
On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents




Abstract
1. Introduction
2. Targeting G-Quadruplex Structures in Cancer
2.1. Casting a Glance over Telomeric G4 Ligands
2.2. Impairment of G4-Mediated Regulation of Gene Expression for Therapeutic Purposes: The Paradigm of MYC
2.3. G4 Ligands with “Promiscuous” Binding Activity and/or Multiple Mechanism of Action
2.4. Targeting G4 for Synthetic Lethality
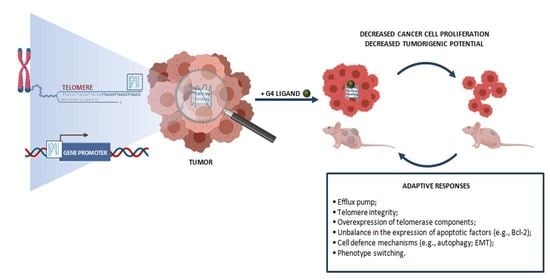
3. Adaptive Responses in Cancer Cells Exposed to G4 Ligands
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shahsavar, K.; Hosseini, M.; Shokri, E.; Xu, G. New insight into G-quadruplexes; diagnosis application in cancer. Anal. Biochem. 2021, 620, 114149. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Hao, Y.; Zheng, K. Kinetics, conformation, stability, and targeting of G-quadruplexes from a physiological perspective. Biochem. Biophys. Res. Commun. 2020, 531, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Hagen, T.; Tatum, N.J.; Hall, J. The diverse structural landscape of quadruplexes. FEBS Lett. 2019, 593, 2083–2102. [Google Scholar] [CrossRef]
- Lejault, P.; Mitteaux, J.; Sperti, F.R.; Monchaud, D. How to untie G-quadruplex knots and why? Cell Chem. Biol. 2021, 28, 436–455. [Google Scholar] [CrossRef]
- Bryan, T.M. G-Quadruplexes at Telomeres: Friend or Foe? Molecules 2020, 25, 3686. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar] [CrossRef]
- Xu, M.; Axhemi, A.; Malgowska, M.; Chen, Y.; Leonard, D.; Srinivasan, S.; Jankowsky, E.; Taylor, D.J. Active and Passive Destabilization of G-Quadruplex DNA by the Telomere POT1-TPP1 Complex. J. Mol. Biol. 2021, 433, 166846. [Google Scholar] [CrossRef]
- Monsen, R.C.; Chakravarthy, S.; Dean, W.L.; Chaires, J.B.; Trent, J.O. The solution structures of higher-order human telomere G-quadruplex multimers. Nucleic Acids Res. 2021, 49, 1749–1768. [Google Scholar] [CrossRef]
- Maizels, N. G4 motifs in human genes. Ann. N. Y. Acad. Sci. 2012, 1267, 53–60. [Google Scholar] [CrossRef]
- Maizels, N.; Gray, L.T. The G4 Genome. PLoS Genet. 2013, 9, e1003468. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef]
- Cimino-Reale, G.; Zaffaroni, N.; Folini, M. Emerging Role of G-quadruplex DNA as Target in Anticancer Therapy. Curr. Pharm. Des. 2016, 22, 6612–6624. [Google Scholar] [CrossRef]
- Sanchez-Martin, V.; Lopez-Pujante, C.; Soriano-Rodriguez, M.; Garcia-Salcedo, J.A. An Updated Focus on Quadruplex Structures as Potential Therapeutic Targets in Cancer. Int. J. Mol. Sci. 2020, 21, 8900. [Google Scholar] [CrossRef]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Pirota, V.; Nadai, M.; Doria, F.; Richter, S. Naphthalene Diimides as Multimodal G-Quadruplex-Selective Ligands. Molecules 2019, 24, 426. [Google Scholar] [CrossRef]
- Awadasseid, A.; Ma, X.; Wu, Y.; Zhang, W. G-quadruplex stabilization via small-molecules as a potential anti-cancer strategy. Biomed. Pharmacother. 2021, 139, 111550. [Google Scholar] [CrossRef]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule–induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef]
- Kharel, P.; Becker, G.; Tsvetkov, V.; Ivanov, P. Properties and biological impact of RNA G-quadruplexes: From order to turmoil and back. Nucleic Acids Res. 2020, 48, 12534–12555. [Google Scholar] [CrossRef]
- Dumas, L.; Herviou, P.; Dassi, E.; Cammas, A.; Millevoi, S. G-Quadruplexes in RNA Biology: Recent Advances and Future Directions. Trends Biochem. Sci. 2021, 46, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Millevoi, S.; Moine, H.; Vagner, S. G-quadruplexes in RNA biology. Wiley Interdiscip. Rev. RNA 2012, 3, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Gala, K.; Khattar, E. Long non-coding RNAs at work on telomeres: Functions and implications in cancer therapy. Cancer Lett. 2021, 502, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.Y.N.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013, 4, 1796. [Google Scholar] [CrossRef]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Henderson, A.; Wu, Y.; Huang, Y.C.; Chavez, E.A.; Platt, J.; Johnson, F.B.; Brosh, R.M.; Sen, D.; Lansdorp, P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014, 42, 860–869. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; Miller, J.; Howat, W.J.; Balasubramanian, S. Elevated Levels of G-Quadruplex Formation in Human Stomach and Liver Cancer Tissues. PLoS ONE 2014, 9, e102711. [Google Scholar] [CrossRef]
- Maizels, N. G4-associated human diseases. EMBO Rep. 2015, 16, 910–922. [Google Scholar] [CrossRef]
- Savva, L.; Georgiades, S.N. Recent Developments in Small-Molecule Ligands of Medicinal Relevance for Harnessing the Anticancer Potential of G-Quadruplexes. Molecules 2021, 26, 841. [Google Scholar] [CrossRef]
- Ou, T.M.; Lu, Y.J.; Tan, J.H.; Huang, Z.S.; Wong, K.Y.; Gu, L.Q. G-quadruplexes: Targets in anticancer drug design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef]
- Collie, G.W.; Parkinson, G.N. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem. Soc. Rev. 2011, 40, 5867. [Google Scholar] [CrossRef]
- Miglietta, G.; Russo, M.; Capranico, G. G-quadruplex–R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020, 48, 11942–11957. [Google Scholar] [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef]
- Li, Q.; Xiang, J.-F.; Yang, Q.-F.; Sun, H.-X.; Guan, A.-J.; Tang, Y.-L. G4LDB: A database for discovering and studying G-quadruplex ligands. Nucleic Acids Res. 2013, 41, D1115–D1123. [Google Scholar] [CrossRef]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of Human Telomerase by a G-Quadruplex-Interactive Compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Henderson, E.; Hardin, C.C.; Walk, S.K.; Tinoco, I.; Blackburn, E.H. Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine·guanine base pairs. Cell 1987, 51, 899–908. [Google Scholar] [CrossRef]
- Berardinelli, F.; Sgura, A.; Facoetti, A.; Leone, S.; Vischioni, B.; Ciocca, M.; Antoccia, A. The G-quadruplex-stabilizing ligand RHPS4 enhances sensitivity of U251MG glioblastoma cells to clinical carbon ion beams. FEBS J. 2018, 285, 1226–1236. [Google Scholar] [CrossRef]
- Berardinelli, F.; Siteni, S.; Tanzarella, C.; Stevens, M.F.; Sgura, A.; Antoccia, A. The G-quadruplex-stabilising agent RHPS4 induces telomeric dysfunction and enhances radiosensitivity in glioblastoma cells. DNA Repair 2015, 25, 104–115. [Google Scholar] [CrossRef]
- Phatak, P.; Cookson, J.C.; Dai, F.; Smith, V.; Gartenhaus, R.B.; Stevens, M.F.G.; Burger, A.M. Telomere uncapping by the G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in vitro and in vivo consistent with a cancer stem cell targeting mechanism. Br. J. Cancer 2007, 96, 1223–1233. [Google Scholar] [CrossRef]
- Leonetti, C.; Scarsella, M.; Riggio, G.; Rizzo, A.; Salvati, E.; D’Incalci, M.; Staszewsky, L.; Frapolli, R.; Stevens, M.F.; Stoppacciaro, A.; et al. G-Quadruplex Ligand RHPS4 Potentiates the Antitumor Activity of Camptothecins in Preclinical Models of Solid Tumors. Clin. Cancer Res. 2008, 14, 7284–7291. [Google Scholar] [CrossRef]
- Biroccio, A.; Porru, M.; Rizzo, A.; Salvati, E.; D’Angelo, C.; Orlandi, A.; Passeri, D.; Franceschin, M.; Stevens, M.F.G.; Gilson, E.; et al. DNA Damage Persistence as Determinant of Tumor Sensitivity to the Combination of Topo I Inhibitors and Telomere-Targeting Agents. Clin. Cancer Res. 2011, 17, 2227–2236. [Google Scholar] [CrossRef]
- Berardinelli, F.; Tanori, M.; Muoio, D.; Buccarelli, M.; di Masi, A.; Leone, S.; Ricci-Vitiani, L.; Pallini, R.; Mancuso, M.; Antoccia, A. G-quadruplex ligand RHPS4 radiosensitizes glioblastoma xenograft in vivo through a differential targeting of bulky differentiated- and stem-cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 311. [Google Scholar] [CrossRef]
- Iachettini, S.; Stevens, M.F.G.; Frigerio, M.; Hummersone, M.G.; Hutchinson, I.; Garner, T.P.; Searle, M.S.; Wilson, D.W.; Munde, M.; Nanjunda, R.; et al. On and off-target effects of telomere uncapping G-quadruplex selective ligands based on pentacyclic acridinium salts. J. Exp. Clin. Cancer Res. 2013, 32, 68. [Google Scholar] [CrossRef]
- Airley, R. Classical anticancer agents. In Cancer Chemotherapy: Basic Science to the Clinic; John Wiley & Sons, Ltd.: West Sussex, UK, 2009; pp. 67–111. ISBN 978-0-470-09255-2. [Google Scholar]
- Clark, G.R.; Pytel, P.D.; Squire, C.J.; Neidle, S. Structure of the First Parallel DNA Quadruplex-Drug Complex. J. Am. Chem. Soc. 2003, 125, 4066–4067. [Google Scholar] [CrossRef]
- Haider, S.M.; Neidle, S.; Parkinson, G.N. A structural analysis of G-quadruplex/ligand interactions. Biochimie 2011, 93, 1239–1251. [Google Scholar] [CrossRef]
- Manet, I.; Manoli, F.; Zambelli, B.; Andreano, G.; Masi, A.; Cellai, L.; Monti, S. Affinity of the anthracycline antitumor drugsDoxorubicin and Sabarubicin for human telomeric G-quadruplex structures. Phys. Chem. Chem. Phys. 2011, 13, 540–551. [Google Scholar] [CrossRef]
- Scaglioni, L.; Mondelli, R.; Artali, R.; Sirtori, F.R.; Mazzini, S. Nemorubicin and doxorubicin bind the G-quadruplex sequences of the human telomeres and of the c-MYC promoter element Pu22. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Barthwal, R.; Raje, S.; Pandav, K. Structural basis for stabilization of human telomeric G-quadruplex [d-(TTAGGGT)] 4 by anticancer drug epirubicin. Bioorg. Med. Chem. 2020, 28, 115761. [Google Scholar] [CrossRef]
- Barthwal, R.; Raje, S.; Pandav, K. Structural basis for stabilization of human telomeric G-quadruplex [d-(TTAGGGT)] 4 by anticancer drug adriamycin. J. Biomol. Struct. Dyn. 2021, 39, 795–815. [Google Scholar] [CrossRef]
- Viglasky, V. Platination of telomeric sequences and nuclease hypersensitive elements of human c-myc and PDGF-A promoters and their ability to form G-quadruplexes. FEBS J. 2009, 276, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Ourliac Garnier, I.; Bombard, S. GG sequence of DNA and the human telomeric sequence react with cis-diammine-diaquaplatinum at comparable rates. J. Inorg. Biochem. 2007, 101, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Heringova, P.; Kasparkova, J.; Brabec, V. DNA adducts of antitumor cisplatin preclude telomeric sequences from forming G quadruplexes. JBIC J. Biol. Inorg. Chem. 2009, 14, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Saker, L.; Ali, S.; Masserot, C.; Kellermann, G.; Poupon, J.; Teulade-Fichou, M.-P.; Ségal-Bendirdjian, E.; Bombard, S. Platinum Complexes Can Bind to Telomeres by Coordination. Int. J. Mol. Sci. 2018, 19, 1951. [Google Scholar] [CrossRef]
- Ju, H.-P.; Wang, Y.-Z.; You, J.; Hou, X.-M.; Xi, X.-G.; Dou, S.-X.; Li, W.; Wang, P.-Y. Folding Kinetics of Single Human Telomeric G-Quadruplex Affected by Cisplatin. ACS Omega 2016, 1, 244–250. [Google Scholar] [CrossRef]
- Zheng, X.; Nie, X.; Fang, Y.; Zhang, Z.; Xiao, Y.; Mao, Z.; Liu, H.; Ren, J.; Wang, F.; Xia, L.; et al. A Cisplatin Derivative Tetra-Pt(bpy) as an Oncotherapeutic Agent for Targeting ALT Cancer. JNCI J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Wang, W.; Hu, S.; Gu, Y.; Yan, Y.; Stovall, D.B.; Li, D.; Sui, G. Human MYC G-quadruplex: From discovery to a cancer therapeutic target. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188410. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Bhattacharya, S.; Dash, J.; Bhattacharya, S. Recent Update on Targeting c-MYC G-Quadruplexes by Small Molecules for Anticancer Therapeutics. J. Med. Chem. 2020, 64, 42–70. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Kuang, G.; Zhang, M.; Kang, S.; Hu, D.; Li, X.; Wei, Z.; Gong, X.; An, L.-K.; Huang, Z.-S.; Shu, B.; et al. Syntheses and Evaluation of New Bisacridine Derivatives for Dual Binding of G-Quadruplex and i-Motif in Regulating Oncogene c-myc Expression. J. Med. Chem. 2020, 63, 9136–9153. [Google Scholar] [CrossRef]
- Gaikwad, S.M.; Phyo, Z.; Arteaga, A.Q.; Gorjifard, S.; Calabrese, D.R.; Connors, D.; Huang, J.; Michalowski, A.M.; Zhang, S.; Liu, Z.-G.; et al. A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma. Cancers 2020, 12, 2952. [Google Scholar] [CrossRef]
- Pandya, N.; Khan, E.; Jain, N.; Satham, L.; Singh, R.; Makde, R.D.; Mishra, A.; Kumar, A. Curcumin analogs exhibit anti-cancer activity by selectively targeting G-quadruplex forming c-myc promoter sequence. Biochimie 2021, 180, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Rigo, R.; Palumbo, M.; Sissi, C. G-quadruplexes in human promoters: A challenge for therapeutic applications. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Huang, Z.-L.; Chen, S.-B.; Wang, C.-X.; Shan, C.; Yin, Q.-K.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Tan, J.-H.; et al. Design, Synthesis, and Evaluation of New Selective NM23-H2 Binders as c-MYC Transcription Inhibitors via Disruption of the NM23-H2/G-Quadruplex Interaction. J. Med. Chem. 2017, 60, 6924–6941. [Google Scholar] [CrossRef] [PubMed]
- Cocco, M.J. Specific interactions of distamycin with G-quadruplex DNA. Nucleic Acids Res. 2003, 31, 2944–2951. [Google Scholar] [CrossRef]
- Pagano, B.; Fotticchia, I.; De Tito, S.; Mattia, C.A.; Mayol, L.; Novellino, E.; Randazzo, A.; Giancola, C. Selective Binding of Distamycin A Derivative to G-Quadruplex Structure [d(TGGGGT)] 4. J. Nucleic Acids 2010, 2010, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Ali, A.; Kamra, M.; Muniyappa, K.; Bhattacharya, S. Specific stabilization of promoter G-Quadruplex DNA by 2,6-disubstituted amidoanthracene-9,10-dione based dimeric distamycin analogues and their selective cancer cell cytotoxicity. Eur. J. Med. Chem. 2020, 195, 112202. [Google Scholar] [CrossRef]
- Wu, R.; Li, L.; Bai, Y.; Yu, B.; Xie, C.; Wu, H.; Zhang, Y.; Huang, L.; Yan, Y.; Li, X.; et al. The long noncoding RNA LUCAT1 promotes colorectal cancer cell proliferation by antagonizing Nucleolin to regulate MYC expression. Cell Death Dis. 2020, 11, 908. [Google Scholar] [CrossRef]
- Ohnmacht, S.A.; Marchetti, C.; Gunaratnam, M.; Besser, R.J.; Haider, S.M.; Di Vita, G.; Lowe, H.L.; Mellinas-Gomez, M.; Diocou, S.; Robson, M.; et al. A G-quadruplex-binding compound showing anti-tumour activity in an in vivo model for pancreatic cancer. Sci. Rep. 2015, 5, 11385. [Google Scholar] [CrossRef]
- Hu, M.H.; Yu, B.Y.; Wang, X.; Jin, G. Drug-like biimidazole derivatives dually target c-MYC/BCL-2 G-quadruplexes and inhibit acute myeloid leukemia. Bioorg. Chem. 2020, 104, 104264. [Google Scholar] [CrossRef]
- Zeng, L.; Wu, Q.; Wang, T.; Li, L.-P.; Zhao, X.; Chen, K.; Qian, J.; Yuan, L.; Xu, H.; Mei, W.-J. Selective stabilization of multiple promoter G-quadruplex DNA by using 2-phenyl-1H-imidazole-based tanshinone IIA derivatives and their potential suppressing function in the metastatic breast cancer. Bioorg. Chem. 2021, 106, 104433. [Google Scholar] [CrossRef]
- Gunaratnam, M.; Swank, S.; Haider, S.M.; Galesa, K.; Reszka, A.P.; Beltran, M.; Cuenca, F.; Fletcher, J.A.; Neidle, S. Targeting Human Gastrointestinal Stromal Tumor Cells with a Quadruplex-Binding Small Molecule. J. Med. Chem. 2009, 52, 3774–3783. [Google Scholar] [CrossRef]
- Tassinari, M.; Cimino-Reale, G.; Nadai, M.; Doria, F.; Butovskaya, E.; Recagni, M.; Freccero, M.; Zaffaroni, N.; Richter, S.N.; Folini, M. Down-Regulation of the Androgen Receptor by G-Quadruplex Ligands Sensitizes Castration-Resistant Prostate Cancer Cells to Enzalutamide. J. Med. Chem. 2018, 61, 8625–8638. [Google Scholar] [CrossRef]
- Paul, R.; Das, T.; Debnath, M.; Chauhan, A.; Dash, J. G-Quadruplex-Binding Small Molecule Induces Synthetic Lethality in Breast Cancer Cells by Inhibiting c-MYC and BCL2 Expression. ChemBioChem 2020, 21, 963–970. [Google Scholar] [CrossRef]
- Parrino, B.; Carbone, A.; Ciancimino, C.; Spanò, V.; Montalbano, A.; Barraja, P.; Cirrincione, G.; Diana, P.; Sissi, C.; Palumbo, M.; et al. Water-soluble isoindolo[2,1-a]quinoxalin-6-imines: In vitro antiproliferative activity and molecular mechanism(s) of action. Eur. J. Med. Chem. 2015, 94, 149–162. [Google Scholar] [CrossRef]
- Wang, K.B.; Elsayed, M.S.; Wu, G.; Deng, N.; Cushman, M.; Yang, D. Indenoisoquinoline Topoisomerase Inhibitors Strongly Bind and Stabilize the MYC Promoter G-Quadruplex and Downregulate MYC. J. Am. Chem. Soc. 2019, 141, 11059–11070. [Google Scholar] [CrossRef]
- Dallavalle, S.; Musso, L.; Artali, R.; Aviñó, A.; Scaglioni, L.; Eritja, R.; Gargallo, R.; Mazzini, S. G-quadruplex binding properties of a potent PARP-1 inhibitor derived from 7-azaindole-1-carboxamide. Sci. Rep. 2021, 11, 3869. [Google Scholar] [CrossRef]
- Salvati, E.; Scarsella, M.; Porru, M.; Rizzo, A.; Iachettini, S.; Tentori, L.; Graziani, G.; D’Incalci, M.; Stevens, M.F.G.; Orlandi, A.; et al. PARP1 is activated at telomeres upon G4 stabilization: Possible target for telomere-based therapy. Oncogene 2010, 29, 6280–6293. [Google Scholar] [CrossRef]
- Li, S.; Topatana, W.; Juengpanich, S.; Cao, J.; Hu, J.; Zhang, B.; Ma, D.; Cai, X.; Chen, M. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- McLuckie, K.I.E.; Di Antonio, M.; Zecchini, H.; Xian, J.; Caldas, C.; Krippendorff, B.-F.; Tannahill, D.; Lowe, C.; Balasubramanian, S. G-Quadruplex DNA as a Molecular Target for Induced Synthetic Lethality in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 9640–9643. [Google Scholar] [CrossRef]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [PubMed]
- Zyner, K.G.; Mulhearn, D.S.; Adhikari, S.; Martínez Cuesta, S.; Di Antonio, M.; Erard, N.; Hannon, G.J.; Tannahill, D.; Balasubramanian, S. Genetic interactions of G-quadruplexes in humans. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Ha, J.; Kang, E.; Cho, S. The role of epithelial–mesenchymal transition-regulating transcription factors in anti-cancer drug resistance. Arch. Pharm. Res. 2021, 44, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Aouali, N.; Renaud, A.; Douarre, C.; Shin-ya, K.; Tazi, J.; Martinez, S.; Trentesaux, C.; Morjani, H.; Riou, J.F. Resistance to senescence induction and telomere shortening by a G-quadruplex ligand inhibitor of telomerase. Cancer Res. 2003, 63, 6149–6153. [Google Scholar] [PubMed]
- Gomez, D.; Aouali, N.; Londoño-Vallejo, A.; Lacroix, L.; Mégnin-Chanet, F.; Lemarteleur, T.; Douarre, C.; Shin-ya, K.; Mailliet, P.; Trentesaux, C.; et al. Resistance to the Short Term Antiproliferative Activity of the G-quadruplex Ligand 12459 Is Associated with Telomerase Overexpression and Telomere Capping Alteration. J. Biol. Chem. 2003, 278, 50554–50562. [Google Scholar] [CrossRef]
- Douarre, C. Overexpression of Bcl-2 is associated with apoptotic resistance to the G-quadruplex ligand 12459 but is not sufficient to confer resistance to long-term senescence. Nucleic Acids Res. 2005, 33, 2192–2203. [Google Scholar] [CrossRef]
- Gomez, D.; Wenner, T.; Brassart, B.; Douarre, C.; O’Donohue, M.-F.; El Khoury, V.; Shin-ya, K.; Morjani, H.; Trentesaux, C.; Riou, J.-F. Telomestatin-induced Telomere Uncapping Is Modulated by POT1 through G-overhang Extension in HT1080 Human Tumor Cells. J. Biol. Chem. 2006, 281, 38721–38729. [Google Scholar] [CrossRef]
- Orlotti, N.I.; Cimino-Reale, G.; Borghini, E.; Pennati, M.; Sissi, C.; Perrone, F.; Palumbo, M.; Daidone, M.G.; Folini, M.; Zaffaroni, N. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy 2012, 8, 1185–1196. [Google Scholar] [CrossRef]
- Recagni, M.; Greco, M.L.; Milelli, A.; Minarini, A.; Zaffaroni, N.; Folini, M.; Sissi, C. Distinct biological responses of metastatic castration resistant prostate cancer cells upon exposure to G-quadruplex interacting naphthalenediimide derivatives. Eur. J. Med. Chem. 2019, 177, 401–413. [Google Scholar] [CrossRef]
- Recagni, M.; Tassinari, M.; Doria, F.; Cimino-Reale, G.; Zaffaroni, N.; Freccero, M.; Folini, M.; Richter, S.N. The Oncogenic Signaling Pathways in BRAF-Mutant Melanoma Cells are Modulated by Naphthalene Diimide-Like G-Quadruplex Ligands. Cells 2019, 8, 1274. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Hamon, F.; Teulade-Fichou, M.-P. Development of a high-throughput G4-FID assay for screening and evaluation of small molecules binding quadruplex nucleic acid structures. Anal. Bioanal. Chem. 2011, 400, 3419–3427. [Google Scholar] [CrossRef]
- Largy, E.; Saettel, N.; Hamon, F.; Dubruille, S.; Teulade-Fichou, M.-P. Screening of a Chemical Library by HT-G4-FID for Discovery of Selective G-quadruplex Binders. Curr. Pharm. Des. 2012, 18, 1992–2001. [Google Scholar] [CrossRef]
- Kaserer, T.; Rigo, R.; Schuster, P.; Alcaro, S.; Sissi, C.; Schuster, D. Optimized Virtual Screening Workflow for the Identification of Novel G-Quadruplex Ligands. J. Chem. Inf. Model. 2016, 56, 484–500. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Nadai, M.; Scalabrin, M.; Talarico, C.; Distinto, S.; Maccioni, E.; Ortuso, F.; Artese, A.; et al. Identification of G-quadruplex DNA/RNA binders: Structure-based virtual screening and biophysical characterization. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1329–1340. [Google Scholar] [CrossRef]
- Parrotta, L.; Ortuso, F.; Moraca, F.; Rocca, R.; Costa, G.; Alcaro, S.; Artese, A. Targeting unimolecular G-quadruplex nucleic acids: A new paradigm for the drug discovery? Expert Opin. Drug Discov. 2014, 9, 1167–1187. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA Polymerase I with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA Synthesis and Solid Tumor Growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef]
- Puig Lombardi, E.; Londoño-Vallejo, A. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2020, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Ruiz, M.; Doria, F.; Belmonte-Reche, E.; De Rache, A.; Campos-Salinas, J.; Lucas, R.; Falomir, E.; Carda, M.; Pérez-Victoria, J.M.; Mergny, J.L.; et al. Synthesis, Binding Properties, and Differences in Cell Uptake of G-Quadruplex Ligands Based on Carbohydrate Naphthalene Diimide Conjugates. Chem. A Eur. J. 2017, 23, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Lopes-Nunes, J.; Carvalho, J.; Antunes, F.; Ribeiro, M.; Campello, M.P.C.; Paulo, A.; Paiva, A.; Salgado, G.F.; Queiroz, J.A.; et al. AS1411 derivatives as carriers of G-quadruplex ligands for cervical cancer cells. Int. J. Pharm. 2019, 568, 118511. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | G4 Target | Tumor Models | Biological (Anticancer) Effects | Ref | |
|---|---|---|---|---|---|
| Cisplatinum derivatives (Tetra-Pt (bpy)) | Telomere | G4 stabilization assessed by FRET (ΔTm = 24.8 °C); binding affinity determined by SPR (KD = 1.13 × 10−7 M); evidence by CD of parallel G4 folding under near-physiological conditions. | Osteosarcoma | -Remarkable cytotoxic activity in vitro (IC50 ~ 15 μM); -Inhibition of telomeric homologous recombination and suppression of ALT activity (reduced ALT-associated promyelocytic leukemia bodies; reduced c-circle DNA; reduced telomere sister chromatin exchanges) in vitro (concentration used 2 μM); accumulation of critically short telomeres after multiple population doublings; -Induction of apoptosis/senescence; -No adverse effect on normal MRC5 fibroblasts in vitro (IC50 = 89.3μM); -Inhibition of xenograft tumor growth in mice (20 mg/kg; i.v.); inhibition of liver metastases. | [56] |
| Isoindoloquinoxalin derivatives | Telomere | G4 stabilizing properties investigated by CD and FRET. | Osteosarcoma | -Cytotoxic activity in vitro (IC50 = 20–30 nM); -Changes in cell cycle phase distribution; induction of apoptosis; -Induction of telomere dysfunctions; -Inhibition of tubulin polymerization. | [76] |
| Antracene derivative (Ant1,5) | Telomere | Analyses on targeted G4 carried out in a previous study. | Melanoma | -Impairment of in vitro cell growth (IC50 = 4–10 μM); -Induction of telomere dysfunctions and of DNA damage; accumulation of p21waf1; -Occurrence of autophagy as a defence mechanism; -Increased cytotoxic activity upon pharmacological inhibition of autophagy. | [92] |
| Bisacridine derivatives (a9) | MYC | G4 stabilization assessed by FRET (ΔTm = 9.9 °C); binding affinity evaluated by SPR (KD = 7.7 μM); interaction with the G4 evaluated by CD, ITC, NMR; native PAGE and molecular docking. | Squamous cell carcinoma | -Down-regulation of MYC expression (Dual-luciferase reporter assay; RT-PCR; Western blotting); -Strong inhibition of in vitro cell growth (IC50 = 1.22 μM); cell cycle perturbations and induction of apoptosis; -Reduction in tumor growth in vivo (15 mg/kg; i.p.); no changes in body weight and no organ toxicity observed. | [60] |
| Benzofuran derivative (D089) | MYC | Analyses on targeted G4 carried out in a previous study. | Multiple myeloma | -Down-regulation of MYC expression (RT-qPCR); - Cytotoxic activity in vitro (IC50 = 11–50 μM); -No remarkable cytotoxic activity in HEK293T cells ectopically expressing MYC under the control of CMV promoter (IC50 = 50μM); -Induction of endoplasmic reticulum stress, senescence and pyroptosis in vitro. | [61] |
| Curcumin derivative (Cur-4) | MYC | G4 stabilization assessed by CD thermal melting (ΔTm ~ 10 °C); binding affinity evaluated by steady-state fluorescence titration (KD = 0.004 × 10−6 M) and by ITC (ΔH1 = 1.46 × 104 cal/mol); increase in lifetime decay for drug-DNA complex analysed by time-correlated single photon counting; docking and molecular dynamic simulation studies. | Cervical carcinoma | -Down-regulation of c-MYC expression (qRT-PCR; Western blotting); -In vitro cytotoxic activity on cells grown as monolayer (IC50 = 5.0 μM); -Low cytotoxic activity in HEK293 cells (IC50 = 64 μM); -Decreased number of living cells in multicellular tumor spheroids with evidence of drug up-take. | [62] |
| 2,6-disubstituted amidoanthracene-9,10-dione based dimeric distamycin analogues (ANMP, ANDP and ANTP) | MYC | G4 stabilization assessed by CD (ΔTm = 3.2–11.1 °C as a function of tested compound) and Taq stop polymerization assay; binding interaction evaluated by UV-vis absorption spectral titration (Ka = 1.4–3.8 106 M−1); fluorescence spectroscopy-based titration, ethidium bromide displacement assay, cyclic voltammetry titration; molecular docking studies. | Cervical carcinoma | -No evidence of pharmacodynamic activity in vitro (MYC expression levels were not assessed); -Cytotoxic activity in vitro (IC50 = 5.3–100 μM); -No cytotoxic activity on normal NIH3T3and HDFa (IC50 > 100 μM) as well as on HEK293T cells (IC50 = 15–43 μM); -Cellular morphological changes and apoptosis induction. | [68] |
| Indenoisoquinolines | MYC | G4 stabilization assessed by FRET (Tm > 5 °C in the presence of the ligand); binding interaction evaluated by NMR titration; signature of a parallel G-quadruplex assessed by CD; binding mode explored by molecular docking; binding selectivity for MYC G4 vs. KRAS G4 assessed by Competition Fluorescence Displacement. | Breast cancer | -Down-regulation of MYC expression (qRT-PCR and Western blot); -Strong topoisomerase I inhibition. | [77] |
| Functionalized naphthalene diimide derivatives (Compound 7) | AR | G4 stabilization assessed by FRET (ΔT½ = 8.3–17 °C), CD and Taq stop polymerization assay; binding affinity determined by SPR (KD = 18 nM). | Metastatic, castration-resistant prostate cancer (mCRPC) | -Down-regulation of AR expression (RT-qPCR and Western blotting); -Remarkable cytotoxic acitivity in vitro; -Significant perturbations in the expression levels of KLK3 and of genes involved in the activation of AR program via feedback mechanisms; -Inhibition of telomerase activity; -Pharmacological synergistic interaction with Enzalutamide (MDV3100). | [74] |
| Biimidazole derivative (BIM-2) | MYC BCL-2 | G4 stabilization assessed by CD (ΔTm = 29.0 °C, MYC; ΔTm = 18.0 °C, BCL-2); binding interaction evaluated by fluorescence titration (KD = 0.75 μM, MYC; KD = 1.53 μM, BCL-2); binding mode assessed by NMR titration; binding mechanism investigated by molecular modelling. | Acute myeloid leukemia | -Down-regulation of MYC and BCL2 expression (end-point RT-PCR; Western blotting); -Cytotoxic activity in vitro (IC50 = 9.2 μM); -No cytotoxic activity on normal BJ fibroblasts (IC50 > 40 μM); -Cell cycle perturbations with a marked increase in cells in the G0/G1 phase; apoptosis induction. | [71] |
| Prolinamide-derived peptidomimetic (Ligand 1) | MYC BCL-2 | G4 stabilization assessed by FRET (ΔTm = 15.0 °C; MYC; ΔTm = 16.0 BCL–2 °C); binding affinity assessed by ITC titration (KD = 1.43 μM; ΔG = −7.98 kcal mol−1, MYC; KD = 2.26 μM; ΔG = −7.70 kcal mol−1, BCL-2); binding interaction assessed by molecular docking. | Breast cancer | -Down-regulation of MYC and BCL-2 expression (Dual luciferase reporter assays; qRT-PCR; Western Blotting); -Cytotoxic activity in vitro (IC50 = 3.8 μM); -No remarkable cytotoxic activity on normal kidney epithelial cells (IC50 > 50 μM); -S-phase cell-cycle arrest, DNA damage and apoptosis induction. | [75] |
| Core-extended naphthalene diimide derivatives | MYC BCL-2 BRAF KIT | G4 stabilization assessed by CD thermal unfolding (Tm > 90 °C in the presence of the ligand) FRET analysis and Taq stop polymerization assay. | Melanoma | -Down-regulation of KIT and BCL-2 protein amounts; no changes in BRAF and MYC protein levels; -Remarkable cytotoxic activity in vitro (IC50 = 9.0 nM and 260 nM) with evidence of G4 occurrence in cells (Immunofluorescence with a G4 specific antibody); -No remarkable cytotoxic effects on normal primary skin fibroblasts (IC50 > 1.000 nM); -Shutdown of RAS/RAF/MAPK and PI3K/AKT signaling pathways; -Cell cycle perturbations; induction of apoptosis (PARP-1 cleavage); Induction of a phenotypic switch in NRAS-mutant melanoma cells. | [94] |
| Imidazole-based tanshinone IIA derivative (Compound 4) | MYC KRAS VEGF BCL-2 Telomere | G4 stabilization assessed by FRET (ΔTm = 7.89 °C, MYC; ΔTm = 5.25 °C, KRAS; ΔTm = 5.27, VEGF; ΔTm = 4.57 °C, BCL-2; ΔTm = 1.76 °C, Telomere); binding interaction evaluated by spectroscopic methods and molecular docking; Influence on G4 conformation assessed by CD. | Metastatic triple-negative Breast cancer | -Down-regulation of MYC, KRAS, VEGF, BCL-2 expression (RT-qPCR) -Cytotoxic activity in vitro (IC50 = 12.8 μM); -No remarkable cytotoxic activity on non-tumorigenic MCF-10A mammary epithelial cells (IC50 = 95.7 μM; safe index (IC50(MCF-10A)/IC50(MDA-MB-231)) = 7.48); -Cell cycle perturbations and inhibition of cell migration and invasion in vitro; -Inhibition of breast cancer growth, metastasis and angiogenesis in an in vivo zebrafish tumor model. | [72] |
| Tetra-substituted naphthalene-diimide derivative (MM41) | BCL-2 K-RAS | G4 stabilization assessed by FRET (ΔTm = 26.4 °C, BCL-2; ΔTm = 22.5 °C, k-RAS1; ΔTm = 19.8 °C, k-RAS2); binding interaction evaluated by molecular modelling. | Pancreatic cancer | -Evidence of pharmacodynamic activity in vivo (reduced BCL-2 and K-RAS protein by Western blotting); -Reduction in tumor xenograft growth in vivo (10–15 mg/kg; i.v.); -No evidence of toxicity determined as absence of body weight loss; -Evidence of no tumor re-growth after > 200 days post-treatment at the dose of 15 mg/kg; -Evidence of tumor drug up-take in vivo (immunofluorescnece on tumor sections). | [70] |
| NDI derivative (Compound 1) | KIT Telomere | G4 stabilization assessed by FRET (ΔTm = 11.2 °C, KIT-1; ΔTm = 29.0 °C, KIT-2; ΔTm = 28.7 °C, Telomere); binding interaction evaluated by molecular docking. | Gastrointestinal stromal tumor | -Cytotoxic activity in vitro (IC50 = 1.62 μM vs. 1.7 μM for Imatinib) -Nearly complete abrogation of KIT expression (RT-PCR; Western blotting) at the IC50 dose; -Potent telomerase activity inhibition at a sub-toxic concentration (modified/TRAP-LIG assay). | [73] |
| Symmetrical-and asymmetrical-substituted naphthalene diimide derivatives | EGFR Telomere | Structural transition of EGFR promoter towards a G4 conformation and stabilization of telomeric G4 evaluated by FRET, ITC and SPR titrations. | Metastatic, castration-resistant prostate cancer (mCRPC) | -Dose-dependent reduction in EGFR protein amounts; -Remarkable cytotoxic activity in vitro (IC50 0.65–5.0 μM as a function of time and cell line); -Interference with RAS/RAF/MAPK and PI3K/AKT signaling pathways; -Time-dependent inhibition of prostate cancer cell growth in vitro (short-term setting); -No major changes in the rate of DU145 cell growth as well as in the amount of EGFR protein upon 60 days of weekly reiterated exposure to subtoxic amounts (½IC50–48h) of the ligands; -Remarkable impairment of PC-3 cell growth associated with an almost complete abrogation of EGFR protein levels upon 60 days of weekly reiterated exposure to subtoxic amounts (½IC50–48h) of the ligands; acquisition of mesenchymal traits and increased telomeric C-circles. | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessandrini, I.; Recagni, M.; Zaffaroni, N.; Folini, M. On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 5947. https://doi.org/10.3390/ijms22115947
Alessandrini I, Recagni M, Zaffaroni N, Folini M. On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents. International Journal of Molecular Sciences. 2021; 22(11):5947. https://doi.org/10.3390/ijms22115947
Chicago/Turabian StyleAlessandrini, Irene, Marta Recagni, Nadia Zaffaroni, and Marco Folini. 2021. "On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents" International Journal of Molecular Sciences 22, no. 11: 5947. https://doi.org/10.3390/ijms22115947
APA StyleAlessandrini, I., Recagni, M., Zaffaroni, N., & Folini, M. (2021). On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents. International Journal of Molecular Sciences, 22(11), 5947. https://doi.org/10.3390/ijms22115947