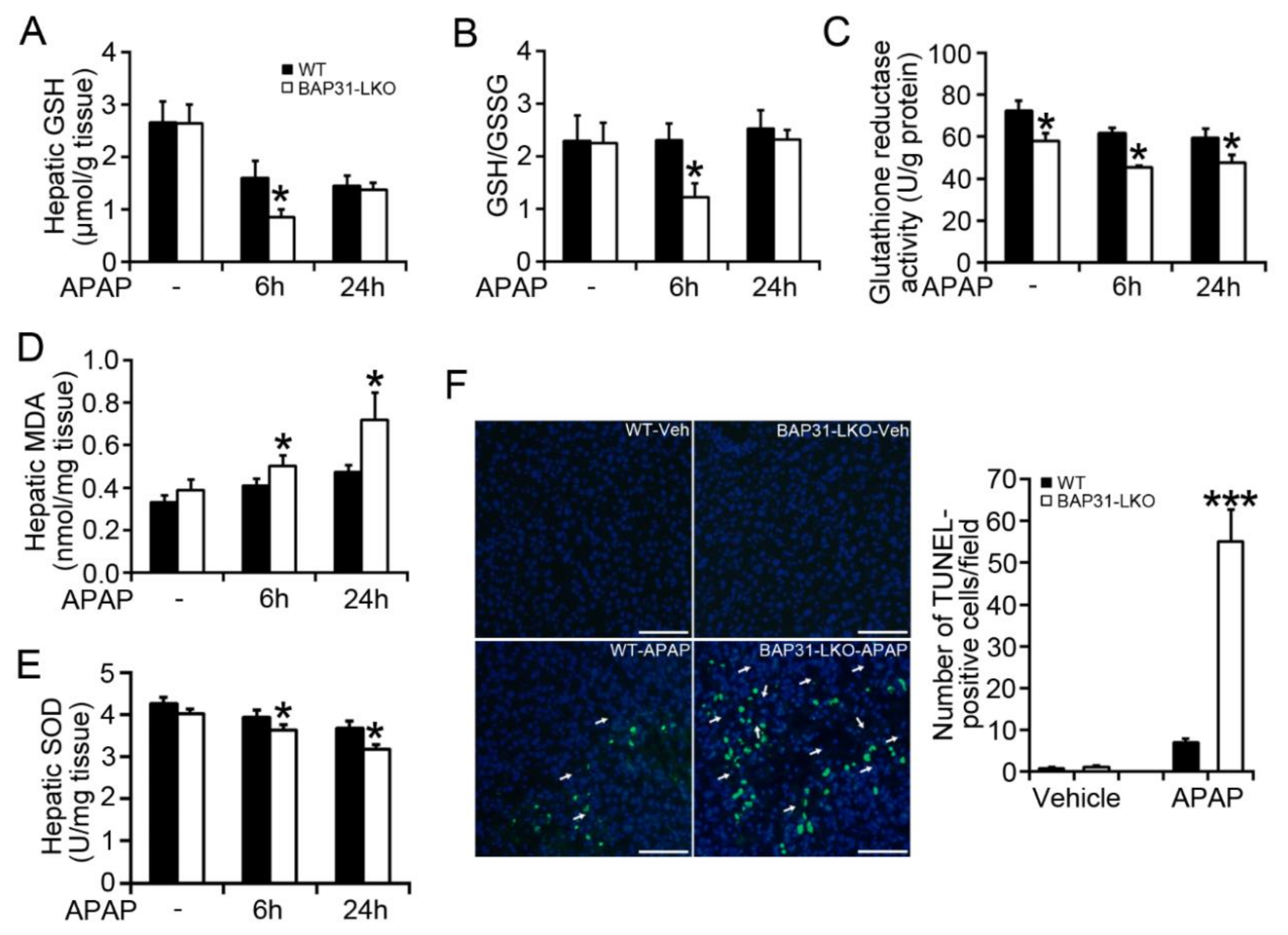
2.2. BAP31-Deficiency Amplified APAP-Induced GSH Depletion and Hepatotoxicity
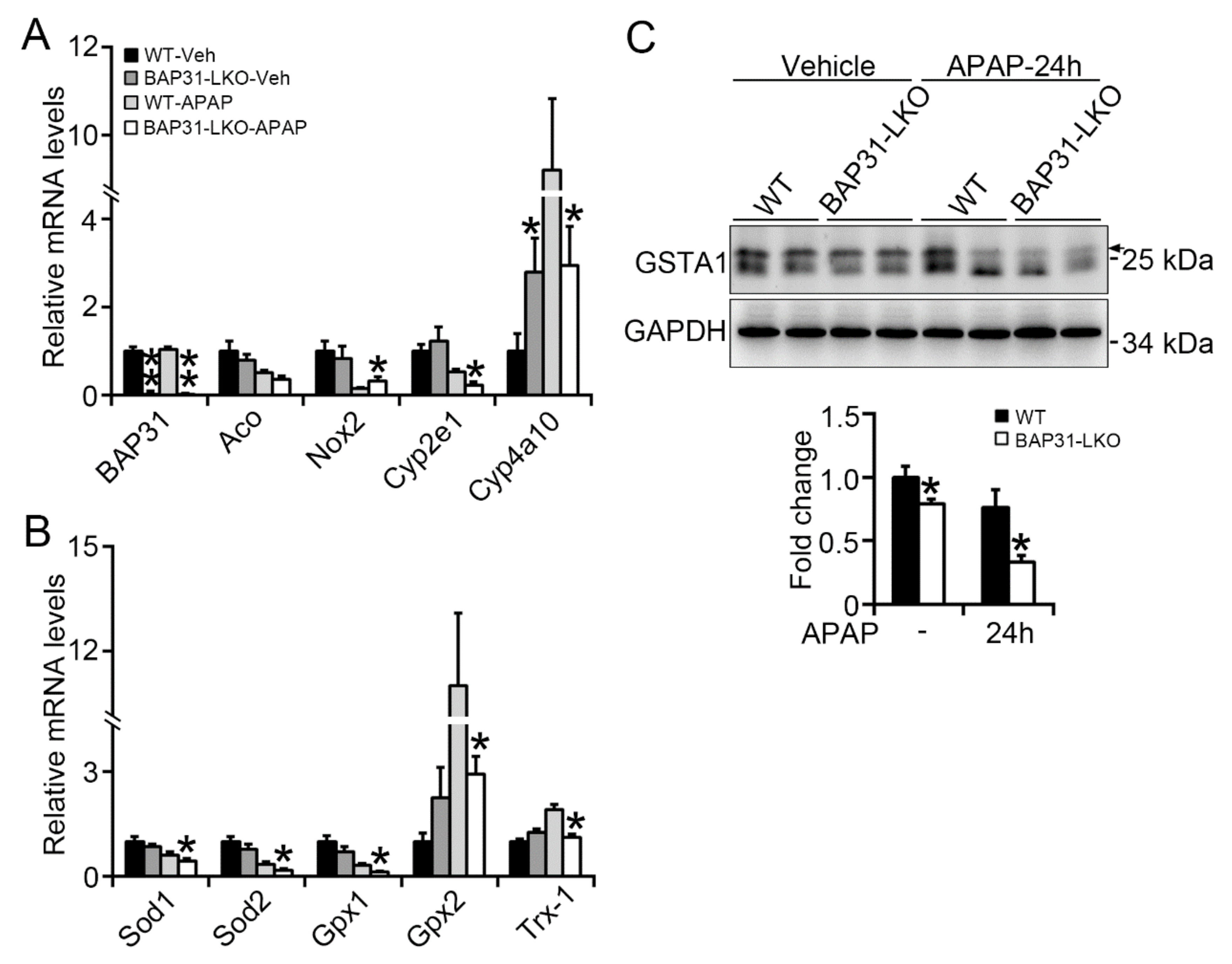
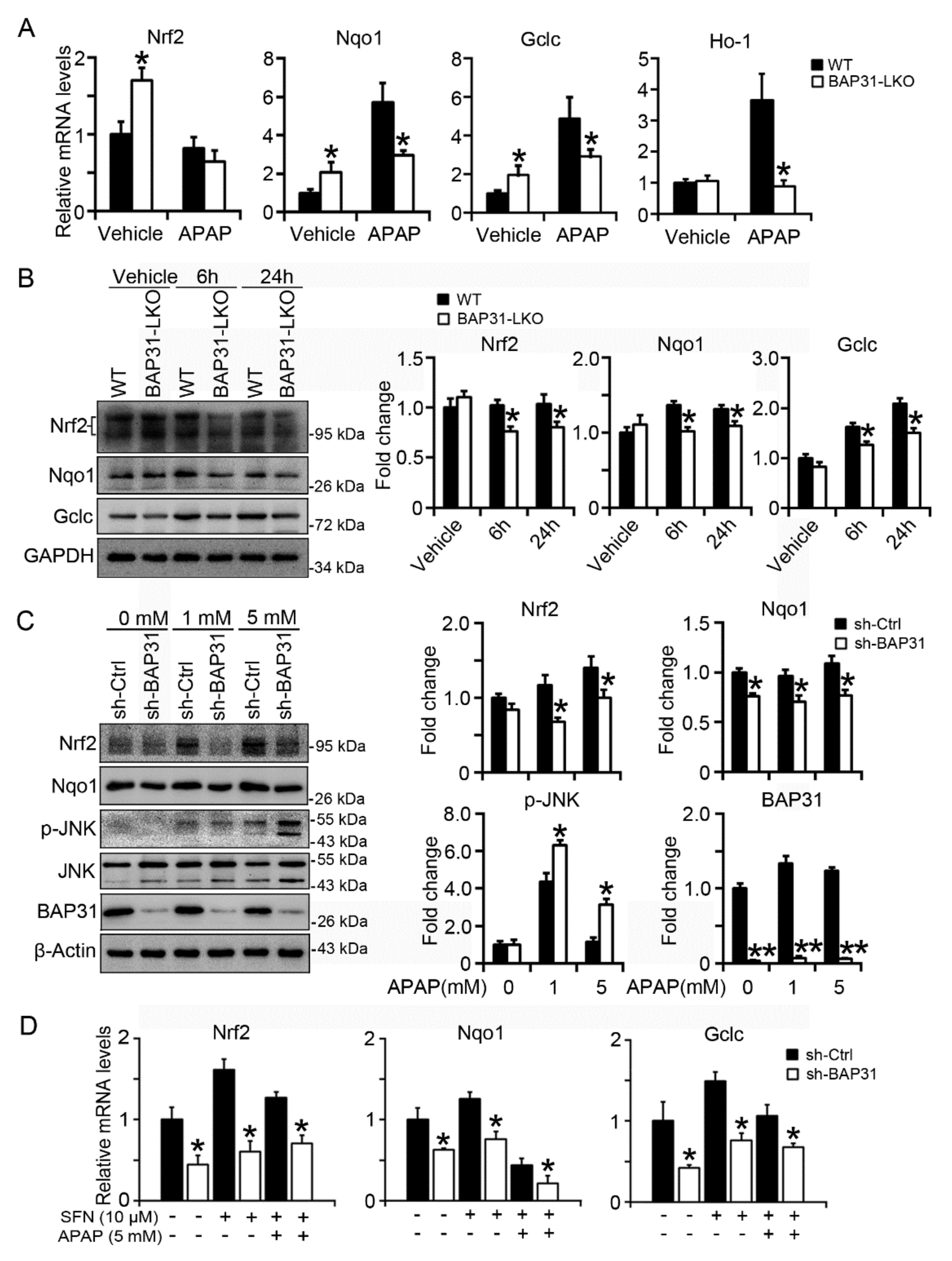
2.3. BAP31-Deficiency Reduced Antioxidant and Detoxification Genes Expression
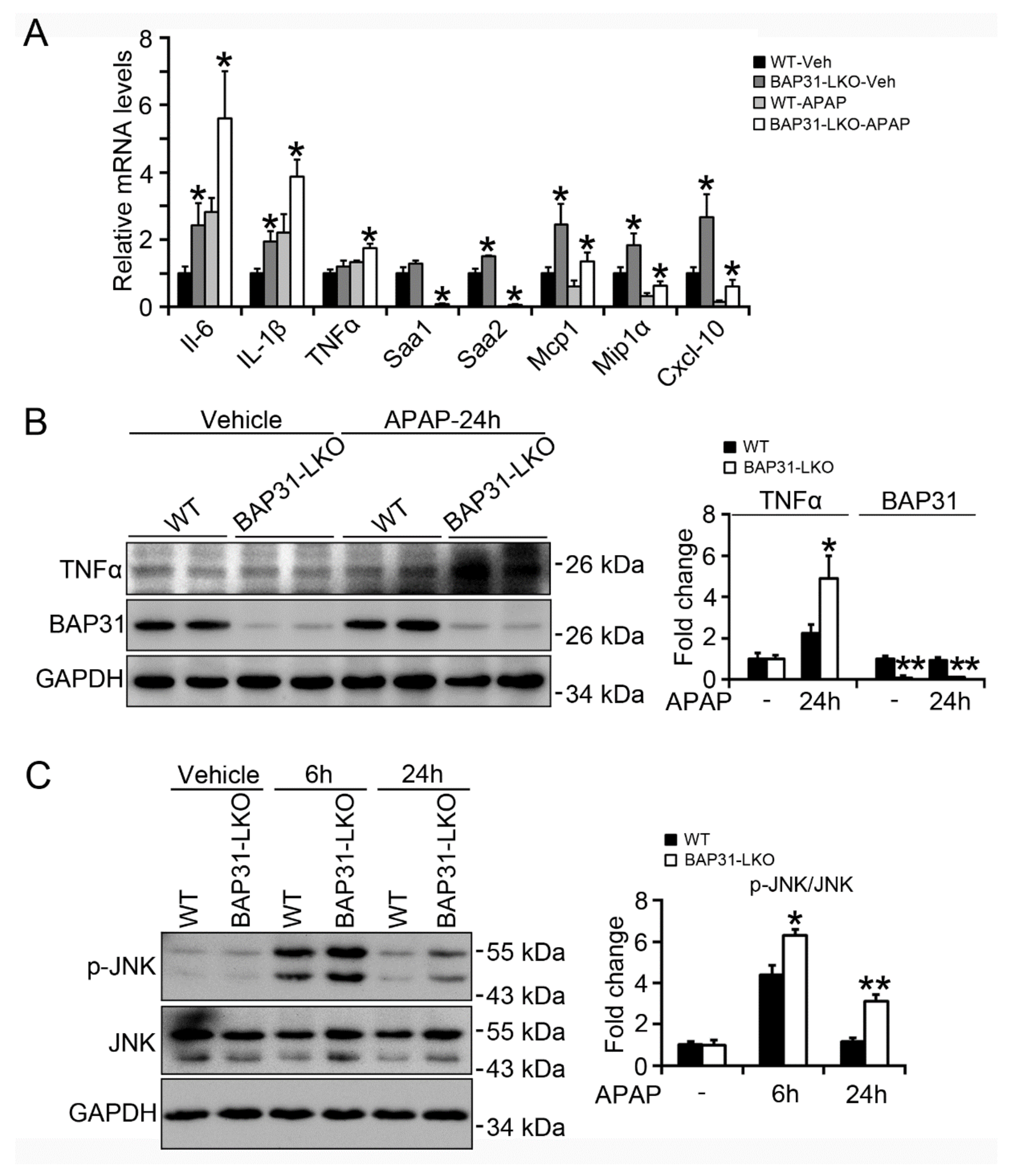
2.4. BAP31-Deficiency Increased APAP-Induced Inflammatory Response
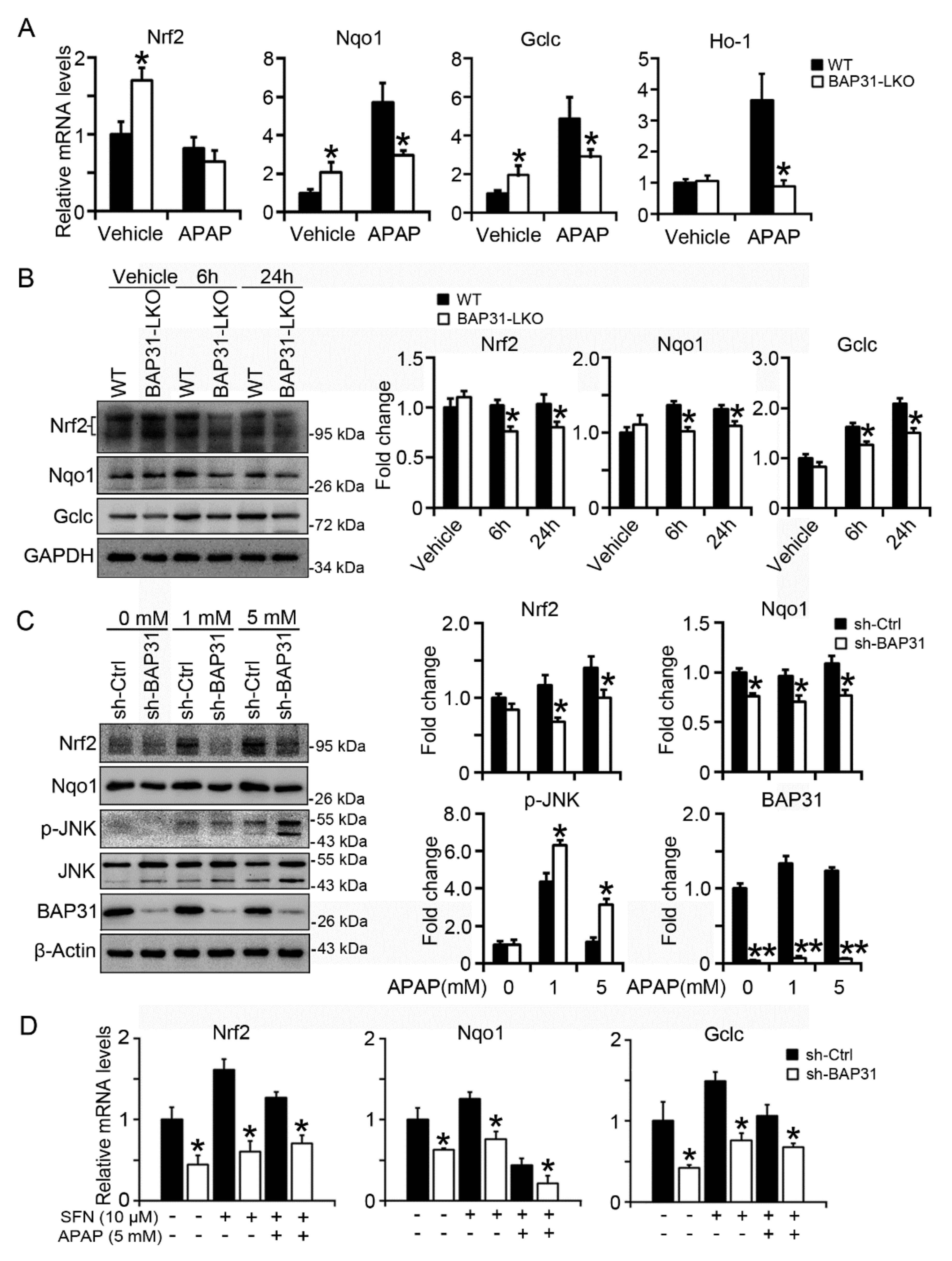
2.5. BAP31-Deficiency Reduced Nrf2 Signaling Activation
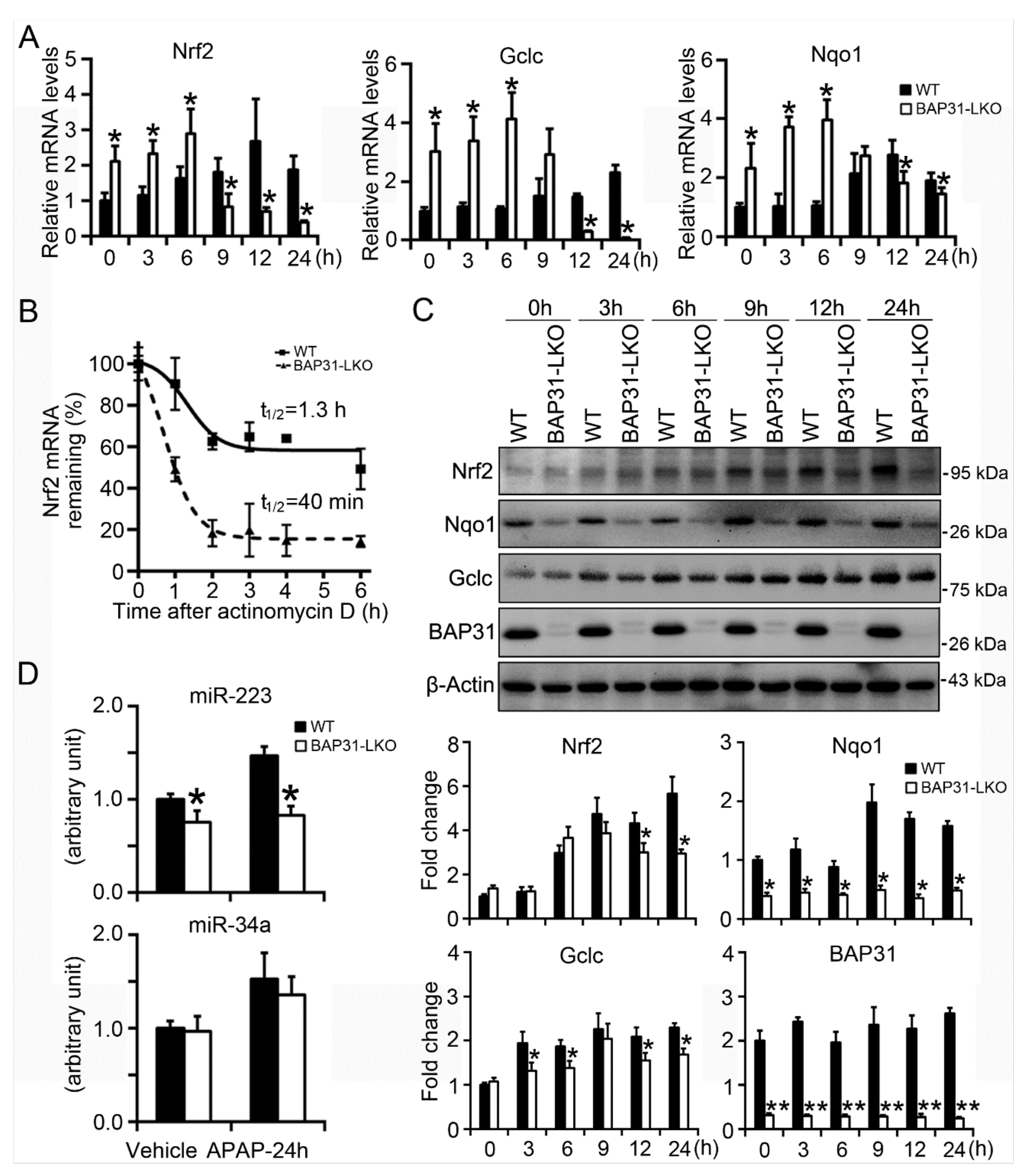
2.6. BAP31-Deficiency Reduced mRNA Stability and Impaired Nrf2 Signaling Induction
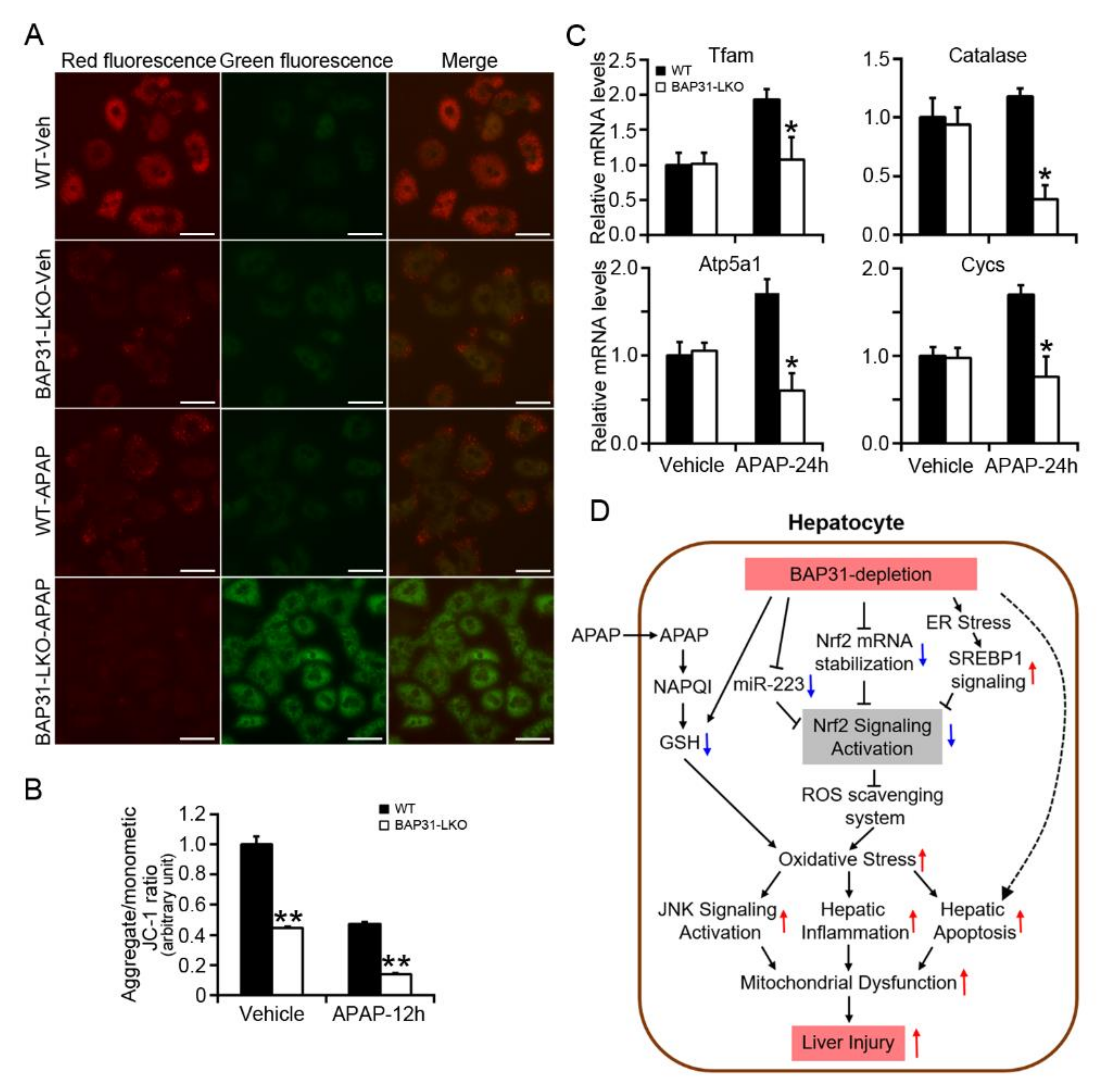
2.7. BAP31-Deficiency Increased Mitochondrial Dysfunction
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. APAP Administration
4.3. Survival Rate Analysis
4.4. Primary Hepatocyte Isolation and Cell Culture
4.5. Liver Enzymes, Glutathione, and Glutathione Reductase Activity Measurement
4.6. Histopathology
4.7. TUNEL Assay
4.8. Assessment of Mitochondrial Membrane Potential
4.9. RNA Extraction, Reverse Transcription, and qRT-PCR
4.10. RNA Stability Assay
4.11. Western Blot Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blieden, M.; Paramore, L.C.; Shah, D.; Ben-Joseph, R. A Perspective on the Epidemiology of Acetaminophen Exposure and Toxicity in the United States. Expert Rev. Clin. Pharmacol. 2014, 7, 341–348. [Google Scholar] [CrossRef]
- Manthripragada, A.D.; Zhou, E.H.; Budnitz, D.S.; Lovegrove, M.C.; Willy, M.E. Characterization of Acetaminophen Overdose-Related Emergency Department Visits and Hospitalizations in the United States. Pharmacoepidemiol. Drug Saf. 2011, 20, 819–826. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Metabolism and Disposition of Acetaminophen: Recent Advances in Relation to Hepatotoxicity and Diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [Green Version]
- Borude, P.; Bhushan, B.; Apte, U. DNA Damage Response Regulates Initiation of Liver Regeneration Following Acetaminophen Overdose. Gene Expr. 2018, 18, 115–123. [Google Scholar] [CrossRef]
- Abe, F.; Van Prooyen, N.; Ladasky, J.J.; Edidin, M. Interaction of Bap31 and MHC Class I Molecules and Their Traffic Out of the Endoplasmic Reticulum. J. Immunol. 2009, 182, 4776–4783. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, M. BAP31 and Its Caspase Cleavage Product Regulate Cell Surface Expression of Tetraspanins and Integrin-mediated Cell Survival. J. Biol. Chem. 2005, 280, 30018–30024. [Google Scholar] [CrossRef] [Green Version]
- Niu, K.; Xu, J.; Cao, Y.; Hou, Y.; Shan, M.; Wang, Y.; Xu, Y.; Sun, M.; Wang, B. BAP31 Is Involved in T Cell Activation through TCR Signal Pathways. Sci. Rep. 2017, 7, 44809. [Google Scholar] [CrossRef]
- Dang, E.; Yang, S.; Song, C.; Jiang, D.; Li, Z.; Fan, W.; Sun, Y.; Tao, L.; Wang, J.; Liu, T.; et al. BAP31, a Newly Defined Cancer/Testis Antigen, Regulates Proliferation, Migration, and Invasion to Promote Cervical Cancer Progression. Cell Death Dis. 2018, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Li, L.Y.; Wang, Y.Q.; Li, Y.Q.; Shan, M.; Sun, S.Z.; Yu, Y.; Wang, B. Hepatocyte-Specific Deletion of BAP31 Promotes SREBP1C Activation, Promotes Hepatic Lipid Accumulation, and Worsens IR in Mice. J. Lipid. Res. 2018, 59, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Fan, Y.; Shan, J.; Xiaoyu, S.; Jialin, X. Induction of Liver Steatosis in BAP31-Deficient Mice Burdened with Tunicamycin-Induced Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasa, M.; Yamagata, T.; Mizuguchi, M.; Itoh, M.; Matsumoto, A.; Hironaka, M.; Honda, A.; Momoi, M.Y.; Shimozawa, N. Contiguous ABCD1 DXS1357E Deletion Syndrome: Report of an Autopsy Case. Neuropathology 2013, 33, 292–298. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Liu, K.H.; Owens, J.A.; Hunter-Chang, S.; Camacho, M.C.; Eboka, R.U.; Chandrasekharan, B.; Baker, N.F.; Darby, T.M.; Robinson, B.S.; et al. Gut-Resident Lactobacilli Activate Hepatic Nrf2 and Protect Against Oxidative Liver Injury. Cell Metab. 2020, 31, 956–968. [Google Scholar] [CrossRef]
- Bataille, A.M.; Manautou, J.E. Nrf2: A Potential Target for New Therapeutics in Liver Disease. Clin. Pharmacol. Ther. 2012, 92, 340–348. [Google Scholar] [CrossRef]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The Mechanism Underlying Acetaminophen-Induced Hepatotoxicity in Humans and Mice Involves Mitochondrial Damage and Nuclear DNA Fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivarini, A.C.; Calegari-Silva, T.C.; Saliba, A.M.; Boaventura, V.S.; Franca-Costa, J.; Khouri, R.; Dierckx, T.; Dias-Teixeira, K.L.; Fasel, N.; Barral, A.M.P.; et al. Systems Approach Reveals Nuclear Factor Erythroid 2-Related Factor 2/Protein Kinase R Crosstalk in Human Cutaneous Leishmaniasis. Front. Immunol. 2017, 8, 1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Rangasamy, T.; Thimmulappa, R.K.; Lee, H.; Osburn, W.O.; Brigelius-Flohe, R.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Glutathione Peroxidase 2, the Major Cigarette Smoke-Inducible Isoform of GPX in Lungs, Is Regulated by Nrf2. Am. J. Respir. Cell Mol. Biol. 2006, 35, 639–650. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Samarghandian, S.; Mohammadinejad, R.; Yaribeygi, H.; Sathyapalan, T.; Sahebkar, A. MicroRNA-Mediated Regulation of Nrf2 Signaling Pathway: Implications in Disease Therapy and Protection against Oxidative Stress. Life Sci. 2020, 244, 117329. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Wang, Y.; Li, H.; Jia, W.; Man, K.; Lo, C.M.; Wang, Y.; Lam, K.S.; Xu, A. Fibroblast Growth Factor 21 Protects against Acetaminophen-Induced Hepatotoxicity by Potentiating Peroxisome Proliferator-Activated Receptor Coactivator Protein-1alpha-Mediated Antioxidant Capacity in Mice. Hepatology 2014, 60, 977–989. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-Mediated Antioxidant Actions of Pro-electrophilic Drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Kay, H.Y.; Kim, Y.W.; Ryu, D.H.; Sung, S.H.; Hwang, S.J.; Kim, S.G. Nrf2-Mediated Liver Protection by Sauchinone, an Antioxidant Lignan, from Acetaminophen Toxicity through the PKCdel-Ta-GSK3beta Pathway. Br. J. Pharmacol. 2011, 163, 1653–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-Specific Deletion of the keap1 Gene Activates Nrf2 and Confers Potent Resistance against Acute Drug Toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Poganik, J.R.; Long, M.J.C.; Disare, M.T.; Liu, X.; Chang, S.H.; Hla, T.; Aye, Y. Post-transcriptional Regulation of Nrf2-mRNA by the mRNA-Binding Proteins HuR and AUF1. FASEB J. 2019, 33, 14636–14652. [Google Scholar] [CrossRef] [Green Version]
- Gong, P.; Cederbaum, A.I. Nrf2 Is Increased by CYP2E1 in Rodent Liver and HepG2 Cells and Protects against Oxidative Stress Caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar] [CrossRef]
- Cacciagli, P.; Sutera-Sardo, J.; Borges-Correia, A.; Roux, J.C.; Dorboz, I.; Desvignes, J.P.; Badens, C.; Delepine, M.; Lathrop, M.; Cau, P.; et al. Mutations in BCAP31 Cause a Severe X-linked Phenotype with Deafness, Dystonia, and Central Hypomyelination and Disorganize the Golgi Apparatus. Am. J. Hum. Genet. 2013, 93, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Hannah, H.-E.; Donglei, Z.; Nhi, N.; David, Y.T.; John, W.H.; Gordon, C.S. BAP31 Interacts with Sec61 Translocons and Promotes Retrotranslocation of CFTRΔF508 via the Derlin-1 Complex. Cell 2008, 133, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Namba, T. BAP31 Regulates Mitochondrial Function via Interaction with Tom40 within ER-Mitochondria Contact Sites. Sci. Adv. 2019, 5, eaaw1386. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 Signaling and ER Stress Crosstalk: Implication for Proteasomal Degradation and Autophagy. Cell. Mol. Life. Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-Dependent Activation of Nrf2 Contributes to Redox Homeostasis and Cell Survival Following Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [Green Version]
- Nigro, D.; Menotti, F.; Cento, A.S.; Serpe, L.; Chiazza, F.; Dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic Administration of Saturated Fats and Fructose Differently Affect SREBP Activity Resulting in Different Modulation of Nrf2 and Nlrp3 Inflammasome Pathways in Mice Liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef]
- Xie, K.; He, X.; Chen, K.; Sakao, K.; Hou, D.X. Ameliorative Effects and Molecular Mechanisms of Vine Tea on Western Diet-Induced NAFLD. Food Funct. 2020, 11, 5976–5991. [Google Scholar] [CrossRef]
- Ding, X.; Jian, T.; Wu, Y.; Zuo, Y.; Li, J.; Lv, H.; Ma, L.; Ren, B.; Zhao, L.; Li, W.; et al. Ellagic Acid Ameliorates Oxidative Stress and Insulin Resistance in High Glucose-Treated HepG2 Cells via miR-223/keap1-Nrf2 Pathway. Biomed. Pharmacother. 2019, 110, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, C.; Ferrer-Mayorga, G.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Martin-Duce, A.; Velasco-Martin, J.P.; Regadera, J.; Fernandez, M.; Alemany, S. Sterile Inflammation in Acetaminophen-induced Liver Injury Is Mediated by Cot/tpl2. J. Biol. Chem. 2013, 288, 15342–15351. [Google Scholar] [CrossRef] [Green Version]
- Woolbright, B.L.; Jaeschke, H. Role of the Inflammasome in Acetaminophen-Induced Liver Injury and Acute Liver Failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Kamo, N.; Ke, B.; Ghaffari, A.A.; Shen, X.d.; Busuttil, R.W.; Cheng, G.; Kupiec-Weglinski, J.W. ASC/Caspase-1/IL-1β Signaling Triggers Inflammatory Responses by Promoting HMGB1 Induction in Liver Ischemia/Reperfusion Injury. Hepatology 2013, 58. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant Stress, Mitochondria, and Cell Death Mechanisms in Drug-Induced Liver Injury: Lessons Learned from Acetaminophen Hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiao, K.; Jia, C.C.; Li, G.X.; Yuan, Q.; Xu, J.K.; Hou, Y.; Wang, B. BAP31 Regulates IRAK1-Dependent Neuroinflammation in Microglia. J. Neuroinflammation 2019, 16, 281. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wu, Y.; Yan, H.Z.; Xia, Z.R.; Wen, W.; Liu, D.Y.; Wan, L.H. Rosmarinic Acid Ameliorates Acetaminophen-Induced Acute Liver Injury in Mice via RACK1/TNF-Alpha Mediated Antioxidant Effect. Pharm. Biol. 2021, 59, 1286–1293. [Google Scholar] [CrossRef]
- Du, K.; Farhood, A.; Jaeschke, H. Mitochondria-Targeted Antioxidant Mito-Tempo Protects Against Acetaminophen Hepatotoxicity. Arch. Toxicol 2017, 91, 761–773. [Google Scholar] [CrossRef]
- Nguyen, G.C.; Sam, J.; Thuluvath, P.J. Hepatitis C Is a Predictor of Acute Liver Injury among Hospitalizations for Acetaminophen Overdose in the United States: A Nationwide Analysis. Hepatology 2008, 48, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Woolbright, B.L.; Jaeschke, H.; Ramachandran, A. Late Protective Effect of Netrin-1 in the Murine Acetaminophen Hepatotoxicity Model. Toxicol. Sci. 2020, 175, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Uzi, D.; Barda, L.; Scaiewicz, V.; Mills, M.; Mueller, T.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Iwawaki, T.; Nahmias, Y.; Xavier, R.; et al. CHOP Is a Critical Regulator of Acetaminophen-Induced Hepatotoxicity. J. Hepatol. 2013, 59, 495–503. [Google Scholar] [CrossRef]
- Xu, J.L.; Kulkarni, S.R.; Li, L.Y.; Slitt, A.L. UDP-Glucuronosyltransferase Expression in Mouse Liver Is Increased in Obesity- and Fasting-Induced Steatosis. Drug Metab. Dispos 2012, 40, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wu, W.; Ou, P.; Wu, M.; Zeng, F.; Zhou, B.; Wu, S. MiR-122-5p Knockdown Protects against APAP-Mediated Liver Injury through Up-Regulating NDRG3. Mol. Cell Biochem 2021, 476, 1257–1267. [Google Scholar] [CrossRef]
- Masuda, Y.; Vaziri, N.D.; Takasu, C.; Li, S.; Robles, L.; Pham, C.; Le, A.; Vo, K.; Farzaneh, S.H.; Stamos, M.J.; et al. Salutary Effect of Pre-treatment with an Nrf2 Inducer on Ischemia Reperfusion Injury in the Rat Liver. Gastroenterol. Hepatol. 2014, 1, 1–7. [Google Scholar] [CrossRef]
- Xiong, W.; Yuan, Z.; Wang, T.; Wu, S.; Xiong, Y.; Yao, Y.; Yang, Y.; Wu, H. Quercitrin Attenuates Acetaminophen-Induced Acute Liver Injury by Maintaining Mitochondrial Complex I Activity. Front. Pharmacol 2021, 12, 586010. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jia, Y.; Cheng, J.; Liu, G.; Song, F. LncRNA NCK1-AS1 Promotes Proliferation and Induces Cell Cycle Progression by Crosstalk NCK1-AS1/miR-6857/CDK1 Pathway. Cell Death Dis. 2018, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Al-Haj, L.; Blackshear, P.J.; Khabar, K.S. Regulation of p21/CIP1/WAF-1 Mediated Cell-Cycle Arrest by RNase L and Tristetraprolin, and Involvement of AU-Rich Elements. Nucleic Acids Res. 2012, 40, 7739–7752. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Lv, X.; Huo, Y.; Hu, X.; Li, X.; Sun, S.; Zhao, X.; Kong, X.; Xu, J. Hepatocyte-Specific Deficiency of BAP31 Amplified Acetaminophen-Induced Hepatotoxicity via Attenuating Nrf2 Signaling Activation in Mice. Int. J. Mol. Sci. 2021, 22, 10788. https://doi.org/10.3390/ijms221910788
Zhao J, Lv X, Huo Y, Hu X, Li X, Sun S, Zhao X, Kong X, Xu J. Hepatocyte-Specific Deficiency of BAP31 Amplified Acetaminophen-Induced Hepatotoxicity via Attenuating Nrf2 Signaling Activation in Mice. International Journal of Molecular Sciences. 2021; 22(19):10788. https://doi.org/10.3390/ijms221910788
Chicago/Turabian StyleZhao, Jie, Xiaotong Lv, Yan Huo, Xiaodi Hu, Xiaochen Li, Shizhuo Sun, Xin Zhao, Xuewei Kong, and Jialin Xu. 2021. "Hepatocyte-Specific Deficiency of BAP31 Amplified Acetaminophen-Induced Hepatotoxicity via Attenuating Nrf2 Signaling Activation in Mice" International Journal of Molecular Sciences 22, no. 19: 10788. https://doi.org/10.3390/ijms221910788
APA StyleZhao, J., Lv, X., Huo, Y., Hu, X., Li, X., Sun, S., Zhao, X., Kong, X., & Xu, J. (2021). Hepatocyte-Specific Deficiency of BAP31 Amplified Acetaminophen-Induced Hepatotoxicity via Attenuating Nrf2 Signaling Activation in Mice. International Journal of Molecular Sciences, 22(19), 10788. https://doi.org/10.3390/ijms221910788