
Transcriptome Analysis in a Primary Human Muscle Cell Differentiation Model for Myotonic Dystrophy Type 1

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Differential Gene Expression in DM1 Myoblasts and Myotubes Compared to Non-Disease Controls
2.2. Pathway Enrichment Analysis: Gene Ontology “Molecular Function”
2.3. Pathway Enrichment Analysis: Gene Ontology “Signalling”
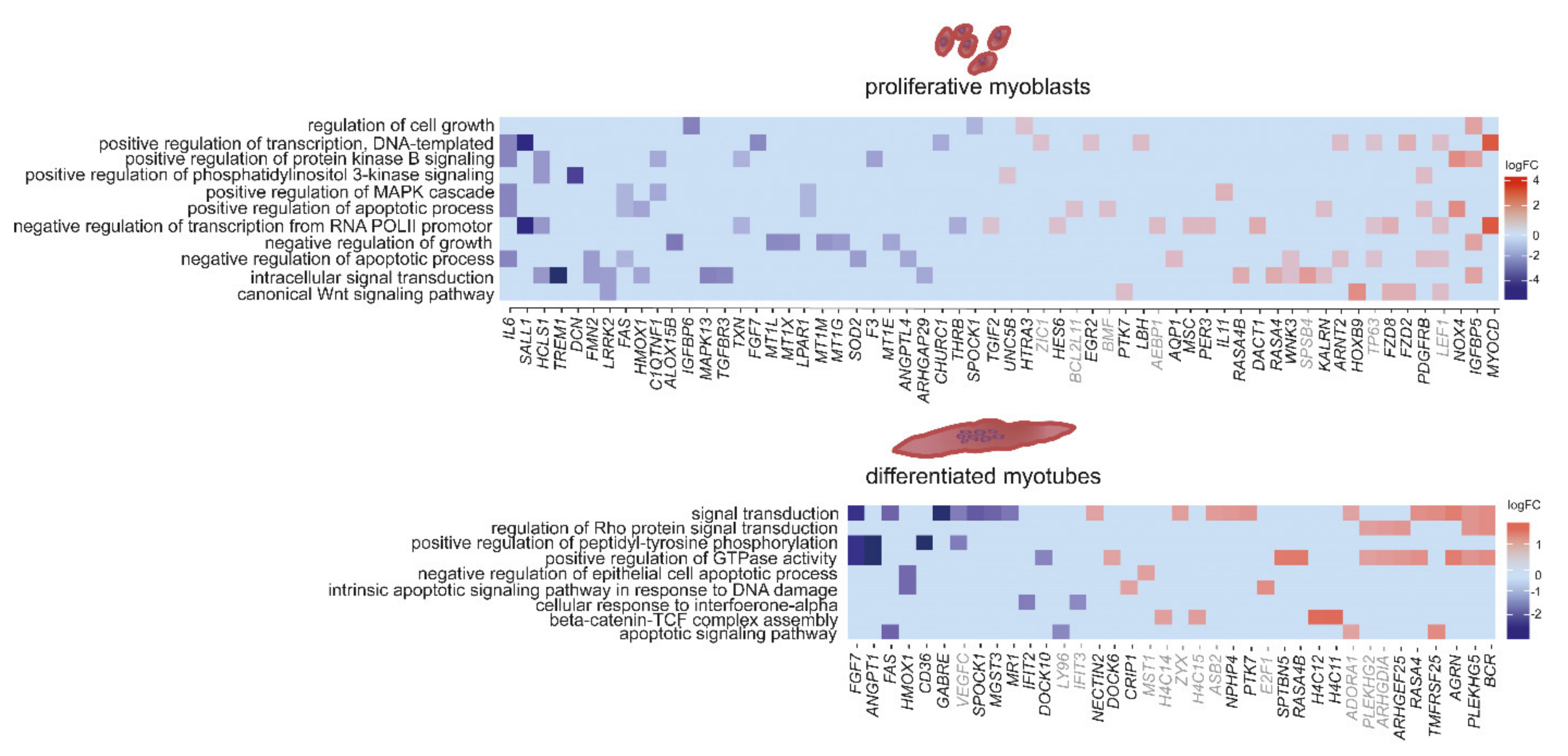
2.4. Pathway Enrichment Analysis: Gene Ontology “Others”
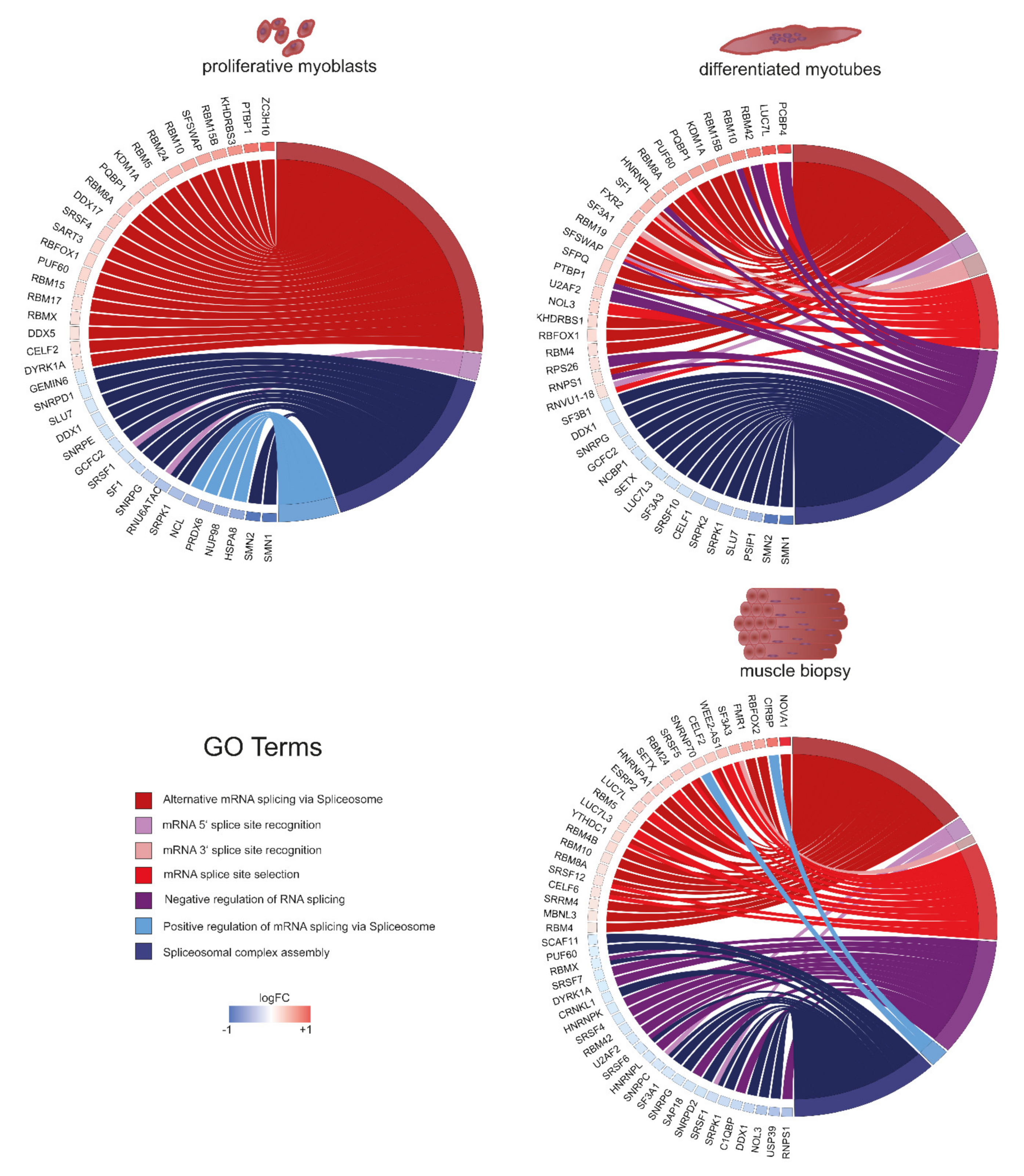
2.5. Splicing as the Basis of Pathway Alterations
3. Discussion
4. Materials and Methods
4.1. Culture of Primary Skeletal Muscle Cells
4.2. Patients and Non-Disease Controls
4.3. Myotube Separation
4.4. RNA Isolation
4.5. RNA Sequencing
4.6. RT-qPCR
4.7. Immunofluorescence Staining and Microscopy
4.8. Quantification of Splicing Alterations
4.9. Bioinformatics
4.10. Muscle Biopsy Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Roelofs, R.; Leroy, B.; Pech, I.; Benzow, K.; Ranum, L.P. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromuscul. Disord. 1999, 9, 19–27. [Google Scholar] [CrossRef]
- Ricker, K.; Koch, M.C.; Lehmann-Horn, F.; Pongratz, D.; Speich, N.; Reiners, K.; Schneider, C.; Moxley, R.T. Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophy. Arch. Neurol. 1995, 52, 25–31. [Google Scholar] [CrossRef]
- Wenninger, S.; Montagnese, F.; Schoser, B. Core Clinical Phenotypes in Myotonic Dystrophies. Front. Neurol. 2018, 9, 303. [Google Scholar] [CrossRef]
- Wheeler, T.M. Myotonic dystrophy: Therapeutic strategies for the future. Neurotherapeutics 2008, 5, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; A Nasser, G.; Ashizawa, T.; De Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G.; French Myotonic Dystrophy Clinical Network. Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry-based study with implications for disease classification. Rev. Neurol. 2016, 172, 572–580. [Google Scholar] [CrossRef]
- De Temmerman, N.; Sermon, K.; Seneca, S.; De Rycke, M.; Hilven, P.; Lissens, W.; Van Steirteghem, A.; Liebaers, I. Intergenerational instability of the expanded CTG repeat in the DMPK gene: Studies in human gametes and preimplantation embryos. Am. J. Hum. Genet. 2004, 75, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.B.; Fenwick, R.G.; Fu, Y.H.; Pizzuti, A.; Caskey, C.T. Relationship between parental trinucleotide GCT repeat length and severity of myotonic dystrophy in offspring. JAMA 1993, 269, 1960–1965. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Willems, P.; Coerwinkel, M.; Nillesen, W.; Smeets, H.; Vits, L.; Höweler, C.; Brunner, H.; Wieringa, B. Gonosomal mosaicism in myotonic dystrophy patients: Involvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperm. Am. J. Hum. Genet. 1994, 54, 575–585. [Google Scholar] [PubMed]
- Rakočević-Stojanović, V.; Savic, D.; Pavlović, S.; Lavrnić, D.; Stević, Z.; Basta, I.; Romac, S.; Apostolski, S. Intergenerational changes of CTG repeat depending on the sex of the transmitting parent in myotonic dystrophy type 1. Eur. J. Neurol. 2005, 12, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Martorell, L.; Cobo, A.M.; Baiget, M.; Naudó, M.; Poza, J.J.; Parra, J. Prenatal diagnosis in myotonic dystrophy type 1. Thirteen years of experience: Implications for reproductive counselling in DM1 families. Prenat. Diagn. 2006, 27, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A.; Johnson, K.; Moxley, R.T. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol. 1994, 35, 104–107. [Google Scholar] [CrossRef]
- Ishii, S.; Nishio, T.; Sunohara, N.; Yoshihara, T.; Takemura, K.; Hikiji, K.; Tsujino, S.; Sakuragawa, N. Small increase in triplet repeat length of cerebellum from patients with myotonic dystrophy. Qual. Life Res. 1996, 98, 138–140. [Google Scholar] [CrossRef]
- Napierala, M.; Krzyzosiak, W.J. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 1997, 272, 31079–31085. [Google Scholar] [CrossRef]
- Dere, R.; Napierala, M.; Ranum, L.P.W.; Wells, R.D. Hairpin Structure-forming Propensity of the (CCTG·CAGG) Tetranucleotide Repeats Contributes to the Genetic Instability Associated with Myotonic Dystrophy Type 2. J. Biol. Chem. 2004, 279, 41715–41726. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice ex-pressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, O.J.; Aagaard, L.; Jensen, T.G.; Damgaard, C.K. Molecular mechanisms in DM1—A focus on foci. Nucleic Acids Res. 2015, 43, 2433–2441. [Google Scholar] [CrossRef]
- Ho, T.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547. [Google Scholar] [CrossRef]
- López-Martínez, A.; Soblechero-Martín, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Du, H.; Cline, M.S.; Osborne, R.J.; Tuttle, D.L.; Clark, T.A.; Donohue, J.P.; Hall, M.P.; Shiue, L.; Swanson, M.S.; Thornton, C.A. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010, 17, 187–193. [Google Scholar] [CrossRef]
- Ebralidze, A.; Wang, Y.; Petkova, V.; Ebralidse, K.; Junghans, R.P. RNA Leaching of Transcription Factors Disrupts Transcription in Myotonic Dystrophy. Science 2004, 303, 383–387. [Google Scholar] [CrossRef]
- Osborne, R.J.; Lin, X.; Welle, S.; Sobczak, K.; O’Rourke, J.R.; Swanson, M.; Thornton, C.A. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 2009, 18, 1471–1481. [Google Scholar] [CrossRef]
- Kalsotra, A.; Singh, R.; Gurha, P.; Ward, A.J.; Creighton, C.J.; Cooper, T.A. The Mef2 Transcription Network Is Disrupted in Myotonic Dystrophy Heart Tissue, Dramatically Altering miRNA and mRNA Expression. Cell Rep. 2014, 6, 336–345. [Google Scholar] [CrossRef]
- Huichalaf, C.; Sakai, K.; Jin, B.; Jones, K.; Wang, G.; Schoser, B.; Schneider-Gold, C.; Sarkar, P.; Pereira-Smith, O.M.; Timchenko, N.; et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 2010, 24, 3706–3719. [Google Scholar] [CrossRef]
- Meola, G.; Jones, K.; Wei, C.; Timchenko, L.T. Dysfunction of protein homeostasis in myotonic dystrophies. Histol. Histopathol. 2013, 28. Available online: http://hdl.handle.net/10201/60219 (accessed on 7 August 2021).
- Cleary, J.D.; Ranum, L.P. Repeat associated non-ATG (RAN) translation: New starts in microsatellite expansion disorders. Curr. Opin. Genet. Dev. 2014, 26, 6–15. [Google Scholar] [CrossRef]
- Gudde, A.; van Heeringen, S.J.; de Oude, A.I.; van Kessel, I.D.G.; Estabrook, J.; Wang, E.T.; Wieringa, B.; Wansink, D.G. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol. 2017, 14, 1374–1388. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef]
- Krol, J.; Fiszer, A.; Mykowska, A.; Sobczak, K.; de Mezer, M.; Krzyzosiak, W.J. Ribonuclease Dicer Cleaves Triplet Repeat Hairpins into Shorter Repeats that Silence Specific Targets. Mol. Cell 2007, 25, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.-P.; Fischer, M.-C.; Jost, B.; Dembélé, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Xu, F.; Li, M.; Wu, S.; Zhang, J.; Wang, A.; Xu, L.; Liu, Y.; Zhu, G. miR-322/-503 rescues myoblast defects in myotonic dystrophy type 1 cell model by targeting CUG repeats. Cell Death Dis. 2020, 11, 891. [Google Scholar] [CrossRef]
- Cappella, M.; Perfetti, A.; Cardinali, B.; Garcia-Manteiga, J.M.; Carrara, M.; Provenzano, C.; Fuschi, P.; Cardani, R.; Renna, L.V.; Meola, G.; et al. High-throughput analysis of the RNA-induced silencing complex in myotonic dystrophy type 1 patients identifies the dysregulation of miR-29c and its target ASB2. Cell Death Dis. 2018, 9, 729. [Google Scholar] [CrossRef]
- Thong, M.-K.; Ambrose, K.K.; Ishak, T.; Lian, L.-H.; Goh, K.-J.; Wong, K.-T.; Ahmad-Annuar, A. Deregulation of microRNAs in blood and skeletal muscles of myotonic dystrophy type 1 patients. Neurol. India 2017, 65, 512–517. [Google Scholar] [CrossRef]
- Fritegotto, C.; Ferrati, C.; Pegoraro, V.; Angelini, C. Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol. Sci. 2017, 38, 619–625. [Google Scholar] [CrossRef]
- Fardaei, M.; Rogers, M.T.; Thorpe, H.M.; Larkin, K.; Hamshere, M.G.; Harper, P.S.; Brook, J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef]
- Fardaei, M.; Larkin, K.; Brook, J.D.; Hamshere, M.G. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001, 29, 2766–2771. [Google Scholar] [CrossRef]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of Splicing Regulated by a CUG-Binding Protein in Myotonic Dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.-S.; Cooper, T.A. Increased Steady-State Levels of CUGBP1 in Myotonic Dystrophy 1 Are Due to PKC-Mediated Hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Charlet, N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.; Cooper, T.A. Loss of the Muscle-Specific Chloride Channel in Type 1 Myotonic Dystrophy Due to Misregulated Alternative Splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Yamashita, Y.; Matsuura, T.; Shinmi, J.; Amakusa, Y.; Masuda, A.; Ito, M.; Kinoshita, M.; Furuya, H.; Abe, K.; Ibi, T.; et al. Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy. J. Hum. Genet. 2012, 57, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef]
- Fugier, C.; Klein, A.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011, 17, 720–725. [Google Scholar] [CrossRef]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Chorney, E.; Crawford Parks, T.E.; Ravel-Chapuis, A.; Klinck, R.; Rocheleau, L.; Pelchat, M.; Chabot, B.; Jasmin, B.J.; Côté, J. Staufen1 Regulates Multiple Alternative Splicing Events either Positively or Negatively in DM1 Indicating Its Role as a Disease Modifier. PLoS Genet. 2016, 12, e1005827. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhuang, Y.; Batra, R.; Thomas, J.D.; Li, M.; Nutter, C.A.; Scotti, M.M.; Carter, H.A.; Wang, Z.J.; Huang, X.-S.; et al. HNRNPA1-induced spliceopathy in a transgenic mouse model of myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2020, 117, 5472–5477. [Google Scholar] [CrossRef]
- Botta, A.; Vallo, L.; Rinaldi, F.; Bonifazi, E.; Amati, F.; Biancolella, M.; Gambardella, S.; Mancinelli, E.; Angelini, C.; Meola, G. Gene expression analysis in myotonic dystrophy: Indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr. 2007, 13, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Beffy, P.; Del Carratore, R.; Masini, M.; Furling, D.; Puymirat, J.; Masiello, P.; Simili, M. Altered signal transduction pathways and induction of autophagy in human myotonic dystrophy type 1 myoblasts. Int. J. Biochem. Cell Biol. 2010, 42, 1973–1983. [Google Scholar] [CrossRef]
- Mastroyiannopoulos, N.P.; Chrysanthou, E.; Kyriakides, T.C.; Uney, J.B.; Mahadevan, M.S.; Phylactou, L.A. The effect of myotonic dystrophy transcript levels and location on muscle differentiation. Biochem. Biophys. Res. Commun. 2008, 377, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Furling, D.; Coiffier, L.; Mouly, V.; Barbet, J.P.; Guily, J.L.S.; Taneja, K.; Gourdon, G.; Junien, C.; Butler-Browne, G.S. Defective satellite cells in congenital myotonic dystrophy. Hum. Mol. Genet. 2001, 10, 2079–2087. [Google Scholar] [CrossRef]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2018, 28, 1312–1321. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Zhu, J.; Sun, B.; Branca, M.; Tang, Y.; Foster, W.; Xiao, X.; Huard, J. Decorin gene transfer promotes muscle cell differentiation and muscle regeneration. Mol. Ther. 2007, 15, 1616–1622. [Google Scholar] [CrossRef]
- Cully, T.R.; Rodney, G.G. Nox4-RyR1-Nox2: Regulators of micro-domain signaling in skeletal muscle. Redox Biol. 2020, 36, 101557. [Google Scholar] [CrossRef] [PubMed]
- Mofarrahi, M.; McClung, J.M.; Kontos, C.D.; Davis, E.C.; Tappuni, B.; Moroz, N.; Pickett, A.E.; Huck, L.; Harel, S.; Danialou, G.; et al. Angiopoietin-1 enhances skeletal muscle regeneration in mice. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R576–R589. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Rosenberg, A.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef]
- Mourtada-Maarabouni, M.; Williams, G.T. RBM5/LUCA-15--tumour suppression by control of apoptosis and the cell cycle? Sci. World J. 2002, 2, 1885–1890. [Google Scholar] [CrossRef]
- Grifone, R.; Saquet, A.; Desgres, M.; Sangiorgi, C.; Gargano, C.; Li, Z.; Coletti, D.; Shi, D.-L. Rbm24 displays dynamic functions required for myogenic differentiation during muscle regeneration. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Ando, S.; Tanaka, M.; Chinen, N.; Nakamura, S.; Shimazawa, M.; Hara, H. SMN Protein Contributes to Skeletal Muscle Cell Maturation Via Caspase-3 and Akt Activation. In Vivo 2020, 34, 3247. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Klein, A.; Gasnier, E.; Jacquemin, V.; Ravassard, P.; Butler-Browne, G.; Mouly, V.; Furling, D. Large CTG Repeats Trigger p16-Dependent Premature Senescence in Myotonic Dystrophy Type 1 Muscle Precursor Cells. Am. J. Pathol. 2009, 174, 1435–1442. [Google Scholar] [CrossRef]
- Meinke, P.; Hintze, S.; Limmer, S.; Schoser, B. Myotonic Dystrophy-A Progeroid Disease? Front. Neurol. 2018, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Hintze, S.; Knaier, L.; Limmer, S.; Schoser, B.; Meinke, P. Nuclear Envelope Transmembrane Proteins in Myotonic Dystrophy Type 1. Front. Physiol. 2018, 9, 1532. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Tomelleri, G.; Filosto, M.; Savio, C.; Rizzuto, N.; Tonin, P. Expression of late myogenic differentiation markers in sarcoplasmic masses of patients with myotonic dystrophy. Neuropathol. Appl. Neurobiol. 2005, 31, 45–52. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. et Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef]
- Hildyard, J.C.; Wells, D.J. Identification and Validation of Quantitative PCR Reference Genes Suitable for Normalizing Expression in Normal and Dystrophic Cell Culture Models of Myogenesis. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Grüning, B.; Diehl, S.; Ramirez, F.; Dündar, F. deepTools: A flexible platform for exploring deep- sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhu, A.; Ibrahim, J.G.; I Love, M. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2018, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Age at Biopsy | Sex | CTG-Repeat Length | |
|---|---|---|---|---|
| DM1-1 | DM1 | 27 | M | 400–600 |
| DM1-2 | DM1 | 34 | M | 240–430 |
| DM1-3 | DM1 | 29 | F | 800–1500 |
| Control-1 | --- | 32 | M | n.d. |
| Control-2 | --- | 49 | F | n.d. |
| Control-3 | --- | 49 | F | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorow, V.; Hintze, S.; Kerr, A.R.W.; Hehr, A.; Schoser, B.; Meinke, P. Transcriptome Analysis in a Primary Human Muscle Cell Differentiation Model for Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 22, 8607. https://doi.org/10.3390/ijms22168607
Todorow V, Hintze S, Kerr ARW, Hehr A, Schoser B, Meinke P. Transcriptome Analysis in a Primary Human Muscle Cell Differentiation Model for Myotonic Dystrophy Type 1. International Journal of Molecular Sciences. 2021; 22(16):8607. https://doi.org/10.3390/ijms22168607
Chicago/Turabian StyleTodorow, Vanessa, Stefan Hintze, Alastair R. W. Kerr, Andreas Hehr, Benedikt Schoser, and Peter Meinke. 2021. "Transcriptome Analysis in a Primary Human Muscle Cell Differentiation Model for Myotonic Dystrophy Type 1" International Journal of Molecular Sciences 22, no. 16: 8607. https://doi.org/10.3390/ijms22168607
APA StyleTodorow, V., Hintze, S., Kerr, A. R. W., Hehr, A., Schoser, B., & Meinke, P. (2021). Transcriptome Analysis in a Primary Human Muscle Cell Differentiation Model for Myotonic Dystrophy Type 1. International Journal of Molecular Sciences, 22(16), 8607. https://doi.org/10.3390/ijms22168607