
Identification of Prognostic and Chemopredictive microRNAs for Non-Small-Cell Lung Cancer by Integrating SEER-Medicare Data

 ,
,
Abstract
:1. Introduction
2. Results
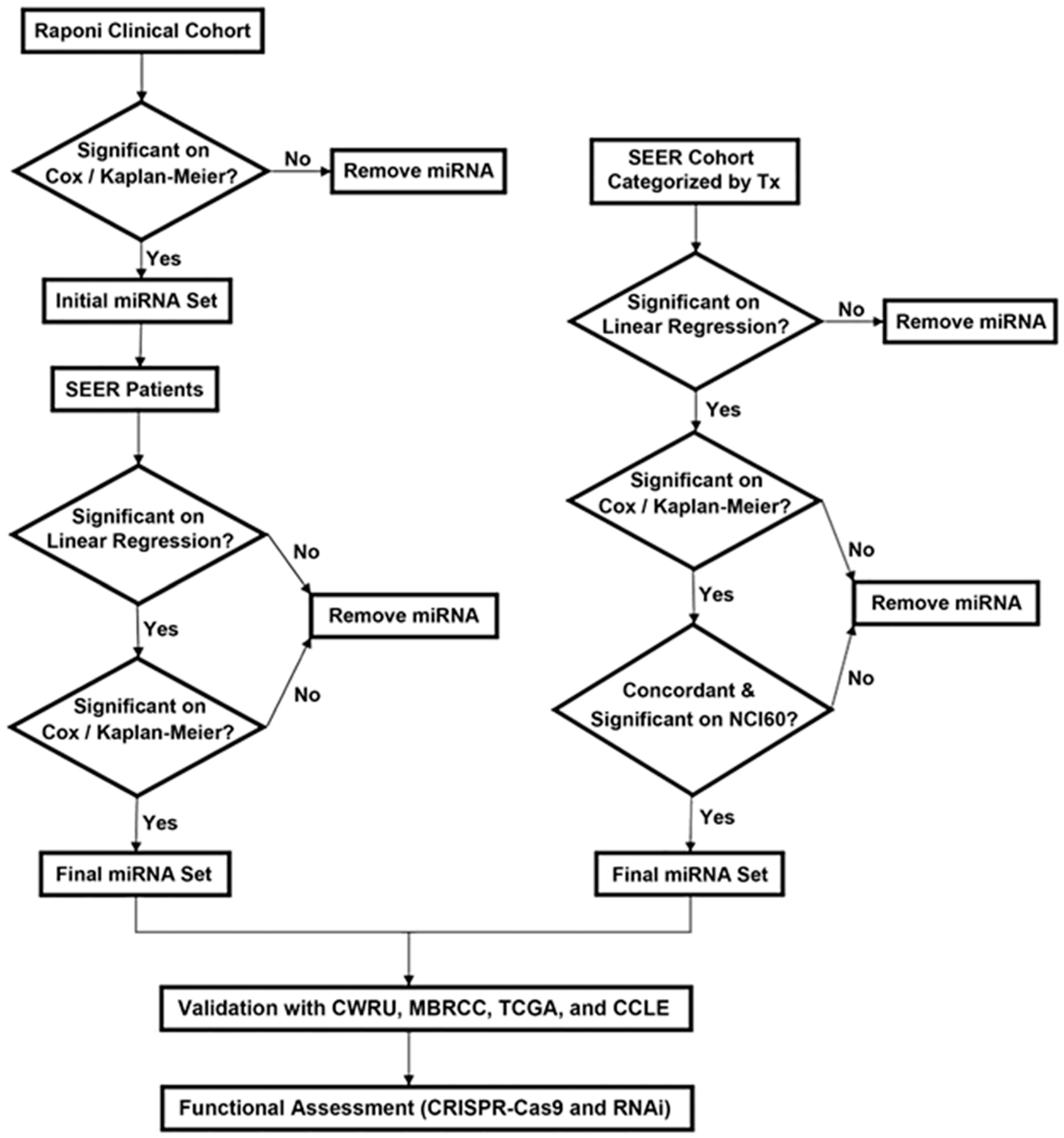
2.1. Identification of Prognostic miRNAs for NSCLC
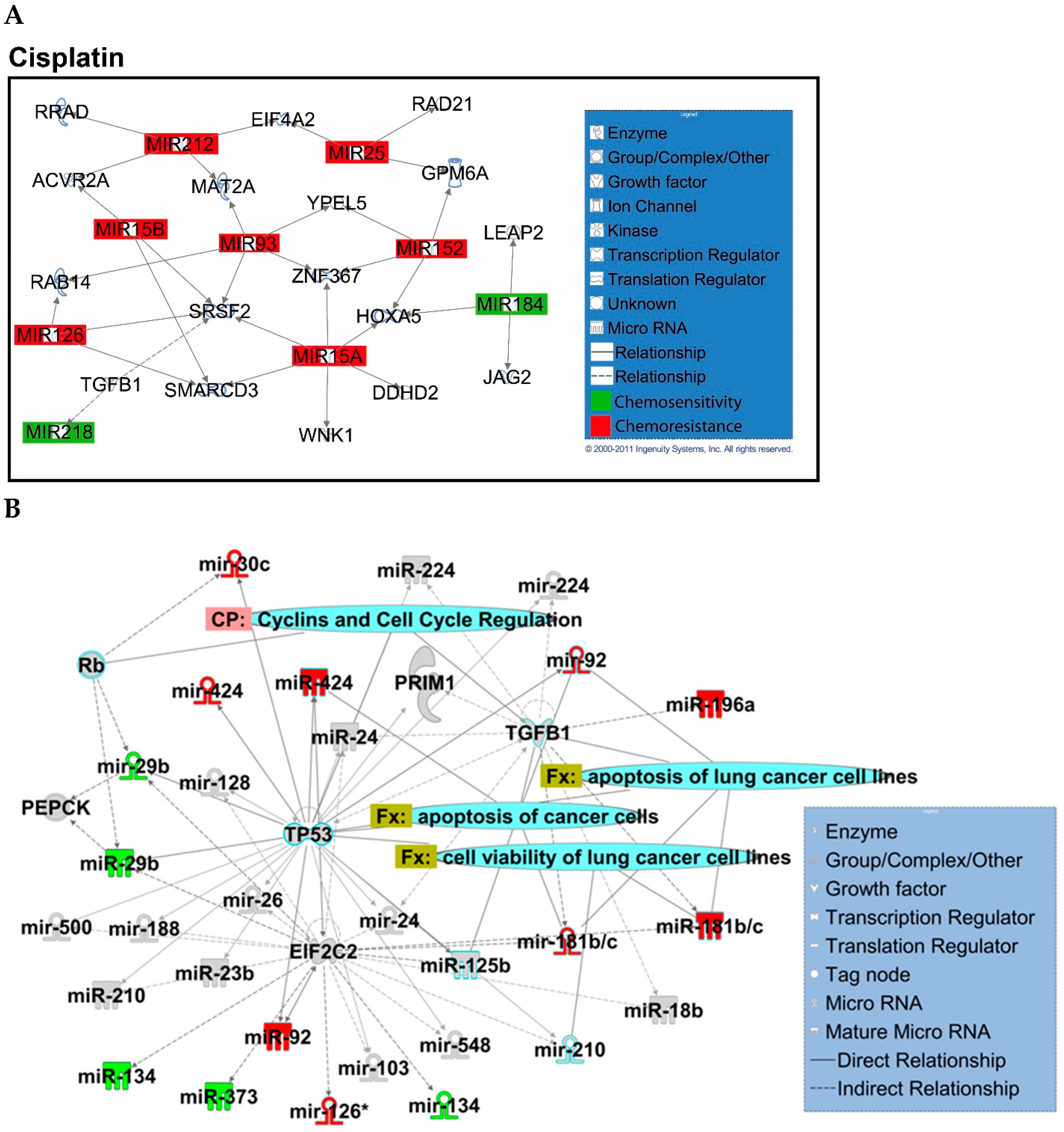
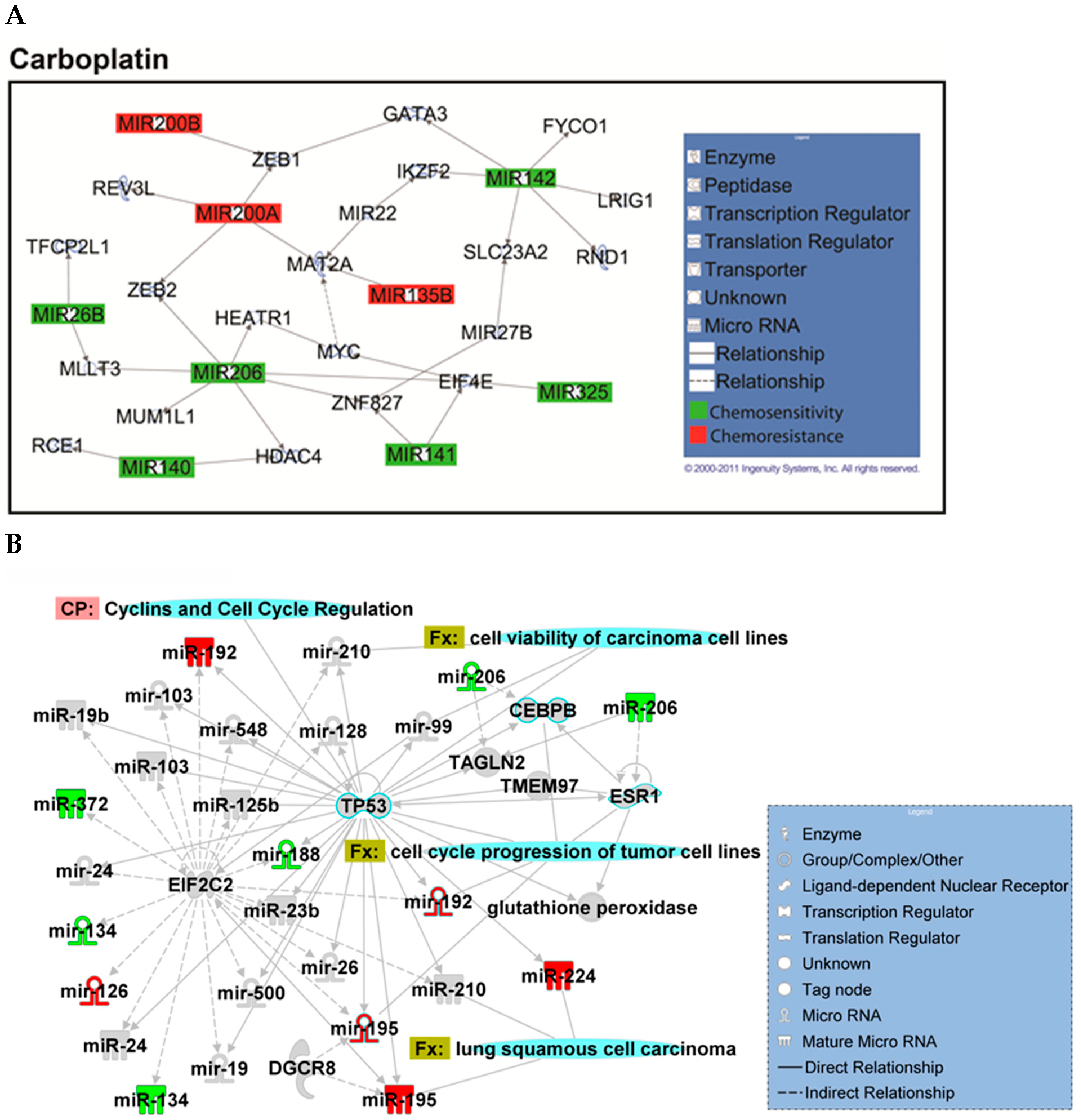
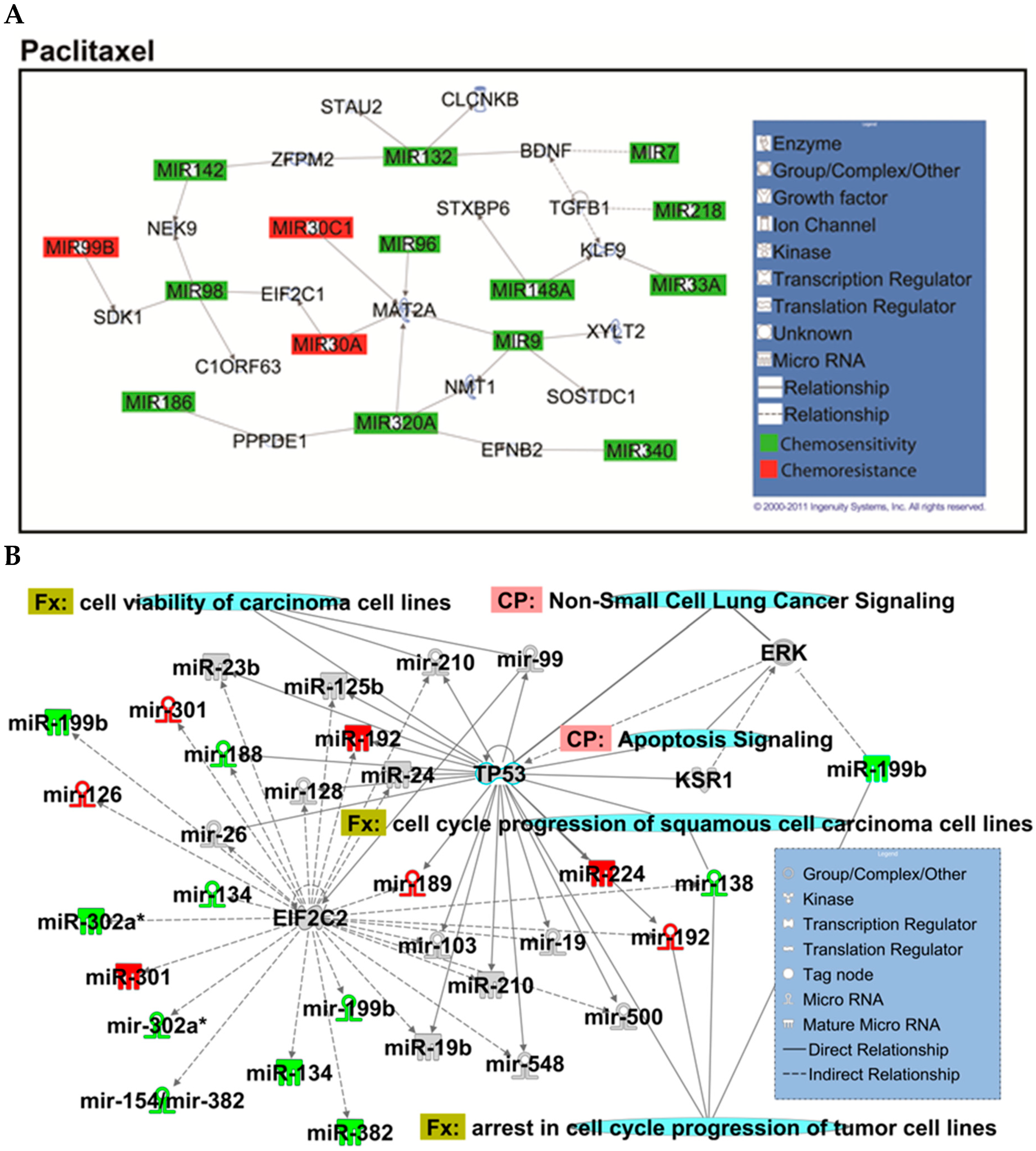
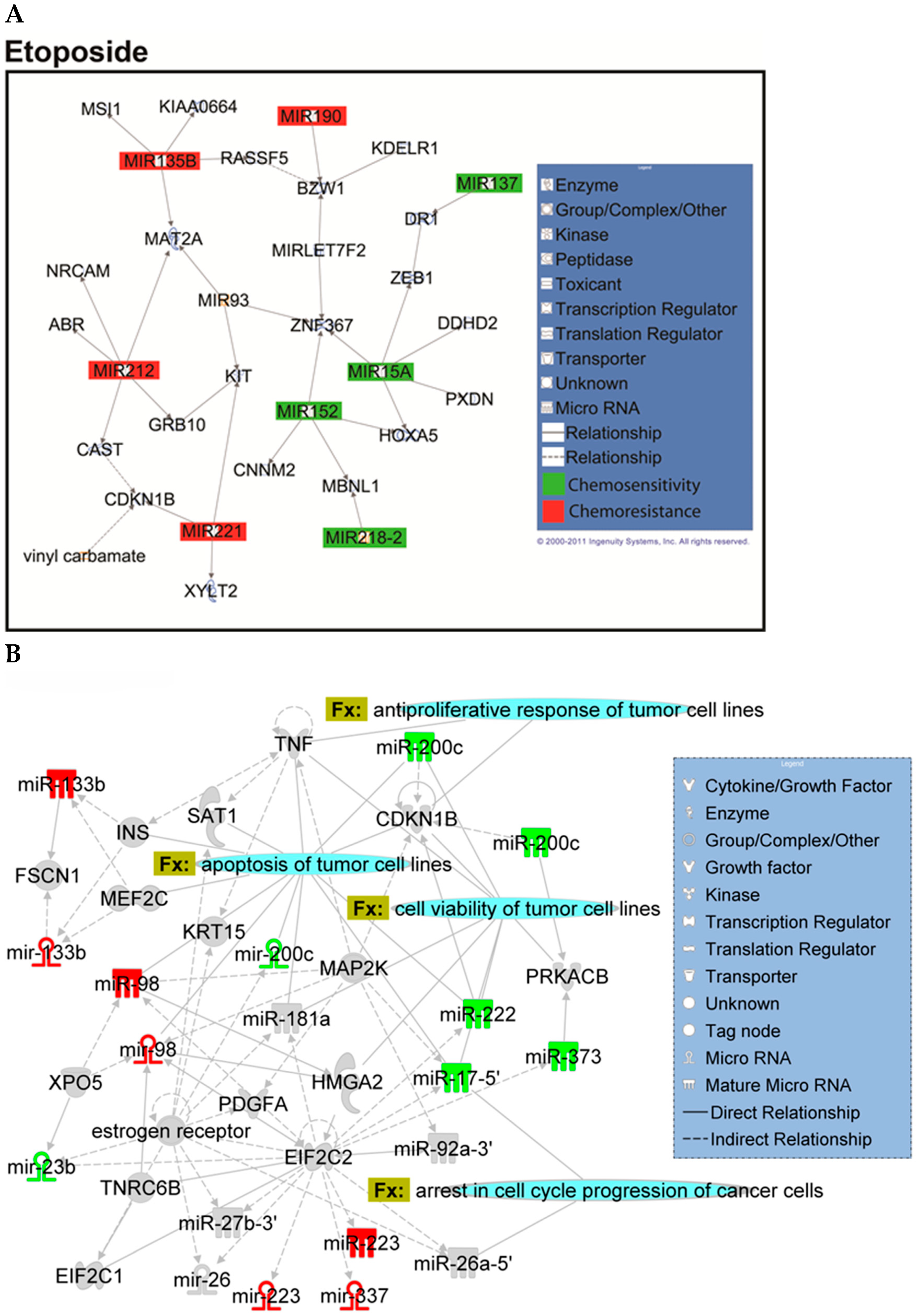
2.2. Identification of Chemopredictive miRNAs for NSCLC
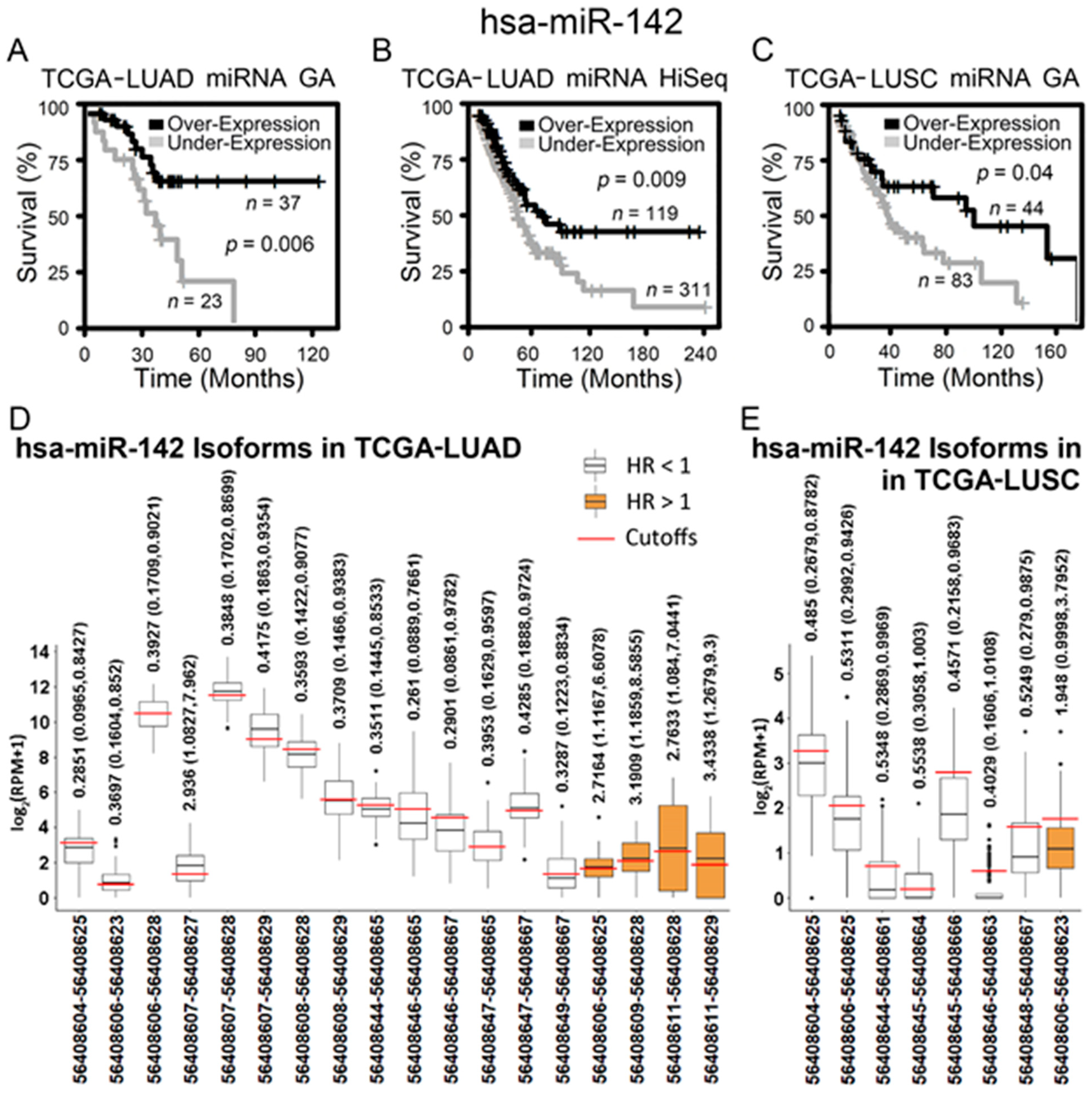
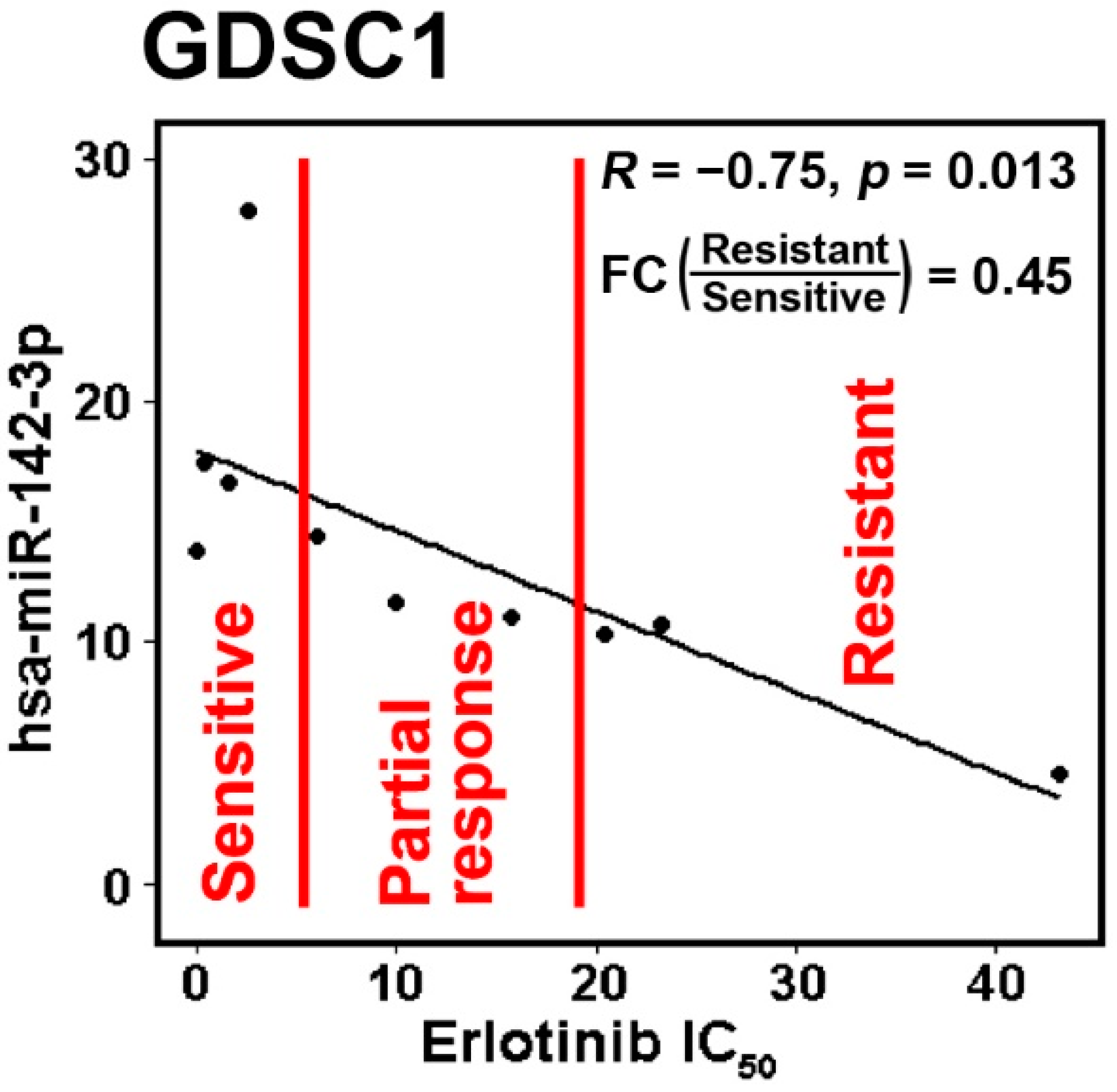
2.3. Hsa-miR-142-3p/Hsa-miR-142 as a Good Prognostic and Chemosensitive Biomarker for NSCLC
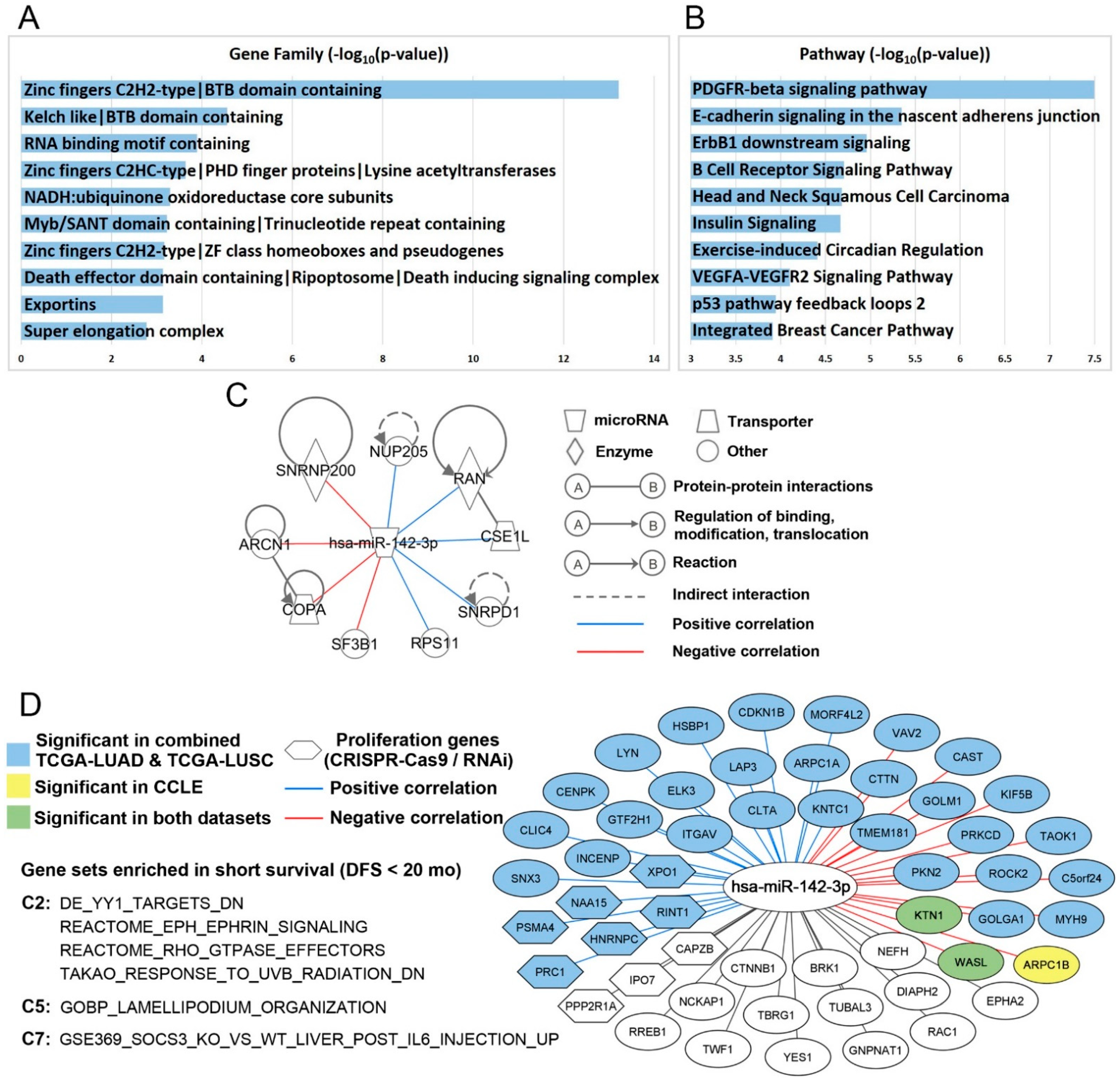
2.4. Hsa-miR-142-3p-Regulated Pathways and Functional Networks in NSCLC
3. Discussion
4. Materials and Methods
4.1. Patient Cohorts Used in Correlation with SEER-Medicare Data
4.2. Validation NSCLC Patient Cohorts
4.3. NCI-60 Cellular Data
4.4. Cancer Cell Line Encyclopedia (CCLE)
4.5. PRISM Drug Response in CCLE
4.6. Genomics of Drug Sensitivity in Cancer (GDSC1/2)
4.7. Identification of Prognostic miRNAs by Correlating with SEER-Medicare Data
4.8. Identification of Chemopredictive miRNA Using Linked SEER-Medicare Data
4.9. Validation of Chemopredictive miRNAs Using the NCI-60 Anticancer Screen
4.10. Validation of Prognostic and Predictive miRNAs Using Independent Patient Cohorts and CCLE
4.11. Analysis of Target Genes of Hsa-miR-142-3p
4.12. CRISPR-Cas9 Assays
4.13. RNAi Functional Assays
4.14. Ingenuity Pathway Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Cancer Stat Facts: Lung and Bronchus Cancer. Available online: https://seer.cancer.gov/statfacts/html/lungb.html (accessed on 26 May 2021).
- Hoffman, P.C.; Mauer, A.M.; Vokes, E.E. Lung cancer. Lancet 2000, 355, 479–485. [Google Scholar] [CrossRef]
- Naruke, T.; Goya, T.; Tsuchiya, R.; Suemasu, K. Prognosis and survival in resected lung carcinoma based on the new international staging system. J. Thorac. Cardiovasc. Surg. 1988, 96, 440–447. [Google Scholar] [CrossRef]
- Vogt, U.; Striehn, E.; Bosse, U.; Klinke, F.; Falkiewicz, B. Lack of squamous cell lung carcinoma in vitro chemosensitivity to various drug regimens in the adenosine triphosphate cell viability chemosensitivity assay. Acta Biochim. Pol. 1999, 46, 299–302. [Google Scholar] [CrossRef]
- Zheng, Y.; Jaklitsch, M.T.; Bueno, R. Neoadjuvant Therapy in Non-Small Cell Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 567–584. [Google Scholar] [CrossRef]
- Crino, L.; Weder, W.; Van, M.J.; Felip, E. Early stage and locally advanced (non-metastatic) non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. 5), v103–v115. [Google Scholar] [CrossRef]
- Byron, E.; Pinder-Schenck, M. Systemic and targeted therapies for early-stage lung cancer. Cancer Control 2014, 21, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Raponi, M.; Dossey, L.; Jatkoe, T.; Wu, X.; Chen, G.; Fan, H.; Beer, D.G. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009, 69, 5776–5783. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivo tumor growth. Breast Cancer Res. 2011, 13, R2. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.L.; Chen, H.Y.; Chang, G.C.; Chen, C.Y.; Chen, H.W.; Singh, S.; Cheng, C.L.; Yu, C.J.; Lee, Y.C.; Chen, H.S.; et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 2008, 13, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hu, Z.; Wang, W.; Ba, Y.; Ma, L.; Zhang, C.; Wang, C.; Ren, Z.; Zhao, Y.; Wu, S.; et al. Identification of ten serum microRNAs from a genome-wide serum microRNA expression profile as novel non-invasive biomarkers for non-small cell lung cancer diagnosis. Int. J. Cancer 2011, 130, 1620–1628. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Schaefer, A.; Steiner, I.; Kempkensteffen, C.; Stephan, C.; Erbersdobler, A.; Jung, K. Robust microRNA stability in degraded RNA preparations from human tissue and cell samples. Clin. Chem. 2010, 56, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Mraz, M.; Malinova, K.; Mayer, J.; Pospisilova, S. MicroRNA isolation and stability in stored RNA samples. Biochem. Biophys. Res. Commun. 2009, 390, 1–4. [Google Scholar] [CrossRef]
- Xi, Y.; Nakajima, G.; Gavin, E.; Morris, C.G.; Kudo, K.; Hayashi, K.; Ju, J. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 2007, 13, 1668–1674. [Google Scholar] [CrossRef] [Green Version]
- Avraham, R.; Yarden, Y. Regulation of signalling by microRNAs. Biochem. Soc. Trans. 2012, 40, 26–30. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. microRNA involvement in human cancer. Carcinogenesis 2012, 33, 1126–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, M. miRNAs as molecular biomarkers of cancer. Expert Rev. Mol. Diagn. 2010, 10, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Markou, A.; Tsaroucha, E.G.; Kaklamanis, L.; Fotinou, M.; Georgoulias, V.; Lianidou, E.S. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin. Chem. 2008, 54, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Rabinowits, G.; Gercel-Taylor, C.; Day, J.M.; Taylor, D.D.; Kloecker, G.H. Exosomal microRNA: A diagnostic marker for lung cancer. Clin. Lung Cancer 2009, 10, 42–46. [Google Scholar] [CrossRef]
- Wang, Q.Z.; Xu, W.; Habib, N.; Xu, R. Potential uses of microRNA in lung cancer diagnosis, prognosis, and therapy. Curr. Cancer Drug Targets 2009, 9, 572–594. [Google Scholar] [CrossRef] [Green Version]
- Świtlik, W.Z.; Szemraj, J. Circulating miRNAs as non-invasive biomarkers for non-small cell lung cancer diagnosis, prognosis and prediction of treatment response. Postepy Hig. Med. Dosw. 2017, 71, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, J. SEER Program.: Comparative Staging Guide for Cancer, Version 1.1 (Rep. No. 93-3640); NIH Publication: Bethesda, MD, USA, 1993. [Google Scholar]
- Stein, W.D.; Litman, T.; Fojo, T.; Bates, S.E. A Serial Analysis of Gene Expression (SAGE) database analysis of chemosensitivity: Comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res. 2004, 64, 2805–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, A.; Jewell, D.A.; Liang, Y.; Ridzon, D.; Moore, J.H.; Chen, C.F.; Ambros, V.R.; Israel, M.A. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007, 67, 2456–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Furuse, K. Platinum/oral etoposide therapy in non-small cell lung cancer. Oncology 1992, 49 (Suppl. 1), 63–69; discussion 70. [Google Scholar] [CrossRef]
- Steuer, C.E.; Behera, M.; Ernani, V.; Higgins, K.A.; Saba, N.F.; Shin, D.M.; Pakkala, S.; Pillai, R.N.; Owonikoko, T.K.; Curran, W.J.; et al. Comparison of Concurrent Use of Thoracic Radiation With Either Carboplatin-Paclitaxel or Cisplatin-Etoposide for Patients With Stage III Non-Small-Cell Lung Cancer: A Systematic Review. JAMA Oncol. 2017, 3, 1120–1129. [Google Scholar] [CrossRef]
- Ramalingam, S.; Belani, C.P. Paclitaxel for non-small cell lung cancer. Expert Opin. Pharmacother. 2004, 5, 1771–1780. [Google Scholar] [CrossRef]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Mohamed, R.; Dukhlallah, D.; Gencheva, M.; Hu, G.; Pearce, M.C.; Kolluri, S.K.; Marsh, C.B.; Eubank, T.D.; Ivanov, A.V.; et al. Molecular Analysis of ZNF71 KRAB in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3752. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [Green Version]
- Pillai, M.M.; Gillen, A.E.; Yamamoto, T.M.; Kline, E.; Brown, J.; Flory, K.; Hesselberth, J.R.; Kabos, P. HITS-CLIP reveals key regulators of nuclear receptor signaling in breast cancer. Breast Cancer Res. Treat. 2014, 146, 85–97. [Google Scholar] [CrossRef]
- Whisnant, A.W.; Bogerd, H.P.; Flores, O.; Ho, P.; Powers, J.G.; Sharova, N.; Stevenson, M.; Chen, C.H.; Cullen, B.R. In-depth analysis of the interaction of HIV-1 with cellular microRNA biogenesis and effector mechanisms. mBio 2013, 4, e000193. [Google Scholar] [CrossRef] [Green Version]
- Haecker, I.; Gay, L.A.; Yang, Y.; Hu, J.; Morse, A.M.; McIntyre, L.M.; Renne, R. Ago HITS-CLIP expands understanding of Kaposi’s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012, 8, e1002884. [Google Scholar] [CrossRef] [Green Version]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Karginov, F.V.; Hannon, G.J. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013, 27, 1624–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottwein, E.; Corcoran, D.L.; Mukherjee, N.; Skalsky, R.L.; Hafner, M.; Nusbaum, J.D.; Shamulailatpam, P.; Love, C.L.; Dave, S.S.; Tuschl, T.; et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 2011, 10, 515–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Watkin, L.B.; Jessen, B.; Wiszniewski, W.; Vece, T.J.; Jan, M.; Sha, Y.; Thamsen, M.; Santos-Cortez, R.L.; Lee, K.; Gambin, T.; et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat. Genet. 2015, 47, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavel, J.; Harter, C.; Wieland, F.T. Reversible dissociation of coatomer: Functional characterization of a beta/delta-coat protein subcomplex. Proc. Natl. Acad. Sci. USA 1998, 95, 2140–2145. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Izumi, K.; Brett, M.; Nishi, E.; Drunat, S.; Tan, E.S.; Fujiki, K.; Lebon, S.; Cham, B.; Masuda, K.; Arakawa, M.; et al. ARCN1 Mutations Cause a Recognizable Craniofacial Syndrome Due to COPI-Mediated Transport Defects. Am. J. Hum. Genet. 2016, 99, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Behrens, P.; Brinkmann, U.; Wellmann, A. CSE1L/CAS: Its role in proliferation and apoptosis. Apoptosis Int. J. Program. Cell Death 2003, 8, 39–44. [Google Scholar] [CrossRef]
- Fujitomo, T.; Daigo, Y.; Matsuda, K.; Ueda, K.; Nakamura, Y. Critical function for nuclear envelope protein TMEM209 in human pulmonary carcinogenesis. Cancer Res. 2012, 72, 4110–4118. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, F.; Tu, Z.; Tang, H.Y.; Zhu, H.; Speicher, D.W.; Zhang, R. Comprehensive analysis of the ubiquitinome during oncogene-induced senescence in human fibroblasts. Cell Cycle 2015, 14, 1540–1547. [Google Scholar] [CrossRef] [Green Version]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Absmeier, E.; Holton, N.; Pietrzyk-Brzezinska, A.J.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Wahl, M.C. The interaction of DNA repair factors ASCC2 and ASCC3 is affected by somatic cancer mutations. Nat. Commun. 2020, 11, 5535. [Google Scholar] [CrossRef] [PubMed]
- Quidville, V.; Alsafadi, S.; Goubar, A.; Commo, F.; Scott, V.; Pioche-Durieu, C.; Girault, I.; Baconnais, S.; Le Cam, E.; Lazar, V.; et al. Targeting the deregulated spliceosome core machinery in cancer cells triggers mTOR blockade and autophagy. Cancer Res. 2013, 73, 2247–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Huang, S.; Zhang, Z.; Qian, X.; Sun, P.; Zhou, X. Pan-cancer analysis on microRNA-associated gene activation. EBioMedicine 2019, 43, 82–97. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Edlund, K.; Holmberg, L.; Kultima, H.G.; Mansouri, L.; Ekman, S.; Bergqvist, M.; Scheibenflug, L.; Lamberg, K.; Myrdal, G.; et al. Gene copy number aberrations are associated with survival in histologic subgroups of non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Jabs, V.; Edlund, K.; Konig, H.; Grinberg, M.; Madjar, K.; Rahnenfuhrer, J.; Ekman, S.; Bergkvist, M.; Holmberg, L.; Ickstadt, K.; et al. Integrative analysis of genome-wide gene copy number changes and gene expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0187246. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjoblom, T.; Bruun, J.; Micke, P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J. Pathol. 2018, 244, 421–431. [Google Scholar] [CrossRef]
- Zhan, P.; Zhang, B.; Xi, G.M.; Wu, Y.; Liu, H.B.; Liu, Y.F.; Xu, W.J.; Zhu, Q.Q.; Cai, F.; Zhou, Z.J.; et al. PRC1 contributes to tumorigenesis of lung adenocarcinoma in association with the Wnt/β-catenin signaling pathway. Mol. Cancer 2017, 16, 108. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, P.; Wen, W.; James, M.A.; Wang, Y.; Bailey-Wilson, J.E.; Amos, C.I.; Pinney, S.M.; Yang, P.; de Andrade, M.; et al. Haplotype and cell proliferation analyses of candidate lung cancer susceptibility genes on chromosome 15q24-25.1. Cancer Res. 2009, 69, 7844–7850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Dharmadhikari, A.V.; Varland, S.; Ma, N.; Domingo, D.; Kleyner, R.; Rope, A.F.; Yoon, M.; Stray-Pedersen, A.; Posey, J.E.; et al. Truncating Variants in NAA15 Are Associated with Variable Levels of Intellectual Disability, Autism Spectrum Disorder, and Congenital Anomalies. Am. J. Hum. Genet. 2018, 102, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Huai, Q.; Zhang, G.; Guo, L.; Song, P.; Xue, X.; Tan, F.; Xue, Q.; Gao, S.; He, J. Elevated Heterogeneous Nuclear Ribonucleoprotein C Expression Correlates With Poor Prognosis in Patients With Surgically Resected Lung Adenocarcinoma. Front. Oncol. 2020, 10, 598437. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Tao, K.; Le Calvez-Kelm, F.; Nguyen-Dumont, T.; Robinot, N.; Hammet, F.; Odefrey, F.; Tsimiklis, H.; Teo, Z.L.; Thingholm, L.B.; et al. Rare mutations in RINT1 predispose carriers to breast and Lynch syndrome-spectrum cancers. Cancer Discov. 2014, 4, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Xu, G.; Wang, L.; Yang, H.; Liu, X.; Zhao, J.; Zhang, H.T. MiR-142-3p represses TGF-β-induced growth inhibition through repression of TGFβR1 in non-small cell lung cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, W.; Zhang, W.; Jia, Y.; Yang, X.; Wang, Y.; Zhang, J. MicroRNA-142-3p/MALAT1 inhibits lung cancer progression through repressing β-catenin expression. Biomed. Pharmacother. 2019, 114, 108847. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [Green Version]
- Filipska, M.; Skrzypski, M.; Czetyrbok, K.; Stokowy, T.; Stasiłojć, G.; Supernat, A.; Jassem, J.; Żaczek, A.J.; Bigda, J. MiR-192 and miR-662 enhance chemoresistance and invasiveness of squamous cell lung carcinoma. Lung Cancer 2018, 118, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.Y.; Zhang, F.; Sun, C.C.; Li, S.J.; Li, G.; Gong, F.Y.; Bo, T.; He, J.; Hua, R.X.; Hu, W.D.; et al. miR-134: A Human Cancer Suppressor? Mol. Ther. Nucleic Acids 2017, 6, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Wei, F.; Zhang, J.; Wang, X.; Li, B. miR-134 inhibits non-small cell lung cancer growth by targeting the epidermal growth factor receptor. J. Cell. Mol. Med. 2016, 20, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Li, S.J.; Li, D.J. Hsa-miR-134 suppresses non-small cell lung cancer (NSCLC) development through down-regulation of CCND1. Oncotarget 2016, 7, 35960–35978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, Y.; Luo, J.; Fu, Z.; Ying, J.; Yu, Y.; Yu, W. miR-134 inhibits epithelial to mesenchymal transition by targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett. 2012, 586, 3761–3765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Huang, P.; Li, Q.; Wang, D.; Xu, C.X. miR-134-5p Promotes Stage I Lung Adenocarcinoma Metastasis and Chemoresistance by Targeting DAB2. Mol. Ther. Nucleic Acids 2019, 18, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloonan, N.; Wani, S.; Xu, Q.; Gu, J.; Lea, K.; Heater, S.; Barbacioru, C.; Steptoe, A.L.; Martin, H.C.; Nourbakhsh, E.; et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011, 12, R126. [Google Scholar] [CrossRef] [Green Version]
- Fossella, F.V. Docetaxel for previously treated non-small-cell lung cancer. Oncology 2002, 16 (Suppl. 6), 45–51. [Google Scholar] [PubMed]
- Arrieta, O.; Barrón, F.; Ramírez-Tirado, L.A.; Zatarain-Barrón, Z.L.; Cardona, A.F.; Díaz-García, D.; Yamamoto Ramos, M.; Mota-Vega, B.; Carmona, A.; Álvarez, M.P.P.; et al. Efficacy and Safety of Pembrolizumab Plus Docetaxel vs Docetaxel Alone in Patients With Previously Treated Advanced Non-Small Cell Lung Cancer: The PROLUNG Phase 2 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 856–864. [Google Scholar] [CrossRef]
- Warren, J.L.; Harlan, L.C.; Fahey, A.; Virnig, B.A.; Freeman, J.L.; Klabunde, C.N.; Cooper, G.S.; Knopf, K.B. Utility of the SEER-Medicare data to identify chemotherapy use. Med. Care 2002, 40 (Suppl. 8), IV55–IV61. [Google Scholar] [CrossRef]
- Potosky, A.L.; Riley, G.F.; Lubitz, J.D.; Mentnech, R.M.; Kessler, L.G. Potential for cancer related health services research using a linked Medicare-tumor registry database. Med. Care 1993, 31, 732–748. [Google Scholar] [CrossRef] [PubMed]
- DepMap. DepMap 20Q2 Public. In Broad; Figshare: London, UK, 2020. [Google Scholar]
- Aguet, F. TOPMed RNA-Seq Pipeline Harmonization Summary. Available online: https://github.com/broadinstitute/gtex-pipeline/blob/master/TOPMed_RNAseq_pipeline.md (accessed on 24 March 2021).
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Ding, Z.; Qian, Y.; Shi, X.; Castranova, V.; Harner, E.J.; Guo, L. Predicting Cancer Drug Response by Proteomic Profiling. Clin. Cancer Res. 2006, 12, 4583–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Ding, Z.; Qian, Y.; Wan, Y.W.; Tosun, K.; Shi, X.; Castranova, V.; Harner, E.J.; Guo, N.L. An integrative genomic and proteomic approach to chemosensitivity prediction. Int. J. Oncol. 2009, 34, 107–115. [Google Scholar] [PubMed] [Green Version]
- Schwartz, L.H.; Litière, S.; de Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, M.S.; Janes, H.; Longton, G.; Leisenring, W.; Newcomb, P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am. J. Epidemiol. 2004, 159, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 19–109. [Google Scholar] [CrossRef]
- Weinstein, J.N. Spotlight on molecular profiling: "Integromic" analysis of the NCI-60 cancer cell lines. Mol. Cancer Ther. 2006, 5, 2601–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Team, R. RStudio: Integrated Development Environment for R, 1.4.1106; R Studio, PBC: Boston, MA, USA, 2021. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Variable | Patient Data Used in Population Correlation Analysis | Validation Patient Cohorts | ||||
|---|---|---|---|---|---|---|
| SEER-Medicare | Clinical Cohort (Raponi et al.) | MBRCC | CWRU | TCGA-LUAD | TCGA-LUSC | |
| Cancer Stage | ||||||
| Stage I | 12,651 (37.3%) | 34 (59.6%) | 32 (51.6%) | 35 (43.8%) | 279 (53.4%) | 245 (48.6%) |
| Stage II | 2662 (7.8%) | 11 (19.3%) | 20 (32.3%) | 39 (48.8%) | 124 (23.8%) | 163 (32.3%) |
| Stage III | 11,514 (34%) | 12 (21.1%) | 8 (12.9%) | 5 (6.3%) | 85 (16.3%) | 85 (16.9%) |
| Stage IV | 5813 (17.1%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 26 (5.0%) | 7 (1.4%) |
| Unstaged/Other | 1257 (3.7%) | 0 (0.0%) | 2 (3.2%) | 1 (1.3%) | 8 (1.5%) | 4 (0.8%) |
| Tumor Grade | ||||||
| Grade 1 | 1462 (4.3%) | 1 (1.8%) | 3 (4.8%) | 2 (2.5%) | N/A | N/A |
| Grade 2 | 13,573 (40.0%) | 28 (49.1%) | 22 (35.5%) | 35 (43.8%) | N/A | N/A |
| Grade 3 | 18,202 (53.7%) | 28 (49.1%) | 27 (43.6%) | 32 (40.0%) | N/A | N/A |
| Grade 4 | 660 (1.9%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | N/A | N/A |
| Ungraded/Other | 0 (0.0%) | 0 (0.0%) | 10 (16.1%) | 11 (13.8%) | N/A | N/A |
| Mean Age (σ) | 71.1 (7.9) | 66.8 (10.7) | 67.8 (9.6) | 69.5 (9.7) | 65.2 (10.0) | 67.3 (8.6) |
| Sex | ||||||
| Male | 22,218 (65.5%) | 39 (68.4%) | 29 (46.8%) | 44 (55%) | 242 (%) | 373 (74.0%) |
| Female | 11,679 (34.5%) | 18 (31.6%) | 31 (50.0%) | 36 (45%) | 280 (%) | 131 (26.0%) |
| Missing | 0 (0.0%) | 0 (0.0%) | 2 (3.2%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Treatment | No Chemotherapy | |
|---|---|---|
| Positive Correlation | Negative Correlation | |
| Surgery | miR-520d*, miR-433, miR-134, miR-382 | miR-328, miR-384, miR-525* |
| Radiation | miR-433 | - |
| Surgery and radiation | miR-453, miR-520d*, miR-134 | miR-197 |
| Any treatment | miR-453, miR-372, miR-142-3p (miR-142), miR-329 (miR-329-2), miR-520d*, miR-433, miR-134, miR-382, miR-493-3p | miR-328, miR-197, miR-384 |
| Treatment | Cisplatin | Carboplatin | Paclitaxel | Etoposide | Any Chemotherapy |
|---|---|---|---|---|---|
| Surgery | miR-29b (miR-29b-1, miR-29b-2) | miR-134, miR-142-3p (miR-142), miR-188, miR-380-5p (miR-380) | miR-134, miR-138 (miR-138-2), miR-142-3p (miR-142), miR-188, miR-380-5p (miR-380) | - | miR-142-3p (miR-142), miR-433 |
| Radiation | - | - | miR-154, miR-302a* | miR-141, miR-17-3p (miR-17), miR-17-5p (miR-17), miR-182, miR-183, miR-19b (miR-19b-1), miR-200c, miR-222, miR-23b | miR-134 |
| Surgery And Radiation | - | miR-134, miR-142-3p (miR-142) | miR-142-3p, miR-220 | - | miR-142-3p (miR-142) |
| Any treatment | miR-129 (miR-129-1, miR-129-2), miR-134, miR-142-3p (miR-142), miR-184, miR-198, miR-370, miR-373, miR-379 | miR-134, miR-142-3p (miR-142), miR-206, miR-33 (miR-33-a), miR-370, miR-372 | miR-134, miR-142-3p (miR-142), miR-199b, miR-370, miR-382 | miR-129 (miR-129-1, miR-129-2), miR-141, miR-142-3p (miR-142), miR-184, miR-218 (miR-218-1), miR-220, miR-335, miR-373, miR-96 | miR-33 (miR-33-a), miR-453, miR-372, miR-142-3p (miR-142), mir-299-3p, miR-329 (miR-329-2), miR-520d*, miR-519a, miR-494, miR-433, miR-134, miR-485-5p (miR-485), miR-518c*, miR-493-3p |
| Treatment | Cisplatin | Carboplatin | Paclitaxel | Etoposide | Any Chemotherapy |
|---|---|---|---|---|---|
| Surgery | miR-126*, miR-136, miR-181b, miR-181c, miR-196a (miR-196a-2), miR-331, miR-375, miR-424, miR-92 (miR-92a-1, miR-92b) | miR-126, miR-192, miR-195, miR-384 | miR-189, miR-192, miR-197, miR-301, miR-328, miR-331, miR-384 | - | miR-197, miR-384 |
| Radiation | miR-384 | - | miR-189 | miR-150, miR-155, miR-192, miR-337, miR-98 | miR-328 |
| Surgery and Radiation | - | miR-192, miR-384 | miR-192, miR-197, miR-328, miR-331, miR-384 | miR-223 | miR-328, miR-197, miR-384 |
| Any treatment | miR-132, miR-181b, miR-30c, miR-30e-3p, miR-324-3p, miR-331, miR-339, miR-384 | miR-126, miR-195, miR-224, miR-384 | miR-126, miR-224, miR-328, miR-331, miR-361, miR-384 | miR-132, miR-133b, miR-155, miR-197, miR-208, miR-214, miR-324-3p, miR-374, miR-423 | miR-328, miR-361, miR-511, miR-197, miR-125a, miR-384, miR-126 |
| Drugs | PRISM | GDSC1 | GDSC2 |
|---|---|---|---|
| Cisplatin | SNRNP200 | SF3B1, RAN | CSE1L |
| Erlotinib | SNRNP200 | CSE1L | |
| Gefitinib | COPA | ||
| Paclitaxel | NUP205 | ||
| Pemetrexed | RAN, SNRPD1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Putila, J.; Raese, R.; Dong, C.; Qian, Y.; Dowlati, A.; Guo, N.L. Identification of Prognostic and Chemopredictive microRNAs for Non-Small-Cell Lung Cancer by Integrating SEER-Medicare Data. Int. J. Mol. Sci. 2021, 22, 7658. https://doi.org/10.3390/ijms22147658
Ye Q, Putila J, Raese R, Dong C, Qian Y, Dowlati A, Guo NL. Identification of Prognostic and Chemopredictive microRNAs for Non-Small-Cell Lung Cancer by Integrating SEER-Medicare Data. International Journal of Molecular Sciences. 2021; 22(14):7658. https://doi.org/10.3390/ijms22147658
Chicago/Turabian StyleYe, Qing, Joseph Putila, Rebecca Raese, Chunlin Dong, Yong Qian, Afshin Dowlati, and Nancy Lan Guo. 2021. "Identification of Prognostic and Chemopredictive microRNAs for Non-Small-Cell Lung Cancer by Integrating SEER-Medicare Data" International Journal of Molecular Sciences 22, no. 14: 7658. https://doi.org/10.3390/ijms22147658
APA StyleYe, Q., Putila, J., Raese, R., Dong, C., Qian, Y., Dowlati, A., & Guo, N. L. (2021). Identification of Prognostic and Chemopredictive microRNAs for Non-Small-Cell Lung Cancer by Integrating SEER-Medicare Data. International Journal of Molecular Sciences, 22(14), 7658. https://doi.org/10.3390/ijms22147658