
Rapid Production and Genetic Stability of Human Mesenchymal Progenitor Cells Derived from Human Somatic Cell Nuclear Transfer-Derived Pluripotent Stem Cells


Abstract
:1. Introduction
2. Results
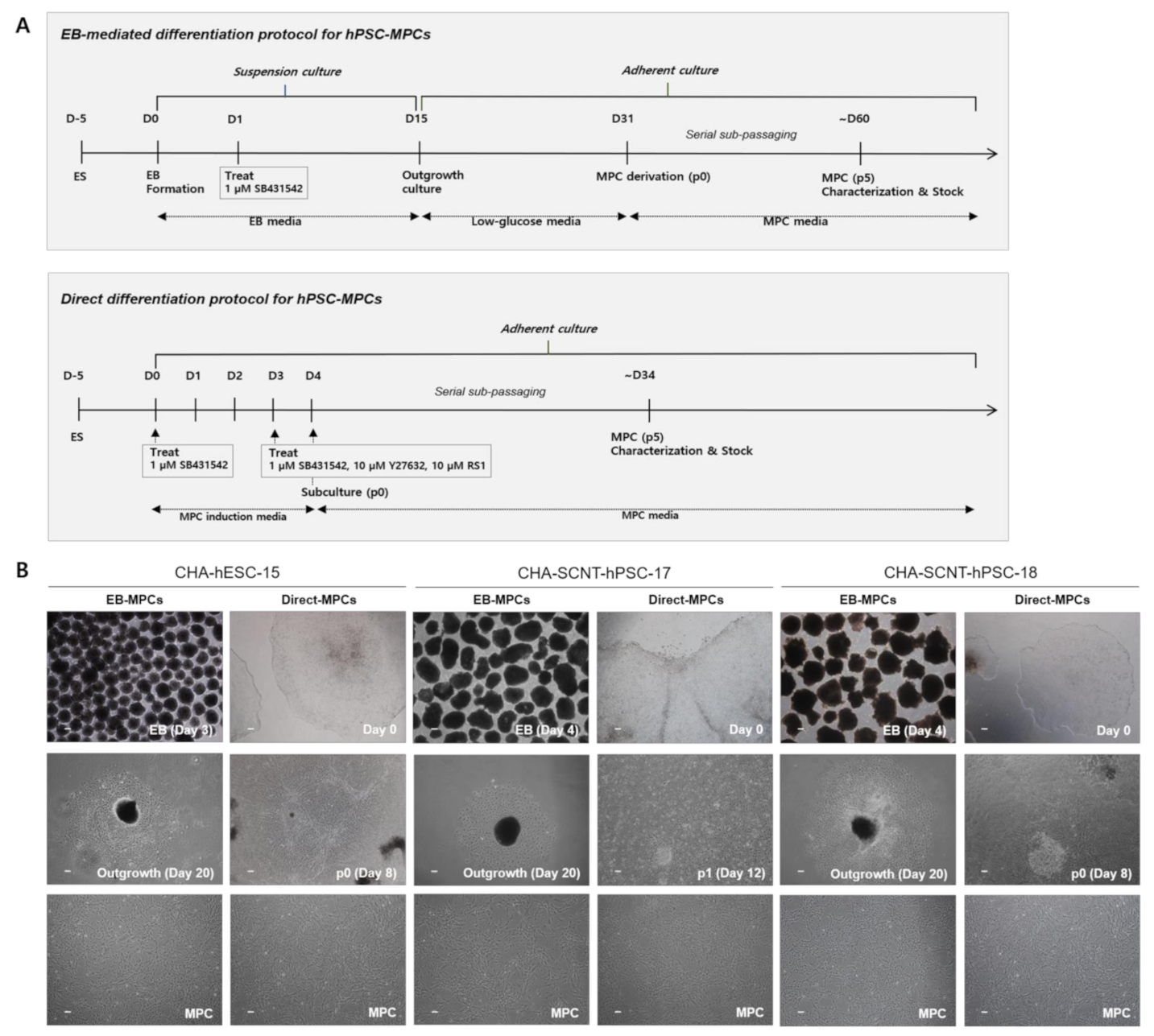
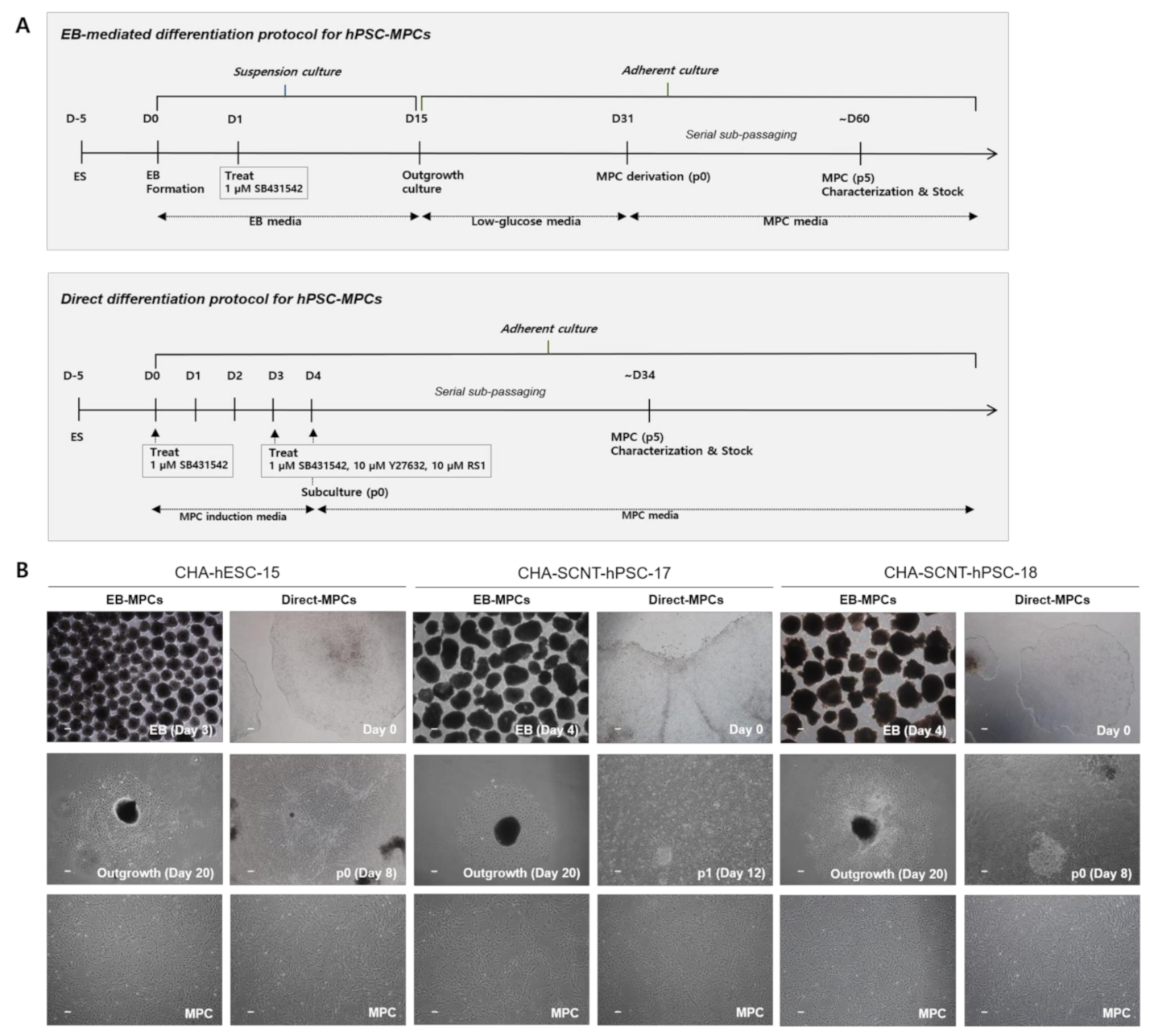
2.1. Differentiation of SCNT-PSCs into MPCs by the 2D-Direct Method
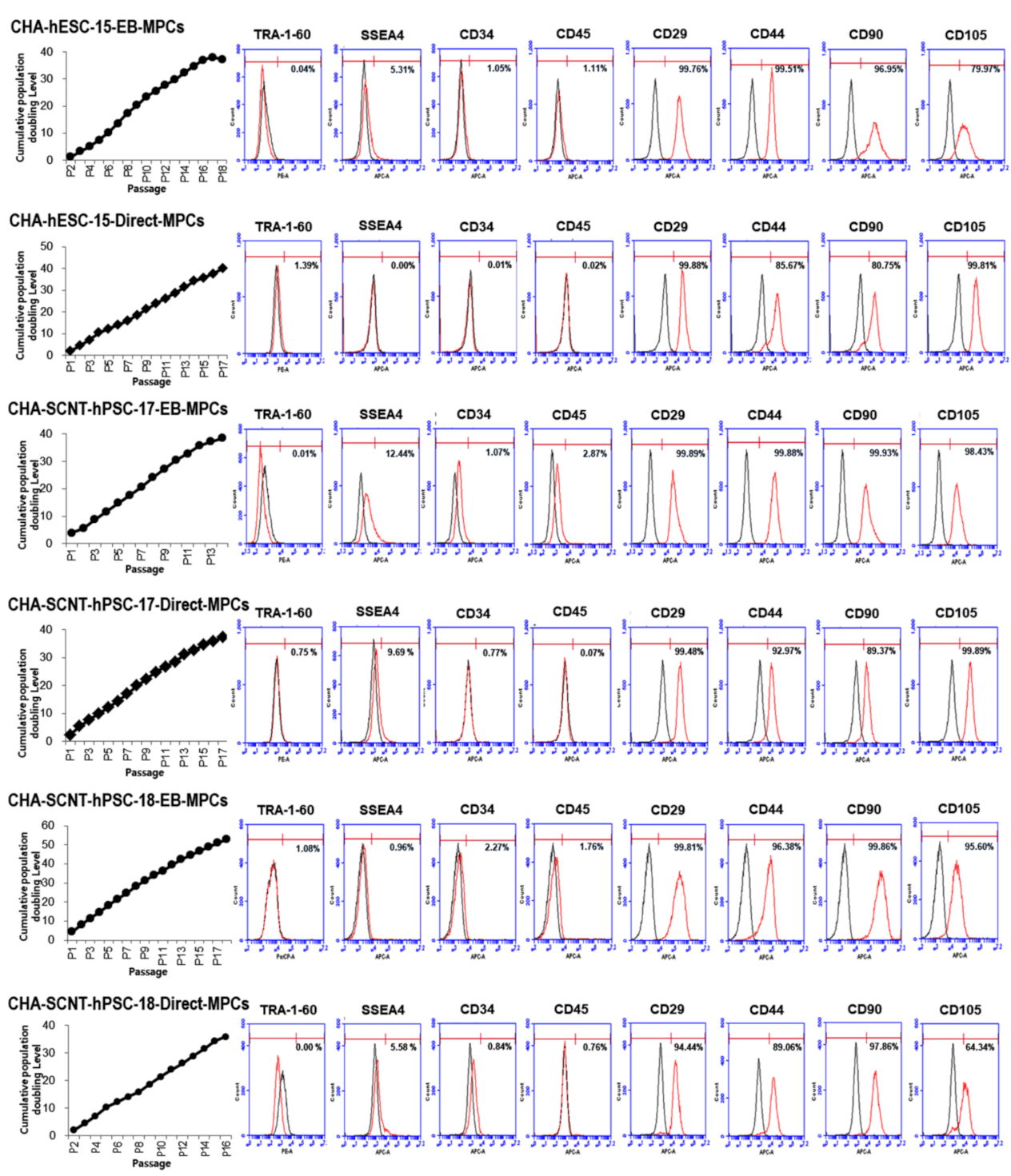
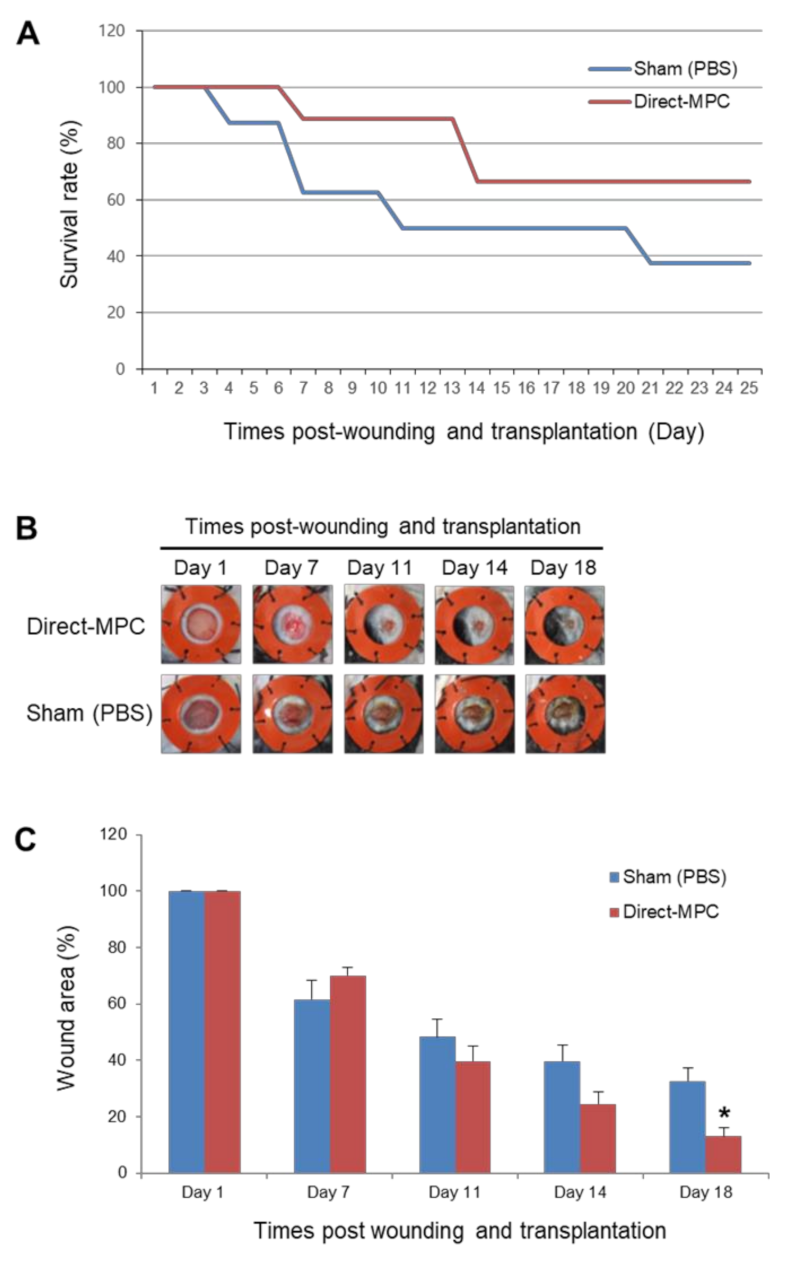
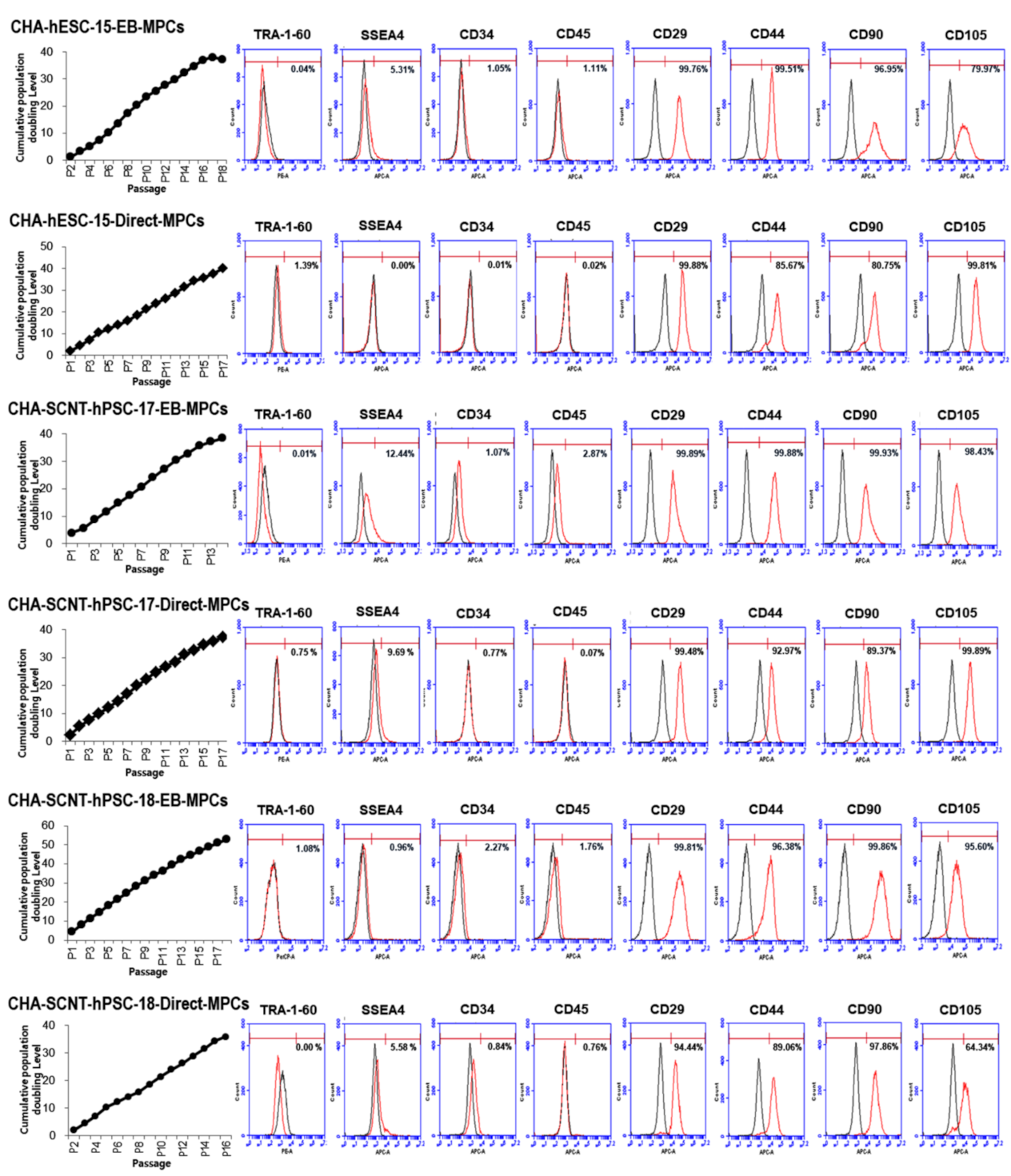
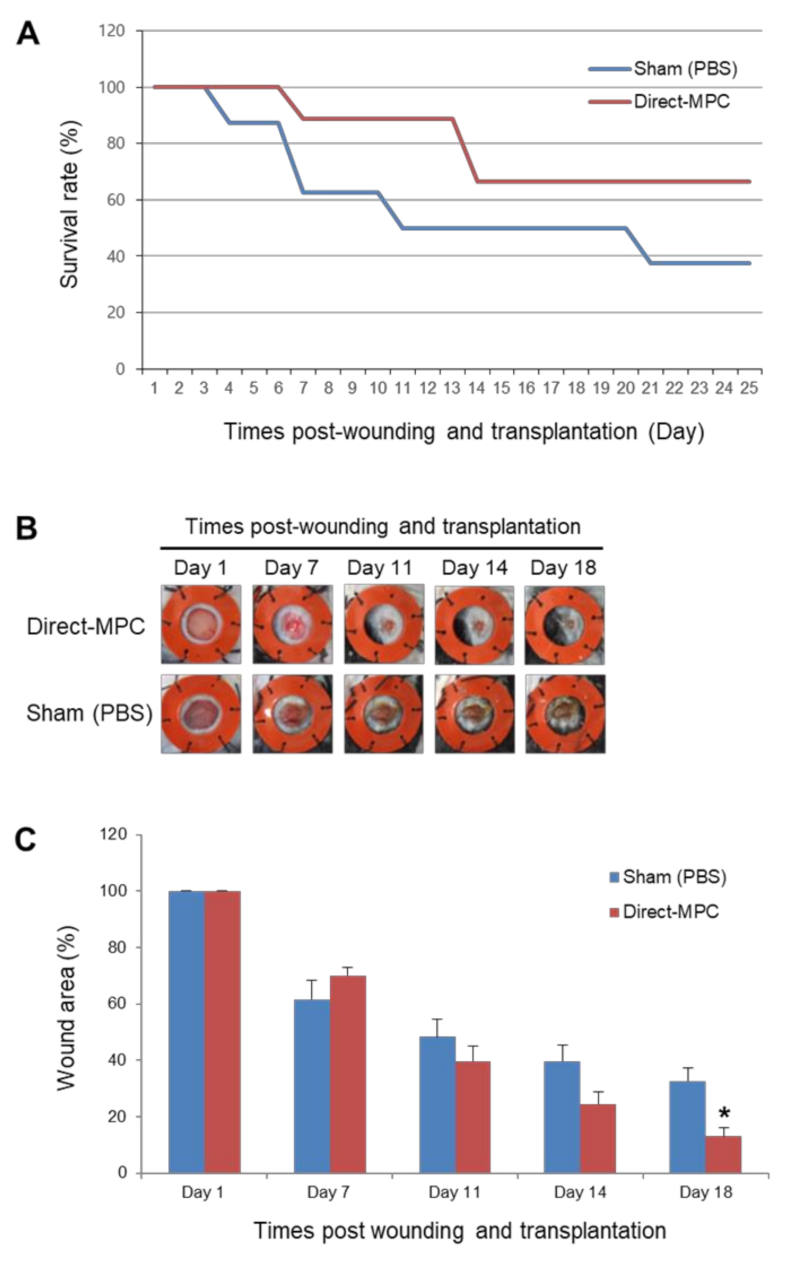
2.2. Differentiation Potential and Functional Analysis of the SCNT-PSC-MPCs
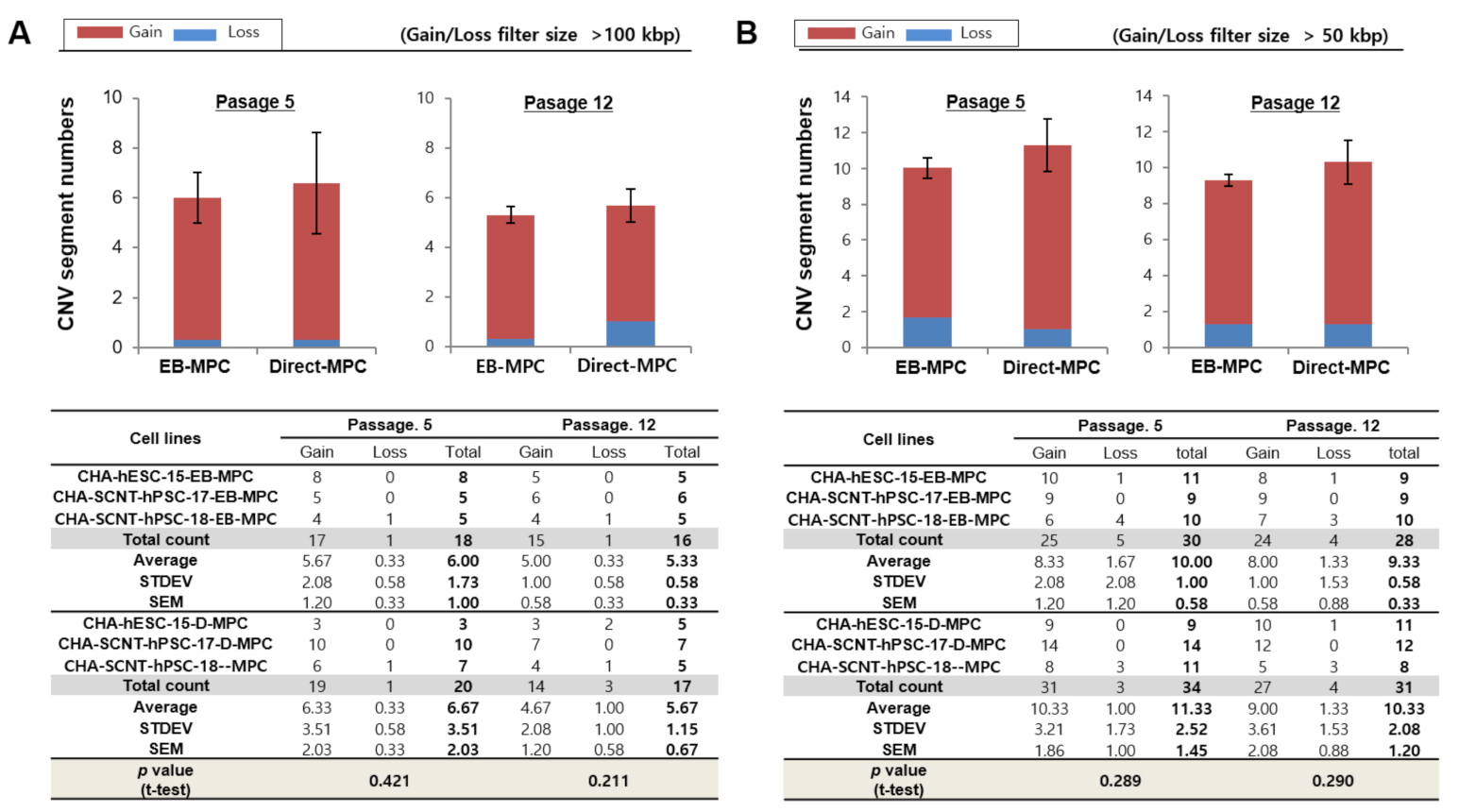
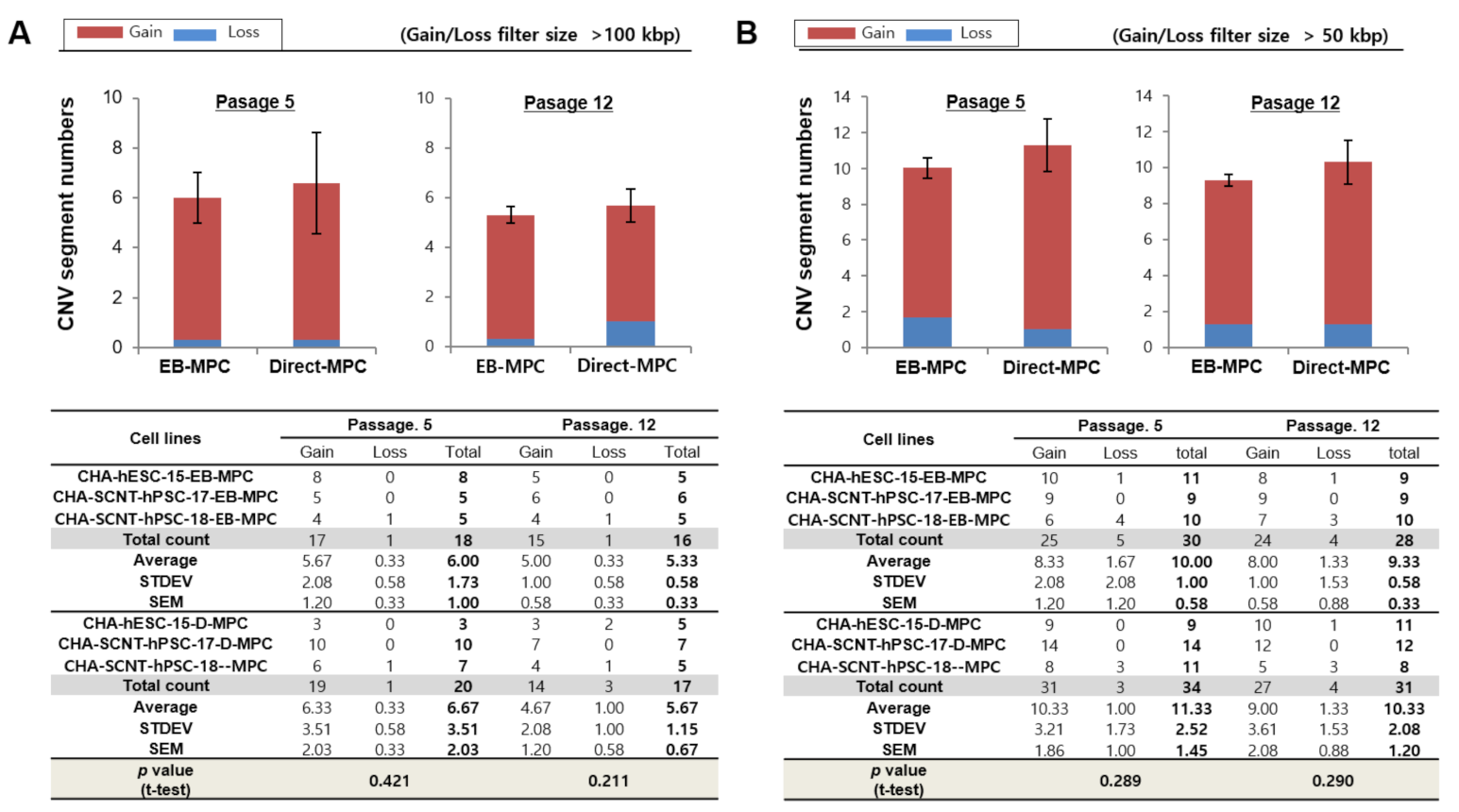
2.3. Array CGH of SCNT-PSCs-MPCs
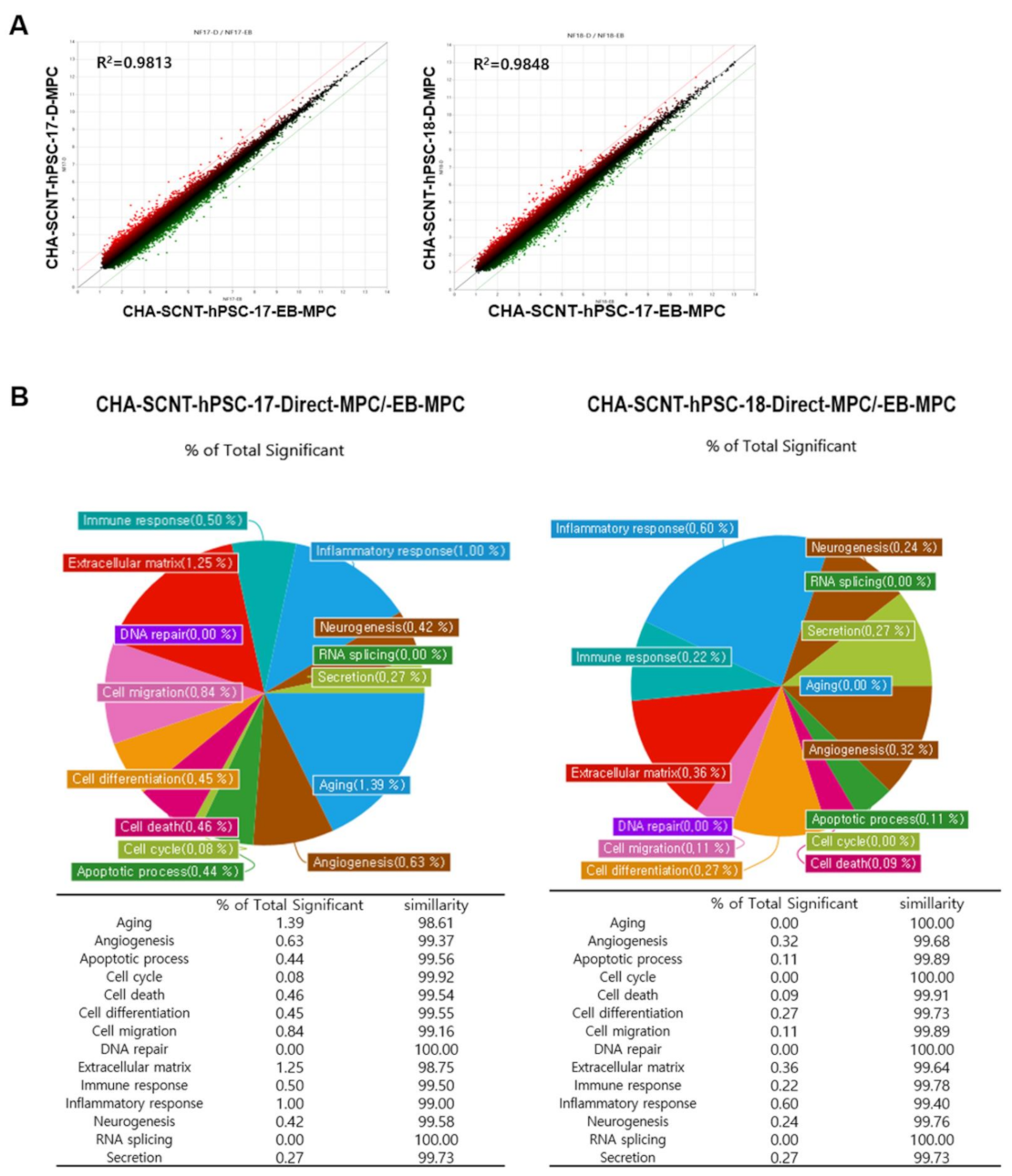
2.4. Analysis of Gene Expression Patterns Using a Microarray of SCNT-PSC-MPCs
2.5. The Teratoma Assay of SCNT-PSCs-MPCs
3. Discussions
4. Materials and Methods
4.1. Ethics Approval
4.2. Human PSCs and Culture
4.3. Differentiation of Human SCNT-PSCs into MPCs
4.4. Growth Kinetics of Human PSC-Derived MPC
4.5. Characterization of Human PSC-Derived MPC
4.6. Teratomas Formation Assay
4.7. Excisional Wound Splinting Model and Cell Transplantation in Type I Diabetes Mouse
4.8. Array-Based Comparative Genomic Hybridization (Array-CGH) for Genetic Stability
4.9. Micro-Array for Transcriptome Profiling
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spees, J.L.; Lee, R.H.; Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res. Ther. 2016, 7, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Chang, C.; Yan, J.; Yao, Z.; Zhang, C.; Li, X.; Mao, H.Q. Effects of Mesenchymal Stem Cell-Derived Paracrine Signals and Their Delivery Strategies. Adv. Healthc. Mater. 2021, 10, e2001689. [Google Scholar] [CrossRef]
- Boregowda, S.V.; Krishnappa, V.; Haga, C.L.; Ortiz, L.A.; Phinney, D.G. A Clinical Indications Prediction Scale Based on TWIST1 for Human Mesenchymal Stem Cells. EBioMedicine 2016, 4, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Menard, C.; Pacelli, L.; Bassi, G.; Dulong, J.; Bifari, F.; Bezier, I.; Zanoncello, J.; Ricciardi, M.; Latour, M.; Bourin, P.; et al. Clinical-grade mesenchymal stromal cells produced under various good manufacturing practice processes differ in their immunomodulatory properties: Standardization of immune quality controls. Stem Cells Dev. 2013, 22, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.; Kluba, T.; Hermanutz-Klein, U.; Bieback, K.; Northoff, H.; Schafer, R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.G.; Galipeau, J.; MSC Committee of the International Society of Cell and Gene Therapy. Manufacturing mesenchymal stromal cells for clinical applications: A survey of Good Manufacturing Practices at U.S. academic centers. Cytotherapy 2019, 21, 782–792. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.P.; Murphy, J.M.; Mahon, B.P. Mesenchymal stem cells avoid allogeneic rejection. J. Inflamm. 2005, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Lee, J.Y.; Park, C.H.; Eum, J.H.; Jung, S.K.; Han, A.R.; Seol, D.W.; Lee, J.S.; Shin, H.S.; Im, J.H.; et al. Cryopreserved Human Oocytes and Cord Blood Cells Can Produce Somatic Cell Nuclear Transfer-Derived Pluripotent Stem Cells with a Homozygous HLA Type. Stem Cell Rep. 2020, 15, 171–184. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.G.; Eum, J.H.; Lee, J.E.; Shim, S.H.; Sepilian, V.; Hong, S.W.; Lee, Y.; Treff, N.R.; Choi, Y.H.; Kimbrel, E.A.; et al. Human somatic cell nuclear transfer using adult cells. Cell Stem Cell 2014, 14, 777–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Lanza, R.P.; Cibelli, J.B.; West, M.D. Prospects for the use of nuclear transfer in human transplantation. Nat. Biotechnol. 1999, 17, 1171–1174. [Google Scholar] [CrossRef]
- Chung, Y.G.; Matoba, S.; Liu, Y.; Eum, J.H.; Lu, F.; Jiang, W.; Lee, J.E.; Sepilian, V.; Cha, K.Y.; Lee, D.R.; et al. Histone Demethylase Expression Enhances Human Somatic Cell Nuclear Transfer Efficiency and Promotes Derivation of Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Yan, L.; Wang, X.; Li, E.; Murphy, K.; Vaccaro, K.; Li, Y.; Xu, R.H. Concise Review: Mesenchymal Stem Cells Derived from Human Pluripotent Cells, an Unlimited and Quality-Controllable Source for Therapeutic Applications. Stem Cells 2019, 37, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Kimbrel, E.A.; Kouris, N.A.; Yavanian, G.J.; Chu, J.; Qin, Y.; Chan, A.; Singh, R.P.; McCurdy, D.; Gordon, L.; Levinson, R.D.; et al. Mesenchymal stem cell population derived from human pluripotent stem cells displays potent immunomodulatory and therapeutic properties. Stem Cells Dev. 2014, 23, 1611–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Zhang, Z.; Jiang, B.; Yan, L.; Park, J.W.; Xu, R.H. Generation of Mesenchymal Stem Cells from Human Embryonic Stem Cells in a Complete Serum-free Condition. Int. J. Biol. Sci. 2018, 14, 1901–1909. [Google Scholar] [CrossRef]
- Zhang, J.; Chan, Y.C.; Ho, J.C.; Siu, C.W.; Lian, Q.; Tse, H.F. Regulation of cell proliferation of human induced pluripotent stem cell-derived mesenchymal stem cells via ether-a-go-go 1 (hEAG1) potassium channel. Am. J. Physiol. Cell Physiol. 2012, 303, C115–C125. [Google Scholar] [CrossRef]
- Jun, S.M.; Park, M.; Lee, J.Y.; Jung, S.; Lee, J.E.; Shim, S.H.; Song, H.; Lee, D.R. Single cell-derived clonally expanded mesenchymal progenitor cells from somatic cell nuclear transfer-derived pluripotent stem cells ameliorate the endometrial function in the uterus of a murine model with Asherman’s syndrome. Cell Prolif. 2019, 52, e12597. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Yoon, S.; Kim, K.P. Combined Ectopic Expression of Homologous Recombination Factors Promotes Embryonic Stem Cell Differentiation. Mol. Ther. 2018, 26, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.R.; Park, J.H.; Shim, S.H.; Hong, K.; La, H.; Park, K.S.; Lee, D.R. Genome stabilization by RAD51-stimulatory compound 1 enhances efficiency of somatic cell nuclear transfer-mediated reprogramming and full-term development of cloned mouse embryos. Cell Prolif. 2021, 54, e13059. [Google Scholar] [CrossRef]
- Liu, S.; de Castro, L.F.; Jin, P.; Civini, S.; Ren, J.; Reems, J.A.; Cancelas, J.; Nayak, R.; Shaw, G.; O’Brien, T.; et al. Manufacturing Differences Affect Human Bone Marrow Stromal Cell Characteristics and Function: Comparison of Production Methods and Products from Multiple Centers. Sci. Rep. 2017, 7, 46731. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kang, M.S.; Park, M.H.; Shim, S.H.; Yoon, T.K.; Chung, H.M.; Lee, D.R. Evaluation of 28 human embryonic stem cell lines for use as unrelated donors in stem cell therapy: Implications of HLA and ABO genotypes. Cell Transplant. 2010, 19, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofalo, V.J.; Allen, R.G.; Pignolo, R.J.; Martin, B.G.; Beck, J.C. Relationship between donor age and the replicative lifespan of human cells in culture: A reevaluation. Proc. Natl. Acad. Sci. USA 1998, 95, 10614–10619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, L.; Prosser, H.C.; Tan, J.T.; Vanags, L.Z.; Ng, M.K.; Bursill, C.A. Murine model of wound healing. J. Vis. Exp. 2013, 75, e50265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ge, J.; Tredget, E.E.; Wu, Y. The mouse excisional wound splinting model, including applications for stem cell transplantation. Nat. Protoc. 2013, 8, 302–309. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHA-hESC-15 | ES (Control) | EB-MPC (p5) | EB-MPC (p12) | Direct-MPC (p5) | Direct-MPC (p12) | ||
|---|---|---|---|---|---|---|---|
| Total SNP | 748,953 | 748,953 | 748,953 | 748,953 | 748,953 | ||
| Called SNP | 731,057 | 725,854 | 729,329 | 722,029 | 728,760 | ||
| All called SNP | 682,461 | 682,461 | 682,461 | 682,461 | 682,461 | ||
| Matched SNP | 682,461 | 678,727 | 679,629 | 675,301 | 678,123 | ||
| Different SNP | 0 | 3,734 | 2,832 | 7,160 | 4,338 | ||
| Matched % | 100% | 99.453% | 99.585% | 98.951% | 99.364% | ||
| CHA-hESC-15 | Different SNP | intron | missense | coding-synonym | Untranslated | Un-known | other |
| EB-MPC (p5) | 3734 | 1437 | 18 | 14 | 18 | 2158 | 89 |
| EB-MPC (p12) | 2832 | 1077 | 11 | 9 | 9 | 1642 | 85 |
| Direct-MPC (p5) | 7160 | 2844 | 20 | 32 | 48 | 4023 | 193 |
| Direct-MPC (p12) | 4338 | 1692 | 19 | 13 | 17 | 2481 | 116 |
| Total | 11,595 | 4581 | 40 | 46 | 68 | 6514 | 346 |
| CHA-SCNT-hPSC-17 | ES (Control) | EB-MPC (p5) | EB-MPC (p12) | Direct-MPC (p5) | Direct-MPC (p12) | ||
|---|---|---|---|---|---|---|---|
| Total SNP | 748,953 | 748,953 | 748,953 | 748,953 | 748,953 | ||
| Called SNP | 717,149 | 721,534 | 713,570 | 720,532 | 716,621 | ||
| All called SNP | 646,014 | 646,014 | 646,014 | 646,014 | 646,014 | ||
| Matched SNP | 646,014 | 632,815 | 629,481 | 632,707 | 628,918 | ||
| Different SNP | 0 | 13,199 | 16,533 | 13,307 | 17,096 | ||
| Matched % | 100% | 97.957% | 97.441% | 97.940% | 99.354% | ||
| CHA-SCNT -hPSC-17 | Different SNP | intron | missense | coding-synonym | Untranslated | Un-known | other |
| EB-MPC (p5) | 13,199 | 4861 | 35 | 35 | 83 | 7830 | 355 |
| EB-MPC (p12) | 16,533 | 6204 | 39 | 56 | 91 | 9720 | 423 |
| Direct-MPC (p5) | 13,307 | 4899 | 33 | 47 | 76 | 7927 | 325 |
| Direct-MPC (p12) | 17,096 | 6467 | 58 | 51 | 106 | 9982 | 432 |
| Total | 30,607 | 11,668 | 88 | 93 | 183 | 17,738 | 837 |
| CHA-SCNT-hPSC-18 | ES (Control) | EB-MPC (p5) | EB-MPC (p12) | Direct-MPC (p5) | Direct-MPC (p12) | ||
|---|---|---|---|---|---|---|---|
| Total SNP | 748,953 | 748,953 | 748,953 | 748,953 | 748,953 | ||
| Called SNP | 736,591 | 728,309 | 734,471 | 736,213 | 729,502 | ||
| All called SNP | 699,127 | 699,127 | 699,127 | 699,127 | 699,127 | ||
| Matched SNP | 699,127 | 699,163 | 697,553 | 697,554 | 694,450 | ||
| Different SNP | 0 | 2964 | 1574 | 1573 | 4667 | ||
| Matched % | 100% | 99.576% | 99.775% | 99.775% | 99.331% | ||
| CHA-SCNT -hPSC-18 | Different SNP | intron | missense | coding-synonym | Untranslated | Un-known | other |
| EB-MPC (p5) | 2964 | 1190 | 12 | 12 | 24 | 1652 | 74 |
| EB-MPC (p12) | 1574 | 575 | 4 | 5 | 17 | 919 | 54 |
| Direct-MPC (p5) | 1573 | 607 | 8 | 4 | 7 | 903 | 44 |
| Direct-MPC (p12) | 4677 | 1850 | 20 | 16 | 29 | 2616 | 146 |
| Total | 7871 | 3158 | 30 | 27 | 62 | 4340 | 254 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, S.K.; Lee, J.E.; Lee, C.W.; Shim, S.H.; Lee, D.R. Rapid Production and Genetic Stability of Human Mesenchymal Progenitor Cells Derived from Human Somatic Cell Nuclear Transfer-Derived Pluripotent Stem Cells. Int. J. Mol. Sci. 2021, 22, 9238. https://doi.org/10.3390/ijms22179238
Jung SK, Lee JE, Lee CW, Shim SH, Lee DR. Rapid Production and Genetic Stability of Human Mesenchymal Progenitor Cells Derived from Human Somatic Cell Nuclear Transfer-Derived Pluripotent Stem Cells. International Journal of Molecular Sciences. 2021; 22(17):9238. https://doi.org/10.3390/ijms22179238
Chicago/Turabian StyleJung, Soo Kyung, Jeoung Eun Lee, Chang Woo Lee, Sung Han Shim, and Dong Ryul Lee. 2021. "Rapid Production and Genetic Stability of Human Mesenchymal Progenitor Cells Derived from Human Somatic Cell Nuclear Transfer-Derived Pluripotent Stem Cells" International Journal of Molecular Sciences 22, no. 17: 9238. https://doi.org/10.3390/ijms22179238
APA StyleJung, S. K., Lee, J. E., Lee, C. W., Shim, S. H., & Lee, D. R. (2021). Rapid Production and Genetic Stability of Human Mesenchymal Progenitor Cells Derived from Human Somatic Cell Nuclear Transfer-Derived Pluripotent Stem Cells. International Journal of Molecular Sciences, 22(17), 9238. https://doi.org/10.3390/ijms22179238