
Infection-Associated Mechanisms of Neuro-Inflammation and Neuro-Immune Crosstalk in Chronic Respiratory Diseases

Abstract
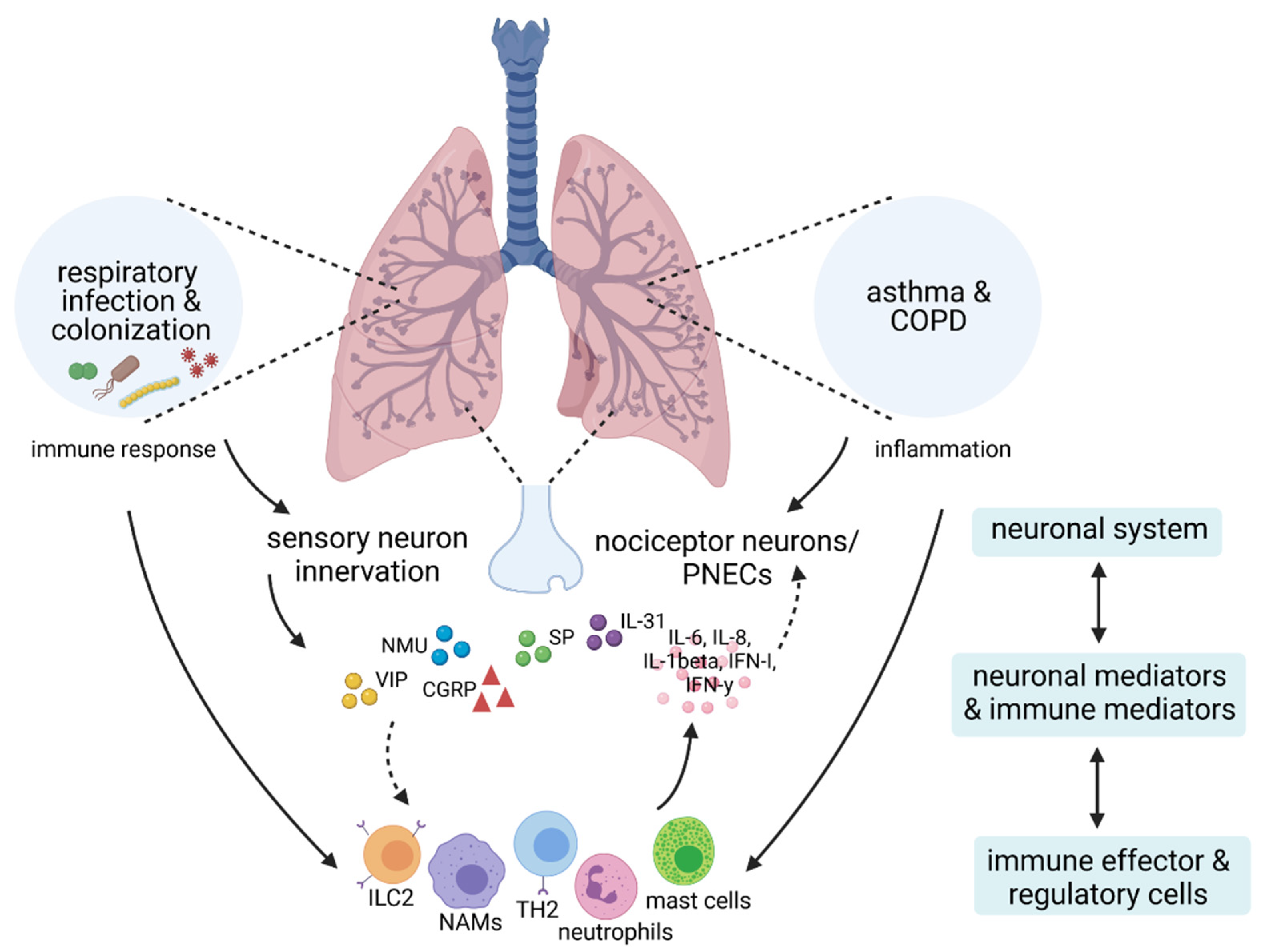
1. Introduction
2. Chronic Respiratory Diseases
2.1. Bronchial Asthma
2.2. Chronic Obstructive Pulmonary Disease
3. Neuro-Immune Interactions in Allergic Asthma and COPD
3.1. Effects of Allergen Challenge on Sensory Nerves and Neuropeptide Release in Asthma
3.2. Neuro-Immune Crosstalk in COPD
4. Neuro-Immune Crosstalk in Respiratory Infection and Microbial Colonization
4.1. Infections with Human Rhinovirus
4.2. Infections with Influenza A Virus
4.3. Infections with Respiratory Syncytial Virus
4.4. Infections and Colonization with Staphylococcus aureus
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bao, Z.; Xiong, J.; Li, W.; Chen, Z.; Shen, H.; Ying, S. Genomic instability in chronic airway inflammatory diseases. Biomed. J. 2015, 38, 117–124. [Google Scholar]
- Calus, L.; Van Zele, T.; Derycke, L.; Krysko, O.; Dutre, T.; Tomassen, P.; Dullaers, M.; Bachert, C.; Gevaert, P. Local inflammation in chronic upper airway disease. Curr. Pharm. Des. 2012, 18, 2336–2346. [Google Scholar] [CrossRef]
- Gautier, C.; Charpin, D. Environmental triggers and avoidance in the management of asthma. J. Asthma Allergy 2017, 10, 47–56. [Google Scholar] [CrossRef]
- Viniol, C.; Vogelmeier, C.F. Exacerbations of copd. Eur. Respir. Rev. 2018, 27, 170103. [Google Scholar] [CrossRef]
- Adeli, M.; El-Shareif, T.; Hendaus, M.A. Asthma exacerbation related to viral infections: An up to date summary. J. Fam. Med. Prim. Care 2019, 8, 2753–2759. [Google Scholar] [CrossRef]
- Grissell, T.V.; Powell, H.; Shafren, D.R.; Boyle, M.J.; Hensley, M.J.; Jones, P.D.; Whitehead, B.F.; Gibson, P.G. Interleukin-10 gene expression in acute virus-induced asthma. Am. J. Respir. Crit. Care Med. 2005, 172, 433–439. [Google Scholar] [CrossRef]
- Stolz, D.; Papakonstantinou, E.; Grize, L.; Schilter, D.; Strobel, W.; Louis, R.; Schindler, C.; Hirsch, H.H.; Tamm, M. Time-course of upper respiratory tract viral infection and copd exacerbation. Eur. Respir. J. 2019, 54, 1900407. [Google Scholar] [CrossRef]
- Jartti, T.; Gern, J.E. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 2017, 140, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Oliver, B.G.; Robinson, P.; Peters, M.; Black, J. Viral infections and asthma: An inflammatory interface? Eur. Respir. J. 2014, 44, 1666–1681. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.D.; Costa, P.S.; Camargos, P.A. Exacerbation of asthma and airway infection: Is the virus the villain? J. Pediatr. 2014, 90, 542–555. [Google Scholar] [CrossRef]
- Tan, K.S.; Lim, R.L.; Liu, J.; Ong, H.H.; Tan, V.J.; Lim, H.F.; Chung, K.F.; Adcock, I.M.; Chow, V.T.; Wang, Y. Respiratory viral infections in exacerbation of chronic airway inflammatory diseases: Novel mechanisms and insights from the upper airway epithelium. Front. Cell Dev. Biol. 2020, 8, 99. [Google Scholar] [CrossRef]
- Diver, S.; Richardson, M.; Haldar, K.; Ghebre, M.A.; Ramsheh, M.Y.; Bafadhel, M.; Desai, D.; Cohen, E.S.; Newbold, P.; Rapley, L.; et al. Sputum microbiomic clustering in asthma and chronic obstructive pulmonary disease reveals a haemophilus-predominant subgroup. Allergy 2020, 75, 808–817. [Google Scholar] [CrossRef]
- Huang, Y.J.; Lynch, S.V. The emerging relationship between the airway microbiota and chronic respiratory disease: Clinical implications. Expert Rev. Respir. Med. 2011, 5, 809–821. [Google Scholar] [CrossRef][Green Version]
- Voisin, T.; Bouvier, A.; Chiu, I.M. Neuro-immune interactions in allergic diseases: Novel targets for therapeutics. Int. Immunol. 2017, 29, 247–261. [Google Scholar] [CrossRef]
- Brain, S.D.; Williams, T.J. Interactions between the tachykinins and calcitonin gene-related peptide lead to the modulation of oedema formation and blood flow in rat skin. Br. J. Pharmacol. 1989, 97, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Maggi, C.A. Tachykinins and calcitonin gene-related peptide (cgrp) as co-transmitters released from peripheral endings of sensory nerves. Prog. Neurobiol. 1995, 45, 1–98. [Google Scholar] [CrossRef]
- Hagiyama, M.; Inoue, T.; Furuno, T.; Iino, T.; Itami, S.; Nakanishi, M.; Asada, H.; Hosokawa, Y.; Ito, A. Increased expression of cell adhesion molecule 1 by mast cells as a cause of enhanced nerve-mast cell interaction in a hapten-induced mouse model of atopic dermatitis. Br. J. Dermatol. 2013, 168, 771–778. [Google Scholar] [CrossRef]
- Le, D.D.; Schmit, D.; Heck, S.; Omlor, A.J.; Sester, M.; Herr, C.; Schick, B.; Daubeuf, F.; Fähndrich, S.; Bals, R.; et al. Increase of mast cell-nerve association and neuropeptide receptor expression on mast cells in perennial allergic rhinitis. Neuroimmunomodulation 2016, 23, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Le, D.D.; Rochlitzer, S.; Fischer, A.; Heck, S.; Tschernig, T.; Sester, M.; Bals, R.; Welte, T.; Braun, A.; Dinh, Q.T. Allergic airway inflammation induces the migration of dendritic cells into airway sensory ganglia. Respir. Res. 2014, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Veres, T.Z.; Rochlitzer, S.; Shevchenko, M.; Fuchs, B.; Prenzler, F.; Nassenstein, C.; Fischer, A.; Welker, L.; Holz, O.; Müller, M.; et al. Spatial interactions between dendritic cells and sensory nerves in allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 2007, 37, 553–561. [Google Scholar] [CrossRef]
- Sawatzky, D.A.; Kingham, P.J.; Court, E.; Kumaravel, B.; Fryer, A.D.; Jacoby, D.B.; McLean, W.G.; Costello, R.W. Eosinophil adhesion to cholinergic nerves via icam-1 and vcam-1 and associated eosinophil degranulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1279–L1288. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis Primers 2015, 1, 15025. [Google Scholar] [CrossRef]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Mims, J.W. Asthma: Definitions and pathophysiology. Int. Forum Allergy Rhinol. 2015, 5 (Suppl. S1), S2–S6. [Google Scholar] [CrossRef]
- Matucci, A.; Vultaggio, A.; Maggi, E.; Kasujee, I. Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question? Respir. Res. 2018, 19, 113. [Google Scholar] [CrossRef]
- Amin, K. The role of mast cells in allergic inflammation. Respir. Med. 2012, 106, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Pathophysiology of allergic inflammation. Immunol. Rev. 2011, 242, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Type 2 inflammation in asthma—Present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Foster, P.S.; Maltby, S.; Rosenberg, H.F.; Tay, H.L.; Hogan, S.P.; Collison, A.M.; Yang, M.; Kaiko, G.E.; Hansbro, P.M.; Kumar, R.K.; et al. Modeling t(h) 2 responses and airway inflammation to understand fundamental mechanisms regulating the pathogenesis of asthma. Immunol. Rev. 2017, 278, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.; Reese, T.A.; Huang, X.; Shinkai, K.; Locksley, R.M. Type 2 immunity is controlled by il-4/il-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J. Exp. Med. 2006, 203, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.G.; Strickland, D.H.; Hales, B.J.; Sly, P.D. Defective respiratory tract immune surveillance in asthma: A primary causal factor in disease onset and progression. Chest 2014, 145, 370–378. [Google Scholar] [CrossRef]
- Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I.; Aldonyte, R. Novel aspects of pathogenesis and regeneration mechanisms in copd. Int. J. Chron. Obstr. Pulmon. Dis. 2015, 10, 995–1013. [Google Scholar]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in copd. Int. J. Chron. Obstr. Pulmon. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Martinez, F.D. Early-life origins of chronic obstructive pulmonary disease. N. Engl. J. Med. 2016, 375, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459. [Google Scholar] [CrossRef]
- Barczyk, A.; Pierzchała, W.; Kon, O.M.; Cosio, B.; Adcock, I.M.; Barnes, P.J. Cytokine production by bronchoalveolar lavage t lymphocytes in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2006, 117, 1484–1492. [Google Scholar] [CrossRef]
- Postma, D.S.; Rabe, K.F. The asthma-copd overlap syndrome. N. Engl. J. Med. 2015, 373, 1241–1249. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir. Med. 2013, 1, 73–83. [Google Scholar] [CrossRef]
- Divo, M.; Cote, C.; de Torres, J.P.; Casanova, C.; Marin, J.M.; Pinto-Plata, V.; Zulueta, J.; Cabrera, C.; Zagaceta, J.; Hunninghake, G.; et al. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 186, 155–161. [Google Scholar] [CrossRef]
- Patel, A.R.; Hurst, J.R. Extrapulmonary comorbidities in chronic obstructive pulmonary disease: State of the art. Expert Rev. Respir. Med. 2011, 5, 647–662. [Google Scholar] [CrossRef]
- Kabata, H.; Artis, D. Neuro-immune crosstalk and allergic inflammation. J. Clin. Investig. 2019, 129, 1475–1482. [Google Scholar] [CrossRef]
- Chang, R.B.; Strochlic, D.E.; Williams, E.K.; Umans, B.D.; Liberles, S.D. Vagal sensory neuron subtypes that differentially control breathing. Cell 2015, 161, 622–633. [Google Scholar] [CrossRef]
- Carr, M.J.; Undem, B.J. Bronchopulmonary afferent nerves. Respirology 2003, 8, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Guyenet, P.G.; Stornetta, R.L.; Bayliss, D.A. Central respiratory chemoreception. J. Comp. Neurol. 2010, 518, 3883–3906. [Google Scholar] [CrossRef]
- Canning, B.J.; Mori, N.; Mazzone, S.B. Vagal afferent nerves regulating the cough reflex. Respir. Physiol. Neurobiol. 2006, 152, 223–242. [Google Scholar] [CrossRef]
- Tränkner, D.; Hahne, N.; Sugino, K.; Hoon, M.A.; Zuker, C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc. Natl. Acad. Sci. USA 2014, 111, 11515–11520. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.; Abdulnour, R.E.; Burkett, P.R.; Lee, S.; Cronin, S.J.; Pascal, M.A.; Laedermann, C.; Foster, S.L.; Tran, J.V.; Lai, N.; et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 2015, 87, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance p in inflammatory disease. J. Cell Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide nmu amplifies ilc2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Green, D.P.; Limjunyawong, N.; Gour, N.; Pundir, P.; Dong, X. A mast-cell-specific receptor mediates neurogenic inflammation and pain. Neuron 2019, 101, 412–420.e3. [Google Scholar] [CrossRef] [PubMed]
- Héron, A.; Dubayle, D. A focus on mast cells and pain. J. Neuroimmunol. 2013, 264, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Babina, M. Mrgprx2 signals its importance in cutaneous mast cell biology: Does mrgprx2 connect mast cells and atopic dermatitis? Exp. Dermatol. 2020, 29, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lee, J.H.; Won, H.K.; Kang, Y.; Song, W.J.; Kwon, H.S.; Cho, Y.S.; Moon, H.B.; Kim, T.B. Clinical significance of serum mrgprx2 as a new biomarker in allergic asthma. Allergy 2020, 75, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Manorak, W.; Idahosa, C.; Gupta, K.; Roy, S.; Panettieri, R., Jr.; Ali, H. Upregulation of mas-related g protein coupled receptor x2 in asthmatic lung mast cells and its activation by the novel neuropeptide hemokinin-1. Respir. Res. 2018, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human mast cell proteome reveals unique lineage, putative functions, and structural basis for cell ablation. Immunity 2020, 52, 404–416.e5. [Google Scholar] [CrossRef]
- Grassin-Delyle, S.; Naline, E.; Buenestado, A.; Risse, P.A.; Sage, E.; Advenier, C.; Devillier, P. Expression and function of human hemokinin-1 in human and guinea pig airways. Respir. Res. 2010, 11, 139. [Google Scholar] [CrossRef]
- Sumpter, T.L.; Ho, C.H.; Pleet, A.R.; Tkacheva, O.A.; Shufesky, W.J.; Rojas-Canales, D.M.; Morelli, A.E.; Larregina, A.T. Autocrine hemokinin-1 functions as an endogenous adjuvant for IgE-mediated mast cell inflammatory responses. J. Allergy Clin. Immunol. 2015, 135, 1019–1030.e8. [Google Scholar] [CrossRef]
- Amin, K.; Janson, C.; Bystrom, J. Role of eosinophil granulocytes in allergic airway inflammation endotypes. Scand. J. Immunol. 2016, 84, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, A.; Hosoki, K.; Toda, M.; Miyake, Y.; Matsushima, Y.; Matsumoto, T.; Boveda-Ruiz, D.; Gil-Bernabe, P.; Nagao, M.; Sugimoto, M.; et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE 2013, 8, e64281. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Gupta, K.; Guo, Q.; Price, R.; Ali, H. Mas-related gene x2 (mrgx2) is a novel g protein-coupled receptor for the antimicrobial peptide ll-37 in human mast cells: Resistance to receptor phosphorylation, desensitization, and internalization. J. Biol. Chem. 2011, 286, 44739–44749. [Google Scholar] [CrossRef]
- Duits, L.A.; Nibbering, P.H.; van Strijen, E.; Vos, J.B.; Mannesse-Lazeroms, S.P.; van Sterkenburg, M.A.; Hiemstra, P.S. Rhinovirus increases human beta-defensin-2 and -3 mrna expression in cultured bronchial epithelial cells. FEMS Immunol. Med. Microbiol. 2003, 38, 59–64. [Google Scholar] [CrossRef]
- Thapaliya, M.; Chompunud Na Ayudhya, C.; Amponnawarat, A.; Roy, S.; Ali, H. Mast cell-specific mrgprx2: A key modulator of neuro-immune interaction in allergic diseases. Curr. Allergy Asthma Rep. 2021, 21, 3. [Google Scholar] [CrossRef]
- Perner, C.; Flayer, C.H.; Zhu, X.; Aderhold, P.A.; Dewan, Z.N.A.; Voisin, T.; Camire, R.B.; Chow, O.A.; Chiu, I.M.; Sokol, C.L. Substance p release by sensory neurons triggers dendritic cell migration and initiates the type-2 immune response to allergens. Immunity 2020, 53, 1063–1077.e7. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. Il-33/il-31 axis in immune-mediated and allergic diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef]
- Ip, W.K.; Wong, C.K.; Li, M.L.; Li, P.W.; Cheung, P.F.; Lam, C.W. Interleukin-31 induces cytokine and chemokine production from human bronchial epithelial cells through activation of mitogen-activated protein kinase signalling pathways: Implications for the allergic response. Immunology 2007, 122, 532–541. [Google Scholar] [CrossRef]
- Kasraie, S.; Niebuhr, M.; Baumert, K.; Werfel, T. Functional effects of interleukin 31 in human primary keratinocytes. Allergy 2011, 66, 845–852. [Google Scholar] [CrossRef]
- Huang, J.; Yue, H.; Jiang, T.; Gao, J.; Shi, Y.; Shi, B.; Wu, X.; Gou, X. Il-31 plays dual roles in lung inflammation in an ova-induced murine asthma model. Biol. Open 2019, 8, bio036244. [Google Scholar] [CrossRef]
- Feld, M.; Garcia, R.; Buddenkotte, J.; Katayama, S.; Lewis, K.; Muirhead, G.; Hevezi, P.; Plesser, K.; Schrumpf, H.; Krjutskov, K.; et al. The pruritus- and th2-associated cytokine il-31 promotes growth of sensory nerves. J. Allergy Clin. Immunol. 2016, 138, 500–508.e4. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.I.; Han, W.C.; Yun, K.J.; Moon, H.B.; Oh, G.J.; Chae, S.C. Identifying polymorphisms in il-31 and their association with susceptibility to asthma. Korean J. Pathol. 2012, 46, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, K.; Irie, H. Interleukin-31 as a clinical target for pruritus treatment. Front. Med. 2021, 8, 638325. [Google Scholar] [CrossRef]
- Sui, P.; Wiesner, D.L.; Xu, J.; Zhang, Y.; Lee, J.; Van Dyken, S.; Lashua, A.; Yu, C.; Klein, B.S.; Locksley, R.M.; et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 2018, 360, eaan8546. [Google Scholar] [CrossRef]
- Undem, B.J.; Kollarik, M. The role of vagal afferent nerves in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Coleridge, J.C.; Coleridge, H.M. Afferent vagal c fibre innervation of the lungs and airways and its functional significance. Rev. Physiol. Biochem. Pharmacol. 1984, 99, 1–110. [Google Scholar]
- Kollarik, M.; Undem, B.J. Mechanisms of acid-induced activation of airway afferent nerve fibres in guinea-pig. J. Physiol. 2002, 543, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Vanzella, L.M.; Bernardo, A.F.B.; Carvalho, T.D.; Vanderlei, F.M.; Silva, A.; Vanderlei, L.C.M. Complexity of autonomic nervous system function in individuals with copd. J. Bras. Pneumol. 2018, 44, 24–30. [Google Scholar] [CrossRef]
- Blake, K.J.; Jiang, X.R.; Chiu, I.M. Neuronal regulation of immunity in the skin and lungs. Trends Neurosci. 2019, 42, 537–551. [Google Scholar] [CrossRef]
- Burian, B.; Storka, A.; Marzluf, B.A.; Yen, Y.C.; Lambers, C.; Robibaro, B.; Vonbank, K.; Mosgoeller, W.; Petkov, V. Vasoactive intestinal peptide (vip) receptor expression in monocyte-derived macrophages from copd patients. Peptides 2010, 31, 603–608. [Google Scholar] [CrossRef]
- Mandal, J.; Roth, M.; Costa, L.; Boeck, L.; Rakic, J.; Scherr, A.; Tamm, M.; Stolz, D. Vasoactive intestinal peptide for diagnosing exacerbation in chronic obstructive pulmonary disease. Respiration 2015, 90, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Miotto, D.; Boschetto, P.; Bononi, I.; Zeni, E.; Cavallesco, G.; Fabbri, L.M.; Mapp, C.E. Vasoactive intestinal peptide receptors in the airways of smokers with chronic bronchitis. Eur. Respir. J. 2004, 24, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Kolahian, S.; Gosens, R. Cholinergic regulation of airway inflammation and remodelling. J. Allergy 2012, 2012, 681258. [Google Scholar] [CrossRef]
- Kistemaker, L.E.; van Os, R.P.; Dethmers-Ausema, A.; Bos, I.S.; Hylkema, M.N.; van den Berge, M.; Hiemstra, P.S.; Wess, J.; Meurs, H.; Kerstjens, H.A.; et al. Muscarinic M3 receptors on structural cells regulate cigarette smoke-induced neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L96–L103. [Google Scholar] [CrossRef] [PubMed]
- Suvas, S. Role of substance p neuropeptide in inflammation, wound healing, and tissue homeostasis. J. Immunol. 2017, 199, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- DeRose, V.; Robbins, R.A.; Snider, R.M.; Spurzem, J.R.; Thiele, G.M.; Rennard, S.I.; Rubinstein, I. Substance p increases neutrophil adhesion to bronchial epithelial cells. J. Immunol. 1994, 152, 1339–1346. [Google Scholar]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Bullone, M.; Carriero, V.; Bertolini, F.; Folino, A.; Mannelli, A.; Di Stefano, A.; Gnemmi, I.; Torchio, R.; Ricciardolo, F.L.M. Elevated serum IgE, oral corticosteroid dependence and il-17/22 expression in highly neutrophilic asthma. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Gu, X.; Karp, P.H.; Brody, S.L.; Pierce, R.A.; Welsh, M.J.; Holtzman, M.J.; Ben-Shahar, Y. Chemosensory functions for pulmonary neuroendocrine cells. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 637–646. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, M.; Wang, X. Changes of calcitonin gene-related peptide content in induced sputum from patients with copd and asthma. Zhonghua Jie He He Hu Xi Za Zhi 1999, 22, 558–561. [Google Scholar]
- Lagomarsino, V.N.; Kostic, A.D.; Chiu, I.M. Mechanisms of microbial-neuronal interactions in pain and nociception. Neurobiol. Pain 2021, 9, 100056. [Google Scholar] [CrossRef]
- Borson, D.B.; Brokaw, J.J.; Sekizawa, K.; McDonald, D.M.; Nadel, J.A. Neutral endopeptidase and neurogenic inflammation in rats with respiratory infections. J. Appl. Physiol. 1989, 66, 2653–2658. [Google Scholar] [CrossRef]
- Hewitt, R.; Farne, H.; Ritchie, A.; Luke, E.; Johnston, S.L.; Mallia, P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther. Adv. Respir. Dis. 2016, 10, 158–174. [Google Scholar] [CrossRef]
- Linden, D.; Guo-Parke, H.; Coyle, P.V.; Fairley, D.; McAuley, D.F.; Taggart, C.C.; Kidney, J. Respiratory viral infection: A potential “missing link” in the pathogenesis of copd. Eur. Respir. Rev. 2019, 28, 180063. [Google Scholar] [CrossRef]
- Ghildyal, R.; Dagher, H.; Donninger, H.; de Silva, D.; Li, X.; Freezer, N.J.; Wilson, J.W.; Bardin, P.G. Rhinovirus infects primary human airway fibroblasts and induces a neutrophil chemokine and a permeability factor. J. Med. Virol. 2005, 75, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Griego, S.D.; Weston, C.B.; Adams, J.L.; Tal-Singer, R.; Dillon, S.B. Role of p38 mitogen-activated protein kinase in rhinovirus-induced cytokine production by bronchial epithelial cells. J. Immunol. 2000, 165, 5211–5220. [Google Scholar] [CrossRef]
- Message, S.D.; Laza-Stanca, V.; Mallia, P.; Parker, H.L.; Zhu, J.; Kebadze, T.; Contoli, M.; Sanderson, G.; Kon, O.M.; Papi, A.; et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and th1/2 cytokine and il-10 production. Proc. Natl. Acad. Sci. USA 2008, 105, 13562–13567. [Google Scholar] [CrossRef]
- Yamaya, M.; Sekizawa, K.; Suzuki, T.; Yamada, N.; Furukawa, M.; Ishizuka, S.; Nakayama, K.; Terajima, M.; Numazaki, Y.; Sasaki, H. Infection of human respiratory submucosal glands with rhinovirus: Effects on cytokine and icam-1 production. Am. J. Physiol. 1999, 277, L362–L371. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, H.; Heaney, L.G.; Cosby, S.L.; McGarvey, L.P. Rhinovirus upregulates transient receptor potential channels in a human neuronal cell line: Implications for respiratory virus-induced cough reflex sensitivity. Thorax 2014, 69, 46–54. [Google Scholar] [CrossRef]
- Grissell, T.V.; Chang, A.B.; Gibson, P.G. Reduced toll-like receptor 4 and substance p gene expression is associated with airway bacterial colonization in children. Pediatr. Pulmonol. 2007, 42, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Doyle, W.J.; Gentile, D.A.; Cohen, S. Emotional style, nasal cytokines, and illness expression after experimental rhinovirus exposure. Brain Behav. Immun. 2006, 20, 175–181. [Google Scholar] [CrossRef]
- Huang, Y.; Smith, D.E.; Ibáñez-Sandoval, O.; Sims, J.E.; Friedman, W.J. Neuron-specific effects of interleukin-1β are mediated by a novel isoform of the il-1 receptor accessory protein. J. Neurosci. 2011, 31, 18048–18059. [Google Scholar] [CrossRef]
- Herrmann, O.; Tarabin, V.; Suzuki, S.; Attigah, N.; Coserea, I.; Schneider, A.; Vogel, J.; Prinz, S.; Schwab, S.; Monyer, H.; et al. Regulation of body temperature and neuroprotection by endogenous interleukin-6 in cerebral ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Penkowa, M.; Giralt, M.; Carrasco, J.; Hadberg, H.; Hidalgo, J. Impaired inflammatory response and increased oxidative stress and neurodegeneration after brain injury in interleukin-6-deficient mice. Glia 2000, 32, 271–285. [Google Scholar] [CrossRef]
- Hedrich, C.M.; Bruck, N.; Paul, D.; Hahn, G.; Gahr, M.; Rösen-Wolff, A. “Mutation negative” familial cold autoinflammatory syndrome (fcas) in an 8-year-old boy: Clinical course and functional studies. Rheumatol. Int. 2012, 32, 2629–2636. [Google Scholar] [CrossRef]
- Barragán-Iglesias, P.; Franco-Enzástiga, Ú.; Jeevakumar, V.; Shiers, S.; Wangzhou, A.; Granados-Soto, V.; Campbell, Z.T.; Dussor, G.; Price, T.J. Type I interferons act directly on nociceptors to produce pain sensitization: Implications for viral infection-induced pain. J. Neurosci. 2020, 40, 3517–3532. [Google Scholar] [CrossRef] [PubMed]
- Kormuth, K.A.; Lin, K.; Qian, Z.; Myerburg, M.M.; Marr, L.C.; Lakdawala, S.S. Environmental persistence of influenza viruses is dependent upon virus type and host origin. mSphere 2019, 4, e00552-19. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Murcia, P.R.; Holmes, E.C. Influenza virus reservoirs and intermediate hosts: Dogs, horses, and new possibilities for influenza virus exposure of humans. J. Virol. 2015, 89, 2990–2994. [Google Scholar] [CrossRef]
- Grebe, K.M.; Takeda, K.; Hickman, H.D.; Bailey, A.L.; Embry, A.C.; Bennink, J.R.; Yewdell, J.W. Cutting edge: Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza a virus pathogenesis. J. Immunol. 2010, 184, 540–544. [Google Scholar] [CrossRef]
- Hosseini, S.; Wilk, E.; Michaelsen-Preusse, K.; Gerhauser, I.; Baumgärtner, W.; Geffers, R.; Schughart, K.; Korte, M. Long-term neuroinflammation induced by influenza a virus infection and the impact on hippocampal neuron morphology and function. J. Neurosci. 2018, 38, 3060–3080. [Google Scholar] [CrossRef]
- Ural, B.B.; Yeung, S.T.; Damani-Yokota, P.; Devlin, J.C.; de Vries, M.; Vera-Licona, P.; Samji, T.; Sawai, C.M.; Jang, G.; Perez, O.A.; et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci. Immunol. 2020, 5, eaax8756. [Google Scholar] [CrossRef]
- Verzele, N.A.J.; Chua, B.Y.; Law, C.W.; Zhang, A.; Ritchie, M.E.; Wightman, O.; Edwards, I.N.; Hulme, K.D.; Bloxham, C.J.; Bielefeldt-Ohmann, H.; et al. The impact of influenza pulmonary infection and inflammation on vagal bronchopulmonary sensory neurons. FASEB J. 2021, 35, e21320. [Google Scholar] [CrossRef]
- Forbester, J.L.; Humphreys, I.R. Genetic influences on viral-induced cytokine responses in the lung. Mucosal Immunol. 2021, 14, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Galic, M.A.; Kentner, A.C.; Reid, A.Y.; Sharkey, K.A.; Pittman, Q.J. Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J. Neurosci. 2015, 35, 4942–4952. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Jorde, I.; Kershaw, O.; Jeron, A.; Bruder, D.; Schreiber, J.; Stegemann-Koniszewski, S. Resolved influenza a virus infection has extended effects on lung homeostasis and attenuates allergic airway inflammation in a mouse model. Microorganisms 2020, 8, 1878. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory syncytial virus--a comprehensive review. Clin. Rev. Allergy Immunol. 2013, 45, 331–379. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Xia, M.; Ingram, D.A.; Kishiyama, J.L.; Kaltreider, H.B.; Byrd, P.K.; Ichikawa, S.; Sreedharan, S.P. Neuropeptide signaling of lymphocytes in immunological responses. Int. Arch. Allergy Immunol. 1995, 107, 202–204. [Google Scholar] [CrossRef]
- Maggi, C.A. The effects of tachykinins on inflammatory and immune cells. Regul. Pept. 1997, 70, 75–90. [Google Scholar] [CrossRef]
- Piedimonte, G.; Rodriguez, M.M.; King, K.A.; McLean, S.; Jiang, X. Respiratory syncytial virus upregulates expression of the substance p receptor in rat lungs. Am. J. Physiol. 1999, 277, L831–L840. [Google Scholar] [CrossRef]
- Ho, W.Z.; Lai, J.P.; Zhu, X.H.; Uvaydova, M.; Douglas, S.D. Human monocytes and macrophages express substance p and neurokinin-1 receptor. J. Immunol. 1997, 159, 5654–5660. [Google Scholar]
- Lai, J.P.; Douglas, S.D.; Ho, W.Z. Human lymphocytes express substance p and its receptor. J. Neuroimmunol. 1998, 86, 80–86. [Google Scholar] [CrossRef]
- Panuska, J.R.; Merolla, R.; Rebert, N.A.; Hoffmann, S.P.; Tsivitse, P.; Cirino, N.M.; Silverman, R.H.; Rankin, J.A. Respiratory syncytial virus induces interleukin-10 by human alveolar macrophages. Suppression of early cytokine production and implications for incomplete immunity. J. Clin. Investig. 1995, 96, 2445–2453. [Google Scholar] [CrossRef] [PubMed]
- Panuska, J.R.; Midulla, F.; Cirino, N.M.; Villani, A.; Gilbert, I.A.; McFadden, E.R., Jr.; Huang, Y.T. Virus-induced alterations in macrophage production of tumor necrosis factor and prostaglandin e2. Am. J. Physiol. 1990, 259, L396–L402. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Humbert, M.; Hanania, N.A.; Zhang, N.; Holgate, S.; Buhl, R.; Bröker, B.M. Staphylococcus aureus and its IgE-inducing enterotoxins in asthma: Current knowledge. Eur. Respir. J. 2020, 55, 1901592. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R. The microbiota, chemical symbiosis, and human disease. J. Mol. Biol. 2014, 426, 3877–3891. [Google Scholar] [CrossRef]
- Jorde, I.; Hildebrand, C.B.; Kershaw, O.; Lücke, E.; Stegemann-Koniszewski, S.; Schreiber, J. Modulation of allergic sensitization and allergic inflammation by Staphylococcus aureus enterotoxin b in an ovalbumin mouse model. Front. Immunol. 2020, 11, 592186. [Google Scholar] [CrossRef]
- Baral, P.; Umans, B.D.; Li, L.; Wallrapp, A.; Bist, M.; Kirschbaum, T.; Wei, Y.; Zhou, Y.; Kuchroo, V.K.; Burkett, P.R.; et al. Nociceptor sensory neurons suppress neutrophil and γδ t cell responses in bacterial lung infections and lethal pneumonia. Nat. Med. 2018, 24, 417–426. [Google Scholar] [CrossRef]
- N’Diaye, A.; Mijouin, L.; Hillion, M.; Diaz, S.; Konto-Ghiorghi, Y.; Percoco, G.; Chevalier, S.; Lefeuvre, L.; Harmer, N.J.; Lesouhaitier, O.; et al. Effect of substance p in Staphylococcus aureus and Staphylococcus epidermidis virulence: Implication for skin homeostasis. Front. Microbiol. 2016, 7, 506. [Google Scholar] [CrossRef]
- Sio, S.W.; Puthia, M.K.; Lu, J.; Moochhala, S.; Bhatia, M. The neuropeptide substance p is a critical mediator of burn-induced acute lung injury. J. Immunol. 2008, 180, 8333–8341. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Asthma | COPD | Respiratory Infections | |
|---|---|---|---|
| SP | |||
| VIP |
| ||
| PNECs |
| ||
| CGRP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camp, B.; Stegemann-Koniszewski, S.; Schreiber, J. Infection-Associated Mechanisms of Neuro-Inflammation and Neuro-Immune Crosstalk in Chronic Respiratory Diseases. Int. J. Mol. Sci. 2021, 22, 5699. https://doi.org/10.3390/ijms22115699
Camp B, Stegemann-Koniszewski S, Schreiber J. Infection-Associated Mechanisms of Neuro-Inflammation and Neuro-Immune Crosstalk in Chronic Respiratory Diseases. International Journal of Molecular Sciences. 2021; 22(11):5699. https://doi.org/10.3390/ijms22115699
Chicago/Turabian StyleCamp, Belinda, Sabine Stegemann-Koniszewski, and Jens Schreiber. 2021. "Infection-Associated Mechanisms of Neuro-Inflammation and Neuro-Immune Crosstalk in Chronic Respiratory Diseases" International Journal of Molecular Sciences 22, no. 11: 5699. https://doi.org/10.3390/ijms22115699
APA StyleCamp, B., Stegemann-Koniszewski, S., & Schreiber, J. (2021). Infection-Associated Mechanisms of Neuro-Inflammation and Neuro-Immune Crosstalk in Chronic Respiratory Diseases. International Journal of Molecular Sciences, 22(11), 5699. https://doi.org/10.3390/ijms22115699