Abstract
Pseudomonas aeruginosa and Sphingobacterium sp. are well known for their ability to decontaminate many environmental pollutants while Geobacillus sp. have been exploited for their thermostable enzymes. This study reports the annotation of genomes of P. aeruginosa S3, Sphingobacterium S2 and Geobacillus EC-3 that were isolated from compost, based on their ability to degrade poly(lactic acid), PLA. Draft genomes of the strains were assembled from Illumina reads, annotated and viewed with the aim of gaining insight into the genetic elements involved in degradation of PLA. The draft genome of Sphinogobacterium strain S2 (435 contigs) was estimated at 5,604,691 bp and the draft genome of P. aeruginosa strain S3 (303 contigs) was estimated at 6,631,638 bp. The draft genome of the thermophile Geobacillus strain EC-3 (111 contigs) was estimated at 3,397,712 bp. A total of 5385 (60% with annotation), 6437 (80% with annotation) and 3790 (74% with annotation) protein-coding genes were predicted for strains S2, S3 and EC-3, respectively. Catabolic genes for the biodegradation of xenobiotics, aromatic compounds and lactic acid as well as the genes attributable to the establishment and regulation of biofilm were identified in all three draft genomes. Our results reveal essential genetic elements that facilitate PLA metabolism at mesophilic and thermophilic temperatures in these three isolates.
1. Introduction
The lactic acid required to produce poly(lactic acid) (PLA) through polycondensation is derived primarily from the microbial fermentation of agro-industrial waste. Frequently, waste by-products from crops like casava, wheat, bran, corn, etc., serve as the fermentable substrates [1,2]. PLA is certified as completely biodegradable under industrial composting conditions [3]. In the last two decades, the biodegradation of PLA has been extensively studied and many microbial species (actinomycete, bacteria, fungus) have been identified with the ability to degrade PLA [4]. Most of the reported bacterial species are from the families Pseudonocardiaceae, Thermomonosporaceae, Micromonosporaceae, Streptosporangiaceae, Bacillaceae and Thermoactinomycetaceae while the fungal species are mainly from the phyla Basidiomycota (Tremellaceae) and Ascomycota (Trichocomaceae, Hypocreaceae) [5,6,7,8,9,10,11].
In our previous study on mesophilic strains, we described four bacterial isolates designated as S1, S2, S3 and S4, able to degrade PLA at ambient temperature [12]. Two of the isolated strains, Sphingobacterium sp. (S2) and P. aeruginosa (S3), were evaluated for their PLA degradation in soil microcosms [13] and are the subject of this study. The genus Sphingobacterium is from the phylum Bacteriodetes, family Sphingobacteriaceae, named with reference to the sphingolipids in their cell wall [14,15]. They are Gram-negative rods, and the GC content of their DNA usually ranges from 35 to 44 mol% [16,17]. Sphingobacterium sp. are found in a range of habitats like soil, forest, compost, activated sludge, rhizosphere, feces, lakes and various food sources [18]. Sphingobacterium have also been reported to have a potential role in the biodegradation of different pollutants, including mixed plastic waste, PAHs, oils and pesticides [19,20,21]. Pseudomonas aeruginosa are Gram-negative bacteria from the γ-subdivision of proteobacteria. They are ubiquitously distributed in soil and aquatic habitats and are well-known opportunistic pathogens [22,23]. They have the ability to thrive in highly diverse and unusual ecological niches with low levels of nutrients. Their metabolic versatility and resistances allow them to survive on a variety of diverse carbon sources and overcome disinfectants and some antibiotics [24,25]. In previous reports, the role of Pseudomonas aeruginosa in the degradation of a variety of different compounds, including polycyclic aromatic hydrocarbons, xenobiotic compounds, oils, dyes and plastics is well documented [26,27,28,29,30]. In a second study we described the isolation of a thermophilic Geobacillus strain isolated with PLA as the sole source of carbon and capable of forming biofilm [31]. Geobacillus sp. are well-described Gram-positive spore forming rods whose thermophilic attributes have been exploited for highly stable enzymatic activities [32,33].
The five PLA degrading bacterial strains reported in our previous studies were isolated from compost and had the ability to degrade PLA at either ambient temperature, in the cases of P. aeruginosa and Sphingomonas sp., [12] or at elevated temperatures in the case of Geobacillus sp. [31]. Interestingly, our P. aeruginosa strains (S3 and S4) were lactate utilizing while the Sphingobacterium S2 and Chryseobacterium S1 strains were unable to utilize lactate when provided as the sole carbon source in minimal media. We also observed that all four isolates could form biofilm on PLA.
The purpose of this study was to analyze the genomes of three isolates (S2, S3 and EC-3) and explore the genetic determinants responsible for conferring their degradation abilities. The genes controlling lactate utilization and biofilm formation and regulation were inferred through comparative sequence analysis at PATRIC [34]. The whole genome sequence analysis for P. aeruginosa sp. is extensive but such data for Sphingobacterium sp. and Geobacillus sp. are limited. To our knowledge this is one of the first reports to give genetic depth to PLA-degrading bacterial strains and provide the basis for further studies.
2. Results
2.1. General Genome Features of Sphingobacterium sp. (S2), P. aeruginosa (S3) and Geobacillus sp. (EC-3)
Table 1 shows the general features for the assembled genomes. The assembly of the draft genome for strain S2 yielded 87 contigs (434,971 maximum length) and a total of 5,445,390 assembled base pairs with 43.66% GC content. There were an estimated 4951 CDS regions, 2864 proteins with functional assignments and several antibiotic resistance genes. The assembled draft genome of S3 had 63 contigs (658,980 maximum length) with a total of 6,509,961 assembled base pairs and was 66.26% GC content. There were an estimated 6239 CDS regions and 4932 proteins with functional assignments and a substantial number of putative antibiotic resistant and virulence determinants. As a thermophile, Geobacillus strain EC-3 had a smaller genome with 111 contigs and a total of 3,397,712 assembled base pairs with the largest contig of 321,190 bp. The G+C content was 52.18% with an estimated 3790 CDS regions and 2806 proteins with functional assignments. Because these were draft sequences, i.e., not closed, the number of chromosomes and plasmids could not be determined.

Table 1.
General genomic features of Sphingobacterium S2, P. aeruginosa S3 and Geobacillus EC-3.
2.2. Genetic Relatedness Based on ANI
The average nucleotide identity (ANI) value describes the similarity between the sequences of the conserved regions of two genomes and measures the genetic relatedness between them [35]. ANI measurements are considered more informative over 16S rRNA gene identity as they are based on a larger number of genes [36] and ANI values ≥ 95% should be considered as the same species [37]. ANI comparisons were done to explore the interspecies genetic relatedness between our three isolates and related species in the public databases. The data point in the upper-right corner of Figure 1 represents S. thalpophilum DSM11723 and S. thalpophilum NCTC11429, both of which showed more than 98% 16S rRNA gene similarity and 98% ANI with strain S2. Strain NCTC11429, isolated from a human wound (NCBI Biosample SAMEA3643323), was the closest match to our strain.
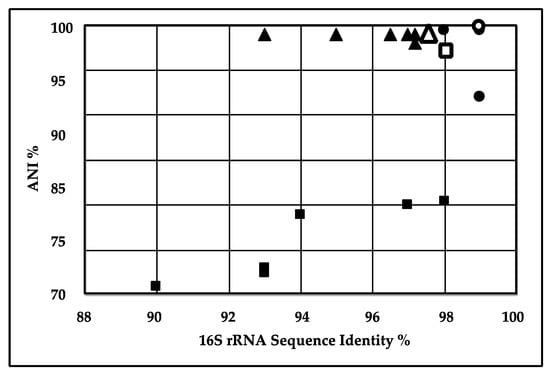
Figure 1.
Correlation between 16S rRNA gene similarity and average nucleotide identity (ANI) of sequenced strains. Sphingobacterium S2 (∎), P. aeruginosa (▲) and Geobacillus EC-3 (●) were compared to their closest relatives by 16S rRNA similarity (abscissa) and ANI (ordinate). The closest relatives, S. thalpophilum DSM11723 ( ), P. aeruginosa PSE305 (△) and G. thermoleovorans CCB_US3_UF5 (
), P. aeruginosa PSE305 (△) and G. thermoleovorans CCB_US3_UF5 ( ), were used in the MAUVE alignments below. Note that the 16S rRNA sequences came from prior sequencing of the 16S rRNA gene [12,31].
), were used in the MAUVE alignments below. Note that the 16S rRNA sequences came from prior sequencing of the 16S rRNA gene [12,31].
), P. aeruginosa PSE305 (△) and G. thermoleovorans CCB_US3_UF5 (), were used in the MAUVE alignments below. Note that the 16S rRNA sequences came from prior sequencing of the 16S rRNA gene [12,31].
In the case of P. aeruginosa strain S3, Figure 1 shows a cluster in the upper-right corner containing closely related strains with more than 98–99% sequence similarity, based on 16S rRNA sequences and 93–97% ANI. P. aeruginosa PSE305 showed the highest ANI value and 16S rRNA gene identity of 97.69% and 99% respectively. Accordingly, P. aeruginosa strain S3 and other strains included in the analysis belong to the same species. The results are consistent with the phylogenetic results based on 16S rRNA gene identity as previously reported [12], but the ANI analysis indicated that our strain was closest to P. aeruginosa PSE305, rather than P. aeruginosa BUP2. Based on 16S rRNA comparative sequence analysis P. aeruginosa O12 PA7 showed 99% sequence similarity to P. aeruginosa strain S3 but had <95% ANI value. For Geobacillus sp. EC-3, Figure 1 shows a cluster in the upper-right corner containing closely related strains with more than 97% sequence similarity based on both 16S rRNA sequences and ANI analysis. The closest completed Geobacillus in the public databases was Geobacillus thermoleovorans strain CCB_US3_UF5. The proximity of Geobacillus sp. EC-3 to G. thermoleovorans (99.4% ANI and 99.8% 16S rRNA) suggests that EC-3 is a thermoleovorans. Sphingobacterium sp. strain S2 and P. aeruginosa strain S3 were isolated from the compost samples, while the most closely related strains, S. thalpophilum DSM11723 and NCTC11429 and P. aeruginosa PSE305, were isolated from human clinical samples. While Pseudomonas aeruginosa is a well-documented pathogen, even Spingobacterium has been identified as a potential opportunistic pathogen [38,39]. Geobacillus EC-3 was also isolated from compost while the closest strain in the database, G. thermoleovorans strain CCB_US3_UF5, was isolated from Ulu hot springs in Malaysia [40].
2.3. MAUVE and MeDuSa Alignments
MAUVE and MeDuSa were used to find sequence homologies of the two isolates with the strains identified as closest in the database by ANI calculations. The alignments shown in Figure 2 were derived by first aligning the SPADE-assembled contigs against reference strains, (S. thalpophilum NCTC11429, P. aeruginosa PSE305 and G. thermoleovorans strain CCB_US3_UF5) with MeDuSa [41], followed by alignment in MAUVE [42] using the MeDuSa ordered scaffolds of the isolates.

Figure 2.
Genomic alignments generated in MeDuSa and MAUVE of Sphingobacterium sp. S2 with Sphingbacterium thalpophilum NCTC11429 (Panel A), P. aeruginosa S3 with P. aeruginosa PSE305 (Panel B) and Geobacillus EC-3 with G. thermoleovorans CCB_US3_UFS (Panel C). Similarly colored blocks indicate assembled regions of two genomes that are highly homologous. Note that the level of homology can vary across the blocks. These blocks are separated by white blocks representing regions with low or no homology. Genome size is indicated as number of base pairs.
The MeDuSa alignment of contigs from the assembled Sphingobacterium S2 against S. thalpophilum NCTC11429 reduced the contig count from 87 to 26. P. aeruginosa PSE305 was used as a reference strain in MeDuSa which reduced the contig count from 63 to 9. MeDuSa reduced the 111 contigs of the Spades assembled Geobacillus sp. EC-3 to 4. Overall, the genomes of these isolates had good synteny with their closest relatives as measured by genome assembly and alignments in MeDuSa and MAUVE. Of particular interest was the assembly and alignments of Geobacillus sp. EC-3. EC-3 appears closest to G. thermoleovorans strain CCB_US3_UF5. However, there are several recent reports of Geobacillus genome projects and at least three isolates, strains ARTRW1, FJAT2391 and KCTC 3570, have a very similar genome organization, as evaluated in MAUVE, in comparison with EC-3 and CCB_US3_UF5, although they were isolated from fairly diverse sources. Strain ARTRW1 was isolated from hot springs in Turkey [43], strain FJAT2391 came from soil in China (Genbank Accession # PRJNA340206), and strain KCTC 3570 (Genbank Accession # PRJNA310809) came from soil near a hot effluent stream in Pennsylvania, USA. The alignment of the genomes of these strains is presented in Figure S1 (Supplemental Information). This conservation of genomic architecture suggests strong selective pressure.
2.4. Genetic Systems and Metabolism
The distribution of identified genes to cell subsystems is shown in Figure 3. The distribution patterns are roughly the same between strains with the exception that a greater percentage of the genomes of S3 and EC-3 are devoted to “Cell Processing” compared to S2, whereas S2 has a greater genetic commitment to “Membrane Transport”.

Figure 3.
Distribution of identified genes to subsystems.
Sphingobacterium spp. are Gram-negative rods, aerobic, exhibiting sliding motility and form yellow-pigmented colonies. Sphingobacterium have been isolated from diverse environments such as soil, water, compost, deserts, blood and urine samples from human patients. A distinctive feature of Sphingobacterium is the presence of sphingolipids at relatively high concentrations in their cell wall [15,44]. In Sphingobacteria S2, 1589 ORFs were assigned metabolic functions. Genes (269) were identified as participating in amino acid metabolism while 329 genes were dedicated to carbohydrate metabolism. Energy metabolism and lipid metabolism had 183 and 149 genes identified, respectively, while 76 genes were assigned to xenobiotic biodegradation and metabolism (Figure 4).

Figure 4.
Distribution of genes to pathway categories in Sphingobacterium sp. S2, P. aeruginosa S3 and Geobacillus sp. EC-3. These data were derived from the pathways evaluation program as implemented in PATRIC [34].
P. aeruginosa is a Gram-negative bacterium able to grow, aerobically, on a wide range of substrates and anaerobically, when nitrate is available as terminal electron acceptor. It is capable of thriving in highly diverse and unusual ecological niches with low availability of nutrients. Its metabolic versatility allows it to use a variety of diverse carbon sources, including certain disinfectants [45]. Moreover, it can synthesize a number of antimicrobial compounds [24,25]. Using genome annotation through PATRIC, 1084 ORFs in P. aeruginosa were assigned to metabolic pathways. Among the major pathways, 532 genes were assigned to amino acid metabolism and 241, 401, 194 and 219 genes were assigned for energy metabolism, carbohydrate metabolism, metabolism of cofactors and vitamins and lipid metabolism, respectively. Genes, 266, were identified as involved with xenobiotics while 136 and 174 genes were linked to nucleotide metabolism and biosynthesis of secondary metabolites.
Geobacillus spp. are Gram-positive spore forming rods that are found widely distributed. They are capable thermophiles and have been routinely isolated from hot springs and compost [40,46]. In strain EC-3, 1662 ORFs were assigned to metabolic functions with 357 devoted to amino acid metabolism, 334 assigned to carbohydrate metabolism, and 169 and 129 assigned to energy and lipid metabolism, respectively. While there are no glaring differences in gene distributions between the three genomes, it is, however, curious to note that Sphingobacterium S2 devotes twice as much genetic power to glycan biosynthesis and metabolism. This indicates that Sphingobacterium can express various enzymes involved in the synthesis and degradation of glycans that may be of value for various biotechnological applications.
2.5. Xenobiotic Biodegradation Metabolism
The role of P. aeruginosa in the degradation of large complex molecules including PAHs, xenobiotic compounds, oil, dyes and plastics is well documented [26,27,28,29,30]. Sphingobacterium spp. have also been reported to have a potential role in the biodegradation of different pollutants, including mixed plastic waste, PAHs, biodegradation of oil and pesticides [19,20,21]. Geobacillus spp. have been identified that can reduce azo dyes and degrade alpha-naphthol and nylon [47,48,49]. Table 2 describes the major pathways and the number of genes related to the biodegradation of different xenobiotic compounds in strains S2, S3 and EC-3. According to sequence analysis, numerous pathways for the biodegradation of xenobiotic compounds were found in the three strains with approximately three times as many in P. aeruginosa S3 as compared to Sphingobacterium sp. S2 and Geobacillus sp. EC-3. This may be due, in part, to the fact that P. aeruginosa is more comprehensively studied compared to Sphingobacterium and Geobacillus. Among all the degradation pathways found in the three strains, the degradation of benzoate in P. aeruginosa had the greatest number of annotated genes. Benzoate, an aromatic compound, has been widely used as a model for the study of the bacterial catabolism of aromatic compounds [50]. Genetic evidence for the ability to degrade 1,4-dichlorobenzene, 2,4-dichlorobenzene and benzoate were detected in all three genomes. Many genes related to the degradation pathway of one of the most important classes of pollutants, PAHs, such as naphthalene, anthracene, 1- and 2-methylnaphthalene, were found in all three strains. Among the halogenated organic compounds, 29 genes were dedicated to tetrachloroethene degradation in P. aeruginosa S3 while only 6 and 8 were found in Sphingobacterium sp. S2 and Geobacillus sp. EC-3, respectively. For aromatic compounds and chlorinated aromatic compounds, pathways for the biodegradation of toluene, trinitrotoluene, xylene degradation, 1,4-Dichlorobenzene degradation and 2,4-Dichlorobenzoate were also found. Genes for the biodegradation of Bisphenol A, one of the most abundantly produced chemicals released into the environment and a serious health concern and environmental pollutant, was also found in P. aeruginosa S3 [51] and Sphingobacterium sp. S2.
Table 2.
Pathways and number of genes involved in aromatic compound metabolism in draft genome sequences of P. aeruginosa S3, Sphingobacterium sp. S2 and Geobacillus sp. EC-3. Unique EC count refers to the number of unique enzymes identified.
2.6. Lactate Metabolism
Lactate utilization as the sole carbon source is a property of many bacteria where a key step of the process is the oxidation of lactate [52,53,54,55,56]. Lactate dehydrogenases found in microbes are of two types, NAD-dependent lactate dehydrogenases (nLDHs) and NAD-independent lactate dehydrogenases (iLDHs), also called respiratory lactate. The latter is usually considered to be the enzyme mainly responsible for the metabolism of lactate as a carbon source [53]. The lactate utilization system is comprised of three main membrane bound proteins: NAD-independent lactate dehydrogenase (l-iLDH), NAD-independent D-lactate dehydrogenase (d-iLDH), and a lactate permease (LldP). Lactate permease is responsible for the uptake of lactate into the cells and lactate dehydrogenases carry out the oxidation of lactate to pyruvate [55,57]. Lactate utilization has been observed in some pathogens [55], stimulating their growth during infections while enhancing the synthesis of pathogenic determinants and increasing resistance against various bactericidal mechanisms [55]. The utilization of lactate by different Pseudomonas strains is well documented [56,58,59,60]. In the sequence analysis of our P. aeruginosa strain S3, a complete cascade of genes was found encoding the machinery for lactate utilization including a lactate permease, both l and d-lactate dehydrogenases and a lactate-responsive regulator LldR (Table 3). This strain was isolated and characterized for its potential to degrade poly(lactic acid) and its ability to utilize lactate as a sole carbon source was established in our previous study [12]. Presence of the lactate utilization machinery found through genome sequencing is consistent with previous observations that both l-iLDH and d-iLDH are present in the single operon and are induced coordinately in Pseudomonas strains and expression of both enzymes is controlled by the presence of an enantiomer of lactate [59].
Table 3.
Genes for lactate metabolism in P. aeruginosa S3, Sphingobacterium sp. S2 and Geobacillus sp. EC-3.
In the genome analysis of our isolate Sphingobacterium sp. S2, we found an incomplete set of genes for lactate metabolism. Both l-lactate dehydrogenase and d-lactate dehydrogenase were present, but no lactate permease was detected (Table 3). Absence of lactate permease is consistent with our previous findings that showed that Sphingobacterium sp. S2 did not utilize lactic acid as the sole source of carbon. Inability to grow on lactic acid had been previously reported in literature for different strains of Sphingobacterium [18,61]. The fact that these strains were isolated, based on their ability to degrade PLA, suggests that a degradation product other than lactate is involved.
Geobacillus sp. EC-3 was capable of growing on lactate as the sole carbon source and we detected lactate dehydrogenase and lactate permease in strain EC-3. In addition, we identified lactate utilization proteins LutA, LutB and LutC, along with the lactate responsive regulator LutR. These lactate utilization proteins are discussed below, relative to biofilm formation.
2.7. Genetic Determinants for Biofilm Formation and Regulation
In contrast to the planktonic lifestyle, cells within a biofilm matrix are in close proximity where secreted enzymes provide optimal returns for the population, especially when targeting the substratum on which the biofilm forms [62]. The phenomenon of microbial biofilm formation is also related to other survival strategies like metal and antimicrobial resistance, tolerance and bioremediation [63,64]. The application of biofilm mediated bioremediation has been found superior to other bioremediation strategies and is being applied in bioremediation of different environmental pollutants [65,66,67,68,69]. Microorganisms that develop a biofilm through attachment and formation of an extracellular protective matrix are physiologically more resilient to environmental changes, making them a logical choice for the remediation of different pollutants. These microbes use different strategies like biosorption, bioaccumulation and biomineralization to slowly degrade compounds [70]. All three of our isolates were capable of forming biofilm on PLA. This was tested because the formation of biofilm on solid polymers that can be degraded for carbon and/or energy is a logical strategy for microbes. Excreted enzymes capable of degradation can be sequestered within the biofilm matrix in close proximity to the polymer and not lost to solution. Therefore, we sought to identify genes involved in biofilm formation in strains S2, S3 and EC-3. P. aeruginosa is a remarkably adept opportunist with striking ability to develop biofilm [22,71,72]. In our previous study, we also observed biofilm formation by our isolate P. aeruginosa strain S3 on the surface of PLA during the process of biodegradation [12]. The phenomenon of biofilm formation on the surface of PLA had previously been reported by other authors as well [37,73,74]. In the genetic analysis of our isolate, we found factors involved in the development of the matrix of P. aeruginosa biofilm and its regulation (Table 4). The genes for three types of exopolysaccharides (EPS), previously reported as involved in the construction of the biofilm matrix of P. aeruginosa, (Pel, Psl and alginate [72,75]) were found in our isolate. These EPS molecules form the protective matrix [76]. Psl is the primary factor in charge of the initiation and maintenance of the biofilm structure by providing cell to cell and cell to surface interactions [77,78,79,80]. It also works as a signaling molecule to the successive events involved in the formation of biofilms and also acts as a defensive layer for different immune and antibiotic attacks [81]. Pel polysaccharide is a glucose-rich extracellular matrix and is involved in the formation of biofilms that are attached to the solid surfaces. It is considered to be less important, compared to Psl [72,80,82,83]. In P. aeruginosa from clinical isolates of CF patients, alginate is produced [23]. Besides its role in maintenance and protection of biofilm structure, it is essential for water and nutrient preservation [84].
Table 4.
Genetic elements involved in biofilm formation and regulation detected in P. aeruginosa S3, Sphingobacterium S2 and Geobacillus EC-3.
Biofilm formation is a multicellular process stimulated by environmental signals and controlled by regulatory networks. During the biofilm formation, cells undergo many phenotypic shifts that are regulated by a large array of genes [85]. In the genome of P. aeruginosa strain S3, several regulatory factors were identified. One of these regulatory factors was the signaling molecule bis-(3′-5′)-cyclic dimeric guanosine monophosphate (c-di-GMP), which is considered to be one of the most significant molecular determinants in biofilm regulation [86]. A c-di-GMP molecule controls the interchange between the planktonic and biofilm-associated lifestyle of bacteria by stimulating the biosynthesis of adhesins and exopolysaccharides during the formation of biofilm [87]. The bacterial cell to cell communication system known as quorum sensing (QS) is involved in the maintenance of many biological processes like biofilm formation, bioluminescence, antibiotic production, virulence factor expression, competence for DNA uptake, and sporulation [88,89]. LasR/LasI, RhlR/RhlI and PQS are the three QS signaling systems employed by P. aeruginosa to control biofilm formation [90,91]. These three QS signaling system were found to be the part of the genome for our isolate.
There is little information in the literature regarding the genetic elements involved in biofilm formation in Sphingobacterium, Genome analysis of our isolate Sphingobacterium sp. S2 showed the presence of some genetic elements that are reported to be involved in biofilm formation in the literature, such as genes for Stage 0 sporulation protein YaaT. This protein is reported to be involved in the sporulation process and biofilm development [92,93]. A response regulator, LuxR involved in the sensory mechanism, and gliding motility protein precursors GldC, GldJ, GldN were also detected in the genome of our strain. These proteins are known for their role in biofilm development [94]. A probable outer membrane protein precursor of OmpA family, considered to be putative adhesins and adhesion-related proteins, were also reported in Sphingobacterium sp. S2 [94].
Geobacillus is very well reported in the literature for biofilm and spore formation [95,96]. Anthranilate phosphoribosyltransferase (EC 2.4.2.18) [97] and 5′-nucleotidase (EC 3.1.3.5), reported to have an impact on virulence and biofilm formation, were found in our Geobacillus sp. EC-3 [98]. Cyclic-di-AMP, well known for its role in the regulation of diverse cellular pathways, including potassium homeostasis, biofilm formation, and cell wall biosynthesis, was also detected in our strain [99].
2.8. Enzymes
The biodegradation of polymers is carried out by two types of enzymes, extracellular enzymes that degrade long chain polymers into short oligomers or subunits that are subsequently carried inside the cell, and intracellular enzymes that further degrade the small, transported units [100,101]. The degradation of synthetic polymers in the environment can be a slow process [102,103]. PLA is a synthetic linear aliphatic polyester of lactic acid monomers joined together by ester linkages [3]. The presence of ester bonds in its backbone, make the polymer sensitive to hydrolysis, both chemically as well as enzymatically [101]. The biodegradation of polyesters is mostly carried out by esterolytic enzymes such as esterases, lipases, or proteases. In the literature, microbial degradation of PLA is reported to be conducted by proteases, lipases, esterases and a few cutinases [4]. Sphingobacterium spp., P. aeruginosa, and Geobacillus spp. had been documented before to have a role in the degradation of different environmental pollutants, such as mixed plastic waste, PAHs, oil, and dyes and pesticides. P. aeruginosa has also been reported to have the potential to degrade PLA nanocomposites [19,26,30,104,105]. In our previous studies we have reported the expression of PLA hydrolyzing lipase and esterase from our isolates P. aeruginosa strain S3 and Sphingobacterium sp. strain S2 [12,97,98]. Genome annotation of S2, S3 and EC-3 reveal the presence of numerous hydrolytic enzymes (Table 5). In the genome of P. aeruginosa S3, 75 different types of proteases, 50 esterases and 25 different types of lipases were detected (i.e., the sum of “Common to Reference Genome” and those “Unique to Strain”).
Table 5.
Different type of hydrolytic enzymes found in the draft genome of P. aeruginosa S3, Sphingobacterium sp. S2, and Geobacillus sp. EC-3.
Similarly, in the genome of Sphingobacterium sp. S2, 36 proteases, 30 esterases and 18 lipases were identified. Of the three genomes, Geobacillus sp. EC-3, the smallest of the three genomes, had the most putative hydrolytic enzymes; EC-3 had 20 lipases, 127 proteases and 72 lipases.
3. Discussion
The present study reports the whole genome sequence analysis of three bacterial strains isolated from compost, P. aeruginosa S3, Sphinogobacterium sp. S2, and Geobacillus sp. EC-3, each possessing the metabolic skill of utilizing a polymeric solid, poly(lactic acid), as the sole source of carbon at ~30 °C or 58 °C. The initial steps in the degradation of solids by bacteria are extracellular, until sufficiently small molecules that can be transported across the outer cell envelope are generated through the activities of secreted enzymes. An obvious microbial strategy for optimizing the effectiveness of secreted enzymes is the migration to a position as proximal to the solid as possible, and nothing is closer than a biofilm. All three of our isolates were able to form biofilm on PLA and all three have putative biofilm markers in their genomes. Genomic analyses of P. aeruginosa strains have a robust literature because of its medical importance and as a consequence, there are many genetic elements that can be identified in P. aeruginosa S3 that are implicated in biofilm formation. These include structural elements (outer membrane matrix proteins), transport mechanisms, and regulatory elements like cyclic di-GMP and three quorum sensing systems. In addition to genetic determinants for biofilm formation, many virulence-associated genes were identified in strain S3.
Regarding Sphingobacterium, a recent description of a Sphingobacterium sp. isolate [106] capable of inhibiting the fungal activity of Fusarium posits that siderophores and chitinases may account for the antifungal activities and, in the genomic analysis of this strain (SJ-25), they identify potential candidate genes. Strain S2 also contains at least four siderophores as well as TonB and an abundance of TonB-dependent receptors. We also detected a chitin binding protein but no indication of excreted chitinases. Curiously, a biofilm related protein, identified as homologous to Stage 0 sporulation protein YaaT, was also detected. Sphingobacterium is a Gram-negative nonsporulating rod in the phylum Bacteroidetes.
Regarding Geobacillus, similar to G. thermodenitrificans T12 [107], there is a respiratory nitrate reductase (alpha, beta, gamma, and delta subunits) as well as CRISPR elements. In addition, UDP-glucose 4-epimerase and mannose-1-phosphate guanylyltransferase, identified in Geobacillus sp. WSUCF1 [108], were also detected in strain EC-3.
Draft genomes of strains S2, S3 and EC-3 were studied to gain an insight into the genetic elements that are involved in the degradation of PLA. The catabolic genes responsible for biodegradation of different xenobiotic compounds, genes responsible for formation and regulation of biofilm, genes for transport and utilization of lactate and several enzymes predicted to be involved in the degradation of many organic pollutants were identified. Of interest was the apparent lack of lactate permease in Sphingobacterium sp. S2, suggesting that an alternative mode of attack on PLA leads to a molecule other than lactate, that is transportable, or that lactate is further modified extracellularly before transport. Finally, Chai, et al. [109] have detected catabolic lactate genes, lutA, lutB and lutC, in Bacillus subtilis and note that the upregulation of these genes is required for growth on lactate as the sole carbon source. Moreover, these genes are under dual regulation, permitting expression in liquid culture as well as within a biofilm. This gene cluster was also found in Geobacillus sp. EC-3. Common features of these three phylogenetically disparate isolates were an abundance of genetic determinants for hydrolytic enzymes and biofilm formation. These three strains were isolated through serial selection on a single carbon source, PLA. The genomic analyses provide insights into the potential array of genes that may be required for the efficient degradation of PLA.
4. Materials and Methods
4.1. Strains and Media
Strains were grown routinely on R2A agar (Difco) or R2broth (Yeast extract 0.5 g/L, Proteose Peptone No. 3 0.5 g/L, Casamino Acids 0.5 g/L, Dextrose 0.5 g/L, Soluble Starch 0.5 g/L, Sodium Pyruvate 0.3 g/L, Dipotassium Phosphate 0.3 g/L, Magnesium Sulfate 0.05 g/L). Strains were stored at −80 °C in 20% glycerol and streaked onto R2A to initiate regrowth. All chemicals and media were purchased from Sigma-Aldrich, Inc., Saint Louis, MO, USA.
4.2. DNA Extraction
Three of our previously isolated PLA degrading bacterial strains, Sphingobacterium sp. strain S2, P. aeruginosa strain S3 and Geobacillus EC-3 (GenBank accession numbers KY432687, KY432688, and MH183212, respectively) were selected for genome sequencing [12]. Strains S2 and S3 were grown separately in 100 mL of LB in a 250 mL Erlenmeyer flask for 16 h in a shaking incubator at 30 °C and 70 rpm. Geobacillus EC-3 was grown in M9 minimal media shaking at 58 °C. Cells were pelleted (10,000 RPM in a Sorvall SS34 rotor, Beckman Coulter Life Sciences, Indianapolis, Indiana USA) and genomic DNA was isolated using MO BIO PowerSoil DNA isolation kit (MO BIO laboratories, Inc., Loker Ave West, Carlsbad, CA, USA). NanoDrop ND-1000 spectrophotometer and ND-1000 V3.1.8 software (Thermo Fisher Scientific Inc., Wilmington, DE, USA) were used to determine DNA concentrations of purified samples and sent for whole genome sequencing at Michigan State University Genomics Facility (MSU-RTSF), East Lansing, MI, USA.
4.3. Genome Sequencing
Libraries for sequencing were prepared using the Illumina TruSeq Nano DNA Library Preparation Kit (Illumina, Inc., San Diego CA, USA) on a Perkin Elmer Sciclone NGS robot (Perkin Elmer, Boston, MA, USA). Before sequencing, the qualities of the libraries were tested and quantification was performed using a combination of Qubit dsDNA HS (Thermo Fisher Scientific, Waltham, MA, USA), Caliper LabChip GX HS DNA (Perkin Elmer, Boston, MA, USA) and Kapa Illumina Library Quantification qPCR assays (Roche Sequencing and Life Science, Wilmington, MA, USA). Libraries were pooled in equimolar quantities and loaded on an Illumina MiSeq standard v2 flow cell with a 2 × 250 bp paired end format and using a v2 500 cycle reagent cartridge. Illumina Real Time Analysis (v1.18.64) was used for base calling and the output was converted to FastQ format with Illumina Bc12fastq (v1.8.4) after demultiplexing (Illumina, Inc., San Diego CA, USA). A total of 6,304,420 reads were obtained for strain S2, 5,800,229 reads for strain S3 and 5,730,761 reads for strain EC-3. The genomic reads have been deposited at the Sequence Read Archives at NCBI with Bioproject IDs of SRP149807 and PRJNA721072.
4.4. Sequence Assembly, Annotation, and Analysis
Assembly of the genomes was performed using the full Spades assembly function within PATRIC (PATRIC 3.4.9) [34] as implemented in the miseq assembly option. This assembly option incorporates BayesHammer algorithms followed by Spades, (Spades version 3.8.). Rast tool kit as implemented in PATRIC [110] was used for the annotation of contigs. The assembled contig file generated from this assembly was used as seed for the Comprehensive Genome Analysis function in PATRIC. The genomes were interrogated for the distribution of specific protein families (PGFams) using the protein family sorter tool on PATRIC. The genomes were compared to their closest reference genomes available on PATRIC to examine the strain-specific unique proteins as well as proteins common to the closest relative using the filter option in protein family sorter tool on PATRIC.
4.5. Average Nucleotide Identity (ANI) for Species Delineation
Isolates were further analyzed using a whole genome based average nucleotide identity (ANI) method to delineate the genomes to their correctly matching relatives. ANI values were calculated using MiSI (microbial species identifier) tool that is publicly available at the Integrated Microbial Genomes (IMG) database [111]. The algorithm used in the original method proposed by Konstantinidis and Tiedje was modified and used to determine ANI between two genomes [112]. The average of the nucleotide identity of the orthologous genes of the pair of genomes was calculated and identified as bidirectional best hits (BBHs) using a similarity search tool accessed in January, 2019, NSimScan (http://www.scidm.org/, accessed on 7 June 2021). The ANI of one genome to another genome is defined as the sum of the %-identity times the alignment length for all best bidirectional hits, divided by the sum of the lengths of the BBH genes. This pairwise calculation is performed in both directions. The strains used for comparison were complete genomes obtained from NCBI and are as follows. For the Sphingobacterium comparisons; S. thalpophilum DSM 11723 (Draft genome of 32 contigs), S. sp. G1-14, S. sp. B29, S. multivorum DSM 11691, S. lactis DSM 22361, S. wenxiniae DSM 22789, S. mizutaii DSM 11724 and S. sp. 21. For the Pseudomonas aeruginosa comparisons; P. aeruginosa PSE302, P. aeruginosa PA96, P. aeruginosa PA01H20, P. aeruginosa DSM 50071 P. aeruginosa PAO1, P. aeruginosa PAK, P. aeruginosa O12 PA7, P. aeruginosa PA_D25, P. aeruginosa PA_D1, P. aeruginosa KU and P. aeruginosa T52373. Regarding Geobacillus sp. comparison; EC-3 Geobacillus thermoleovorans CCB_US3_UF5, Geobacillus thermoleovorans strain SGAir0734, Geobacillus thermocatenulatus strain BGSC 93A1, Geobacillus sp. A8 and Geobacillus kaustophilus NBRC 102445 were used.
4.6. Comparative Alignments Using MeDuSa and MAUVE
For the comparative alignment of the genomes with their reference genomes and their visualization, MeDuSa [41] was used to reduce the number of contigs through comparison with the gene order of the closest strain. This was followed by alignment with MAUVE [42] to a reference strain to provide an estimate of alignment similarity. The reference strain selected was the closest finished genome in the public database. Alignments in MAUVE were also performed with contigs directly from the Spades assembly. P. aeruginosa PSE305 was used as a reference for strain S3 (ANI of 98.22) while Sphingobacterium thalpophilum NCTC11429 (ANI of 98.46) was selected as a reference for Sphingobacterium sp. S2. According to the “Similar Genome Finder” function implemented in “PATRIC”, G. thermoleovorans strains B23 and CCB_US3_UF5 from public databases were closest to EC-3. This function uses Mash/MinHash [113] to determine genome similarities. Strain CCB_US3_UF5 was used as the reference strain in MAUVE as this genome has been finished (1 chromosome) and has an ANI similarity of 99.9%.
Supplementary Materials
The following is available online at https://www.mdpi.com/article/10.3390/ijms22147385/s1, Figure S1: MAUVE alignment of five Geobacillus genomes.
Author Contributions
Conceptualization, S.M.S., E.C.-A., T.L.M. and R.A.; methodology, S.M.S., E.C.-A., T.L.M. and R.A.; software, S.M.S., E.C.-A., T.L.M. and R.A.; validation, S.M.S., E.C.-A., T.L.M. and R.A.; formal analysis, S.M.S., E.C.-A., T.L.M. and R.A.; resources, T.L.M. and R.A.; data curation, T.L.M.; writing—original draft preparation, S.M.S. and T.L.M.; writing—review and editing, S.M.S., E.C.-A., T.L.M., R.A. and A.A.S.; supervision, T.L.M. and R.A.; project administration, T.L.M. and R.A.; funding acquisition, S.M.S., T.L.M. and R.A. All authors have read and agreed to the published version of the manuscript.
Funding
This project was partially supported by the USDA National Institute of Food and Agriculture and Michigan State University AgBioResearch, Hatch project number MICL02665 and internal funding from the Department of Microbiology and Molecular Genetics.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Sequences for the three genomes have been deposited at the Sequence Read Archive at NCBI with the accession numbers SRP149807 and PRJNA721072.
Acknowledgments
RA thanks the support of the SoP, MSU. TLM and RA thank the support of the MSU Genomics Facility and the Ribosomal Database Project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Castro-Aguirre, E.; Iniguez-Franco, F.; Samsudin, H.; Fang, X.; Auras, R. Poly (lactic acid)—Mass production, processing, industrial applications, and end of life. Adv. Drug Deliv. Rev. 2016, 107, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Dorgan, J.R.; Lehermeier, H.J.; Palade, L.I.; Cicero, J. Macromolecular symposia. In Polylactides: Properties and Prospects of an Environmentally Benign Plastic from Renewable Resources; Wiley Online Library: Hackensack, NJ, USA, 2001; pp. 55–66. [Google Scholar]
- Auras, R.; Harte, B.; Selke, S. An overview of polylactides as packaging materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Ren, Y.; Wang, X. New advances in the biodegradation of Poly (lactic) acid. Int. Biodeterior. Biodegrad. 2017, 117, 215–223. [Google Scholar] [CrossRef]
- Copinet, A.; Legin-Copinet, E.; Erre, D. Compostability of co-extruded starch/poly (lactic acid) polymeric material degradation in an activated inert solid medium. Materials 2009, 2, 749–764. [Google Scholar] [CrossRef]
- Jarerat, A.; Pranamuda, H.; Tokiwa, Y. Poly (l-lactide)-degrading activity in various actinomycetes. Macromol. Biosci. 2002, 2, 420–428. [Google Scholar] [CrossRef]
- Jarerat, A.; Tokiwa, Y. Degradation of Poly (l-lactide) by a Fungus. Macromol. Biosci. 2001, 1, 136–140. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Robson, G.D. The influence of biotic and abiotic factors on the rate of degradation of poly (lactic) acid (PLA) coupons buried in compost and soil. Polym. Degrad. Stab. 2013, 98, 2063–2071. [Google Scholar] [CrossRef]
- Masaki, K.; Kamini, N.R.; Ikeda, H.; Iefuji, H. Cutinase-like enzyme from the yeast Cryptococcus sp. strain S-2 hydrolyzes polylactic acid and other biodegradable plastics. Appl. Environ. Microbiol. 2005, 71, 7548–7550. [Google Scholar] [CrossRef]
- Saadi, Z.; Rasmont, A.; Cesar, G.; Bewa, H.; Benguigui, L. Fungal degradation of poly (l-lactide) in soil and in compost. J. Polym. Environ. 2012, 20, 273–282. [Google Scholar] [CrossRef]
- Watanabe, M.; Kawai, F.; Tsuboi, S.; Nakatsu, S.; Ohara, H. Study on enzymatic hydrolysis of polylactic acid by endogenous depolymerization model. Macromol. Theory Simul. 2007, 16, 619–626. [Google Scholar] [CrossRef]
- Satti, S.M.; Shah, A.A.; Auras, R.; Marsh, T.L. Isolation and characterization of bacteria capable of degrading poly (lactic acid) at ambient temperature. Polym. Degrad. Stab. 2017, 144, 392–400. [Google Scholar] [CrossRef]
- Satti, S.M.; Shah, A.A.; Marsh, T.L.; Auras, R. Biodegradation of Poly(lactic acid) in Soil Microcosms at Ambient Temperature: Evaluation of Natural Attenuation, Bio-augmentation and Bio-stimulation. J. Polym. Environ. 2018, 26, 3848–3857. [Google Scholar] [CrossRef]
- Sreenivas, A.; Reddy, G.S.; Shivaji, S. Draft genome sequence of a psychrophilic bacterium, Sphingomonas antarcticum, isolated from the soils of Schirmacher oasis, Antarctica. Genome Announc. 2014, 2, e00696-14. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, E.; Kaneko, T.; Yano, I.; Moss, C.W.; Miyoshi, N. Sphingobacterium gen. nov., Sphingobacterium spiritivorum comb. nov., Sphingobacterium multivorum comb. nov., Sphingobacterium mizutae sp. nov., and Flavobacterium indologenes sp. nov.: Glucose-nonfermenting gram-negative rods in CDC groups IIK-2 and IIb. Int. J. Syst. Evol. Microbiol. 1983, 33, 580–598. [Google Scholar] [CrossRef]
- He, X.; Xiao, T.; Kuang, H.; Lan, X.; Tudahong, M.; Osman, G.; Fang, C.; Rahman, E. Sphingobacterium shayense sp. nov., isolated from forest soil. Int. J. Syst. Evol. Microbiol. 2010, 60, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, H.; Zhang, C.-X.; Yang, S.-Y.; Liu, X.-H.; Zhang, K.-Y.; Lai, R. Sphingobacterium siyangense sp. nov., isolated from farm soil. Int. J. Syst. Evol. Microbiol. 2008, 58, 1458–1462. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahmed, I.; Ehsan, M.; Sin, Y.; Paek, J.; Khalid, N.; Hayat, R.; Chang, Y.H. Sphingobacterium pakistanensis sp. nov., a novel plant growth promoting rhizobacteria isolated from rhizosphere of Vigna mungo. Antonie Van Leeuwenhoek 2014, 105, 325–333. [Google Scholar] [CrossRef]
- Abraham, J.; Silambarasan, S. Biodegradation of chlorpyrifos and its hydrolyzing metabolite 3,5,6-trichloro-2-pyridinol by Sphingobacterium sp. JAS3. Process. Biochem. 2013, 48, 1559–1564. [Google Scholar] [CrossRef]
- Haiyan, R.; Shulan, J.; ud din Ahmad, N.; Dao, W.; Chengwu, C. Degradation characteristics and metabolic pathway of 17α-ethynylestradiol by Sphingobacterium sp. JCR5. Chemosphere 2007, 66, 340–346. [Google Scholar] [CrossRef]
- Noparat, P.; Maneerat, S.; Saimmai, A. Application of biosurfactant from Sphingobacterium spiritivorum AS43 in the biodegradation of used lubricating oil. Appl. Biochem. Biotechnol. 2014, 172, 3949–3963. [Google Scholar] [CrossRef]
- Campa, M.; Bendinelli, M.; Friedman, H. Pseudomonas aeruginosa as an Opportunistic Pathogen; Plenum Press: New York, NY, USA, 1993. [Google Scholar]
- Govan, J.R.; Deretic, V. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 1996, 60, 539–574. [Google Scholar] [CrossRef] [PubMed]
- Bodey, G.P.; Bolivar, R.; Fainstein, V.; Jadeja, L. Infections caused by Pseudomonas aeruginosa. Rev. Infect. Dis. 1983, 5, 279–313. [Google Scholar] [CrossRef]
- Kung, V.L.; Ozer, E.A.; Hauser, A.R. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2010, 74, 621–641. [Google Scholar] [CrossRef]
- Das, D.; Baruah, R.; Roy, A.S.; Singh, A.K.; Boruah, H.P.D.; Kalita, J.; Bora, T.C. Complete genome sequence analysis of Pseudomonas aeruginosa N002 reveals its genetic adaptation for crude oil degradation. Genomics 2015, 105, 182–190. [Google Scholar] [CrossRef]
- Das, N.; Chandran, P. Microbial degradation of petroleum hydrocarbon contaminants: An overview. Biotechnol. Res. Int. 2011, 2011, 13. [Google Scholar] [CrossRef]
- Howard, G.T. Biodegradation of polyurethane: A review. Int. Biodeterior. Biodegrad. 2002, 49, 245–252. [Google Scholar] [CrossRef]
- Kahraman, H.; Geckil, H. Degradation of benzene, toluene and xylene by Pseudomonas aeruginosa engineered with the Vitreoscilla hemoglobin gene. Eng. Life Sci. 2005, 5, 363–368. [Google Scholar] [CrossRef]
- Nachiyar, C.V.; Rajkumar, G.S. Degradation of a tannery and textile dye, Navitan Fast Blue S5R by Pseudomonas aeruginosa. World J. Microbiol. Biotechnol. 2003, 19, 609–614. [Google Scholar] [CrossRef]
- Castro-Aguirre, E.; Auras, R.; Selke, S.; Rubino, M.; Marsh, T. Enhancing the biodegradation rate of poly (lactic acid) films and PLA bio-nanocomposites in simulated composting through bioaugmentation. Polym. Degrad. Stab. 2018, 154, 46–54. [Google Scholar] [CrossRef]
- Hussein, A.H.; Lisowska, B.K.; Leak, D.J. The genus Geobacillus and their biotechnological potential. Adv. Appl. Microbiol. 2015, 92, 1–48. [Google Scholar] [PubMed]
- Margaryan, A.; Shahinyan, G.; Hovhannisyan, P.; Panosyan, H.; Birkeland, N.-K.; Trchounian, A. Geobacillus and Anoxybacillus spp. from terrestrial geothermal springs worldwide: Diversity and biotechnological applications. In Extremophiles in Eurasian Ecosystems: Ecology, Diversity, and Applications; Springer: Berlin/Heidelberg, Germany, 2018; pp. 119–166. [Google Scholar]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Qin, Q.L.; Xie, B.B.; Yu, Y.; Shu, Y.L.; Rong, J.C.; Zhang, Y.J.; Zhao, D.L.; Chen, X.L.; Zhang, X.Y.; Chen, B. Comparative genomics of the marine bacterial genus Glaciecola reveals the high degree of genomic diversity and genomic characteristic for cold adaptation. Environ. Microbiol. 2014, 16, 1642–1653. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Castro, J.C.; Kyrpides, N.C.; Cole, J.R.; Tiedje, J.M.; Konstantinidis, K.T. How much do rRNA gene surveys underestimate extant bacterial diversity? Appl. Environ. Microbiol. 2018, 84, e00014-18. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Freney, J.; Hansen, W.; Ploton, C.; Meugnier, H.; Madier, S.; Bornstein, N.; Fleurette, J. Septicemia caused by Sphingobacterium multivorum. J. Clin. Microbiol. 1987, 25, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Lambiase, A.; Rossano, F.; Piazza, O.; Del Pezzo, M.; Catania, M.R.; Tufano, R. Typing of Pseudomonas aeruginosa isolated from patients with VAP in an intensive care unit. New Microbiol. 2009, 32, 277. [Google Scholar]
- Sakaff, M.K.L.M.; Rahman, A.Y.A.; Saito, J.A.; Hou, S.; Alam, M. Complete genome sequence of the thermophilic bacterium Geobacillus thermoleovorans CCB_US3_UF5. Am. Soc. Microbiol. 2012, 194. [Google Scholar] [CrossRef]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.-F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Oztug, M.; Cebeci, A.; Mumcu, H.; Akgoz, M.; Karaguler, N.G. Whole-Genome Sequence of Geobacillus thermoleovorans ARTRW1, Isolated from Armutlu Geothermal Spring, Turkey. Microbiol. Resour. Announc. 2020, 9, e00269-20. [Google Scholar] [CrossRef]
- Naka, T.; Fujiwara, N.; Yano, I.; Maeda, S.; Doe, M.; Minamino, M.; Ikeda, N.; Kato, Y.; Watabe, K.; Kumazawa, Y. Structural analysis of sphingophospholipids derived from Sphingobacterium spiritivorum, the type species of genus Sphingobacterium. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2003, 1635, 83–92. [Google Scholar] [CrossRef]
- Sagripanti, J.-L.; Bonifacino, A. Resistance of Pseudomonas aeruginosa to liquid disinfectants on contaminated surfaces before formation of biofilms. J. AOAC Int. 2000, 83, 1415–1422. [Google Scholar] [CrossRef]
- Banat, I.M.; Marchant, R. Geobacillus activities in soil and oil contamination remediation. In Endospore-Forming Soil Bacteria; Springer: Berlin/Heidelberg, Germany, 2011; pp. 259–270. [Google Scholar]
- Giedraityte, G.; Kalėdienė, L. Catechol 1, 2-dioxygenase from α-naphthol degrading thermophilic Geobacillus sp. strain: Purification and properties. Cent. Eur. J. Biol. 2009, 4, 68–73. [Google Scholar] [CrossRef]
- Leja, K.; Lewandowicz, G. Polymer biodegradation and biodegradable polymers-a review. Pol. J. Environ. Stud. 2010, 19, 255–266. [Google Scholar]
- Sugiura, W.; Yoda, T.; Matsuba, T.; Tanaka, Y.; Suzuki, Y. Expression and characterization of the genes encoding azoreductases from Bacillus subtilis and Geobacillus stearothermophilus. Biosci. Biotechnol. Biochem. 2006, 70, 1655–1665. [Google Scholar] [CrossRef][Green Version]
- Carmona, M.; Zamarro, M.T.; Blázquez, B.; Durante-Rodríguez, G.; Juárez, J.F.; Valderrama, J.A.; Barragán, M.J.; García, J.L.; Díaz, E. Anaerobic catabolism of aromatic compounds: A genetic and genomic view. Microbiol. Mol. Biol. Rev. 2009, 73, 71–133. [Google Scholar] [CrossRef]
- Eio, E.J.; Kawai, M.; Tsuchiya, K.; Yamamoto, S.; Toda, T. Biodegradation of bisphenol A by bacterial consortia. Int. Biodeterior. Biodegrad. 2014, 96, 166–173. [Google Scholar] [CrossRef]
- Erwin, A.; Gotschlich, E. Oxidation of D-lactate and L-lactate by Neisseria meningitidis: Purification and cloning of meningococcal D-lactate dehydrogenase. J. Bacteriol. 1993, 175, 6382–6391. [Google Scholar] [CrossRef]
- Garvie, E.I. Bacterial lactate dehydrogenases. Microbiol. Rev. 1980, 44, 106. [Google Scholar] [CrossRef] [PubMed]
- Gibello, A.; Collins, M.; Dominguez, L.; Fernandez-Garayzabal, J.; Richardson, P. Cloning and analysis of the L-lactate utilization genes from Streptococcus iniae. Appl. Environ. Microbiol. 1999, 65, 4346–4350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Gao, C.; Ma, C.; Xu, P. Microbial lactate utilization: Enzymes, pathogenesis, and regulation. Trends Microbiol. 2014, 22, 589–599. [Google Scholar] [CrossRef]
- Ma, C.; Gao, C.; Qiu, J.; Hao, J.; Liu, W.; Wang, A.; Zhang, Y.; Wang, M.; Xu, P. Membrane-bound l-and d-lactate dehydrogenase activities of a newly isolated Pseudomonas stutzeri strain. Appl. Microbiol. Biotechnol. 2007, 77, 91–98. [Google Scholar] [CrossRef]
- Núñez, M.A.F.; Kwon, O.; Wilson, T.H.; Aguilar, J.; Baldoma, L.; Lin, E.C. Transport of l-lactate, d-lactate, and glycolate by the LldP and GlcA membrane carriers of Escherichia coli. Biochem. Biophys. Res. Commun. 2002, 290, 824–829. [Google Scholar] [CrossRef]
- Brown, P.; Tata, R. Glycollate inhibition of growth of Pseudomonas aeruginosa on lactate medium. Microbiology 1987, 133, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Hu, C.; Zheng, Z.; Ma, C.; Jiang, T.; Dou, P.; Zhang, W.; Che, B.; Wang, Y.; Lv, M. Lactate utilization is regulated by the FadR-type regulator LldR in Pseudomonas aeruginosa. J. Bacteriol. 2012, 194, 2687–2692. [Google Scholar] [CrossRef]
- O’Brien, R. Metabolism of D-and L-lactate by Pseudomonas putida. Aust. J. Biol. Sci. 1977, 30, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, L.-L.; Xu, C.-K.; Xi, J.-Q.; Yang, F.-X.; Zhou, F.; Zhou, Y.; Mo, M.-H.; Li, W.-J. Sphingobacterium nematocida sp. nov., a nematicidal endophytic bacterium isolated from tobacco. Int. J. Syst. Evol. Microbiol. 2012, 62, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, K.K. What drives bacteria to produce a biofilm? FEMS Microbiol. Lett. 2004, 236, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.; Ceri, H.; Turner, R.J. Multimetal resistance and tolerance in microbial biofilms. Nat. Rev. Microbiol. 2007, 5, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Paul, A. Microbial extracellular polymeric substances: Central elements in heavy metal bioremediation. Indian J. Microbiol. 2008, 48, 49. [Google Scholar] [CrossRef] [PubMed]
- Dash, H.R.; Das, S. Bioremediation potential of mercury by Bacillus species isolated from marine environment and wastes of steel industry. Bioremediat. J. 2014, 18, 204–212. [Google Scholar] [CrossRef]
- Decho, A.W. Microbial biofilms in intertidal systems: An overview. Cont. Shelf Res. 2000, 20, 1257–1273. [Google Scholar] [CrossRef]
- Mangwani, N.; Shukla, S.; Kumari, S.; Rao, T.; Das, S. Characterization of Stenotrophomonas acidaminiphila NCW-702 biofilm for implication in the degradation of polycyclic aromatic hydrocarbons. J. Appl. Microbiol. 2014, 117, 1012–1024. [Google Scholar] [CrossRef]
- Shimada, K.; Itoh, Y.; Washio, K.; Morikawa, M. Efficacy of forming biofilms by naphthalene degrading Pseudomonas stutzeri T102 toward bioremediation technology and its molecular mechanisms. Chemosphere 2012, 87, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Paul, D.; Jain, R.K. Biofilms: Implications in bioremediation. Trends Microbiol. 2006, 14, 389–397. [Google Scholar] [CrossRef]
- Barkay, T.; Schaefer, J. Metal and radionuclide bioremediation: Issues, considerations and potentials. Curr. Opin. Microbiol. 2001, 4, 318–323. [Google Scholar] [CrossRef]
- Kyaw, B.M.; Champakalakshmi, R.; Sakharkar, M.K.; Lim, C.S.; Sakharkar, K.R. Biodegradation of low density polythene (LDPE) by Pseudomonas species. Indian J. Microbiol. 2012, 52, 411–419. [Google Scholar] [CrossRef]
- Ryder, C.; Byrd, M.; Wozniak, D.J. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr. Opin. Microbiol. 2007, 10, 644–648. [Google Scholar] [CrossRef]
- Richert, A.; Walczak, M.; Brzezinska, M.S. The influence of modified polyhexamethylene guanidine PHMG on the biodegradation of polylactide. Int. Biodeterior. Biodegrad. 2013, 84, 97–103. [Google Scholar] [CrossRef]
- Walczak, M.; Brzezinska, M.S.; Sionkowska, A.; Michalska, M.; Jankiewicz, U.; Deja-Sikora, E. Biofilm formation on the surface of polylactide during its biodegradation in different environments. Colloids Surf. B Biointerfaces 2015, 136, 340–345. [Google Scholar] [CrossRef]
- Ghafoor, A.; Hay, I.D.; Rehm, B.H. Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 2011, 77, 5238–5246. [Google Scholar] [CrossRef] [PubMed]
- Parsek, M.R.; Singh, P.K. Bacterial biofilms: An emerging link to disease pathogenesis. Annu. Rev. Microbiol. 2003, 57, 677–701. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Conover, M.; Lu, H.; Parsek, M.R.; Bayles, K.; Wozniak, D.J. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009, 5, e1000354. [Google Scholar] [CrossRef]
- Ma, L.; Jackson, K.D.; Landry, R.M.; Parsek, M.R.; Wozniak, D.J. Analysis of Pseudomonas aeruginosa conditional psl variants reveals roles for the psl polysaccharide in adhesion and maintaining biofilm structure postattachment. J. Bacteriol. 2006, 188, 8213–8221. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lu, H.; Sprinkle, A.; Parsek, M.R.; Wozniak, D.J. Pseudomonas aeruginosa Psl is a galactose-and mannose-rich exopolysaccharide. J. Bacteriol. 2007, 189, 8353–8356. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, Y.; Liu, Y.; Zhang, J.; Ulstrup, J.; Molin, S. Distinct roles of extracellular polymeric substances in Pseudomonas aeruginosa biofilm development. Environ. Microbiol. 2011, 13, 1705–1717. [Google Scholar] [CrossRef]
- Wei, Q.; Ma, L.Z. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2013, 14, 20983–21005. [Google Scholar] [CrossRef]
- Friedman, L.; Kolter, R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J. Bacteriol. 2004, 186, 4457–4465. [Google Scholar] [CrossRef]
- Friedman, L.; Kolter, R. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 2004, 51, 675–690. [Google Scholar] [CrossRef]
- Sutherland, I.W. Biofilm exopolysaccharides: A strong and sticky framework. Microbiology 2001, 147, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, M.; Bangera, M.G.; Bumgarner, R.E.; Parsek, M.R.; Teitzel, G.M.; Lory, S.; Greenberg, E. Gene expression in Pseudomonas aeruginosa biofilms. Nature 2001, 413, 860–864. [Google Scholar] [CrossRef]
- Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009, 7, 263–273. [Google Scholar] [CrossRef]
- Wu, Y.; Arumugam, K.; Tay, M.Q.X.; Seshan, H.; Mohanty, A.; Cao, B. Comparative genome analysis reveals genetic adaptation to versatile environmental conditions and importance of biofilm lifestyle in Comamonas testosteroni. Appl. Microbiol. Biotechnol. 2015, 99, 3519–3532. [Google Scholar] [CrossRef]
- Fuqua, C.; Winans, S.C.; Greenberg, E.P. Census and consensus in bacterial ecosystems: The LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu. Rev. Microbiol. 1996, 50, 727–751. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Greenberg, E.P. A network of networks: Quorum-sensing gene regulation in Pseudomonas aeruginosa. Int. J. Med. Microbiol. 2006, 296, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Venturi, V. Regulation of quorum sensing in Pseudomonas. FEMS Microbiol. Rev. 2006, 30, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Carabetta, V.J.; Tanner, A.W.; Greco, T.M.; Defrancesco, M.; Cristea, I.M.; Dubnau, D. A complex of YlbF, YmcA and YaaT regulates sporulation, competence and biofilm formation by accelerating the phosphorylation of Spo0A. Mol. Microbiol. 2013, 88, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, S.; Asai, K.; Ogasawara, N.; Takeuchi, M.; Sato, T. Mutation in yaaT leads to significant inhibition of phosphorelay during sporulation in Bacillus subtilis. J. Bacteriol. 2002, 184, 5545–5553. [Google Scholar] [CrossRef]
- Levipan, H.A.; Avendaño-Herrera, R. Different phenotypes of mature biofilm in Flavobacterium psychrophilum share a potential for virulence that differs from planktonic state. Front. Cell. Infect. Microbiol. 2017, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Somerton, B.; Flint, S.; Palmer, J.; Brooks, J.; Lindsay, D. Preconditioning with cations increases the attachment of Anoxybacillus flavithermus and Geobacillus species to stainless steel. Appl. Environ. Microbiol. 2013, 79, 4186–4190. [Google Scholar] [CrossRef]
- Zhao, Y.; Caspers, M.P.; Metselaar, K.I.; de Boer, P.; Roeselers, G.; Moezelaar, R.; Groot, M.N.; Montijn, R.C.; Abee, T.; Kort, R. Abiotic and microbiotic factors controlling biofilm formation by thermophilic sporeformers. Appl. Environ. Microbiol. 2013, 79, 5652–5660. [Google Scholar] [CrossRef] [PubMed]
- Noor, H.; Satti, S.M.; ud Din, S.; Farman, M.; Hasan, F.; Khan, S.; Badshah, M.; Shah, A.A. Insight on esterase from Pseudomonas aeruginosa strain S3 that depolymerize poly (lactic acid) (PLA) at ambient temperature. Polym. Degrad. Stab. 2020, 174, 109096. [Google Scholar] [CrossRef]
- Satti, S.M.; Abbasi, A.M.; Marsh, T.L.; Auras, R.; Hasan, F.; Badshah, M.; Farman, M.; Shah, A.A. Statistical optimization of lipase production from Sphingobacterium sp. strain S2 and evaluation of enzymatic depolymerization of poly (lactic acid) at mesophilic temperature. Polym. Degrad. Stab. 2019, 160, 1–13. [Google Scholar] [CrossRef]
- Wang, X.; Davlieva, M.; Reyes, J.; Panesso, D.; Arias, C.A.; Shamoo, Y. A novel phosphodiesterase of the GdpP family modulates cyclic di-AMP levels in response to cell membrane stress in daptomycin-resistant enterococci. Antimicrob. Agents Chemother. 2017, 61, e01422-16. [Google Scholar] [CrossRef]
- Gu, J.-D. Microbiological deterioration and degradation of synthetic polymeric materials: Recent research advances. Int. Biodeterior. Biodegrad. 2003, 52, 69–91. [Google Scholar] [CrossRef]
- Shah, A.A.; Hasan, F.; Hameed, A.; Ahmed, S. Biological degradation of plastics: A comprehensive review. Biotechnol. Adv. 2008, 26, 246–265. [Google Scholar] [CrossRef]
- Albertsson, A.-C. The shape of the biodegradation curve for low and high density polyethenes in prolonged series of experiments. Eur. Polym. J. 1980, 16, 623–630. [Google Scholar] [CrossRef]
- Albertsson, A.C.; Barenstedt, C.; Karlsson, S. Abiotic degradation products from enhanced environmentally degradable polyethylene. Acta Polym. 1994, 45, 97–103. [Google Scholar] [CrossRef]
- Muenmee, S.; Chiemchaisri, W.; Chiemchaisri, C. Microbial consortium involving biological methane oxidation in relation to the biodegradation of waste plastics in a solid waste disposal open dump site. Int. Biodeterior. Biodegrad. 2015, 102, 172–181. [Google Scholar] [CrossRef]
- Shimpi, N.; Borane, M.; Mishra, S.; Kadam, M. Biodegradation of polystyrene (PS)-poly (lactic acid) (PLA) nanocomposites using Pseudomonas aeruginosa. Macromol. Res. 2012, 20, 181–187. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, H.; Xing, Y.-T.; Li, N.; Wang, S.; Sun, J.-Q. Complete genome sequence of Sphingobacterium psychroaquaticum strain SJ-25, an aerobic bacterium capable of suppressing fungal pathogens. Curr. Microbiol. 2020, 77, 115–122. [Google Scholar] [CrossRef]
- Aas, M.J.; Vriesendorp, B.; van de Weijer, A.H.; van der Oost, J.; van Kranenburg, R. Complete genome sequence of Geobacillus thermodenitrificans T12, a potential host for biotechnological applications. Curr. Microbiol. 2018, 75, 49–56. [Google Scholar]
- Wang, J.; Goh, K.M.; Salem, D.R.; Sani, R.K. Genome analysis of a thermophilic exopolysaccharide-producing bacterium-Geobacillus sp. WSUCF1. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Chai, Y.; Kolter, R.; Losick, R. A widely conserved gene cluster required for lactate utilization in Bacillus subtilis and its involvement in biofilm formation. J. Bacteriol 2009, 191, 2423–2430. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).