
Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
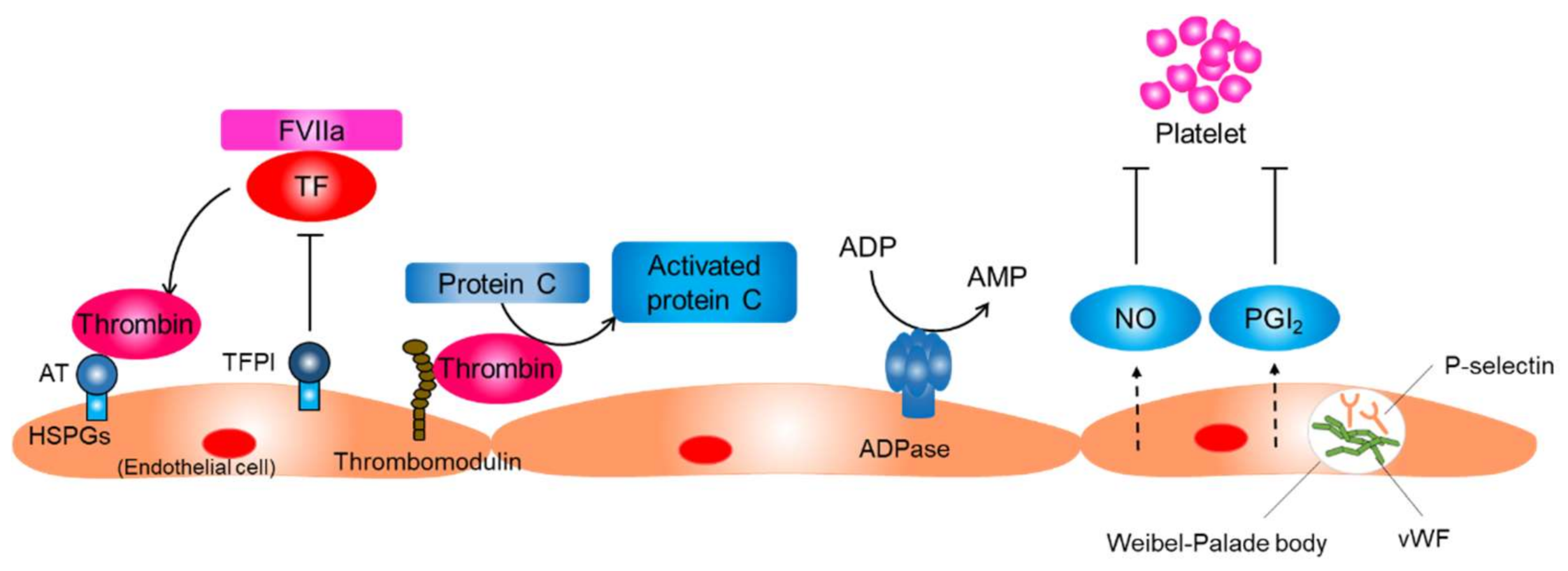
2. Functions of Resting Endothelial Cells
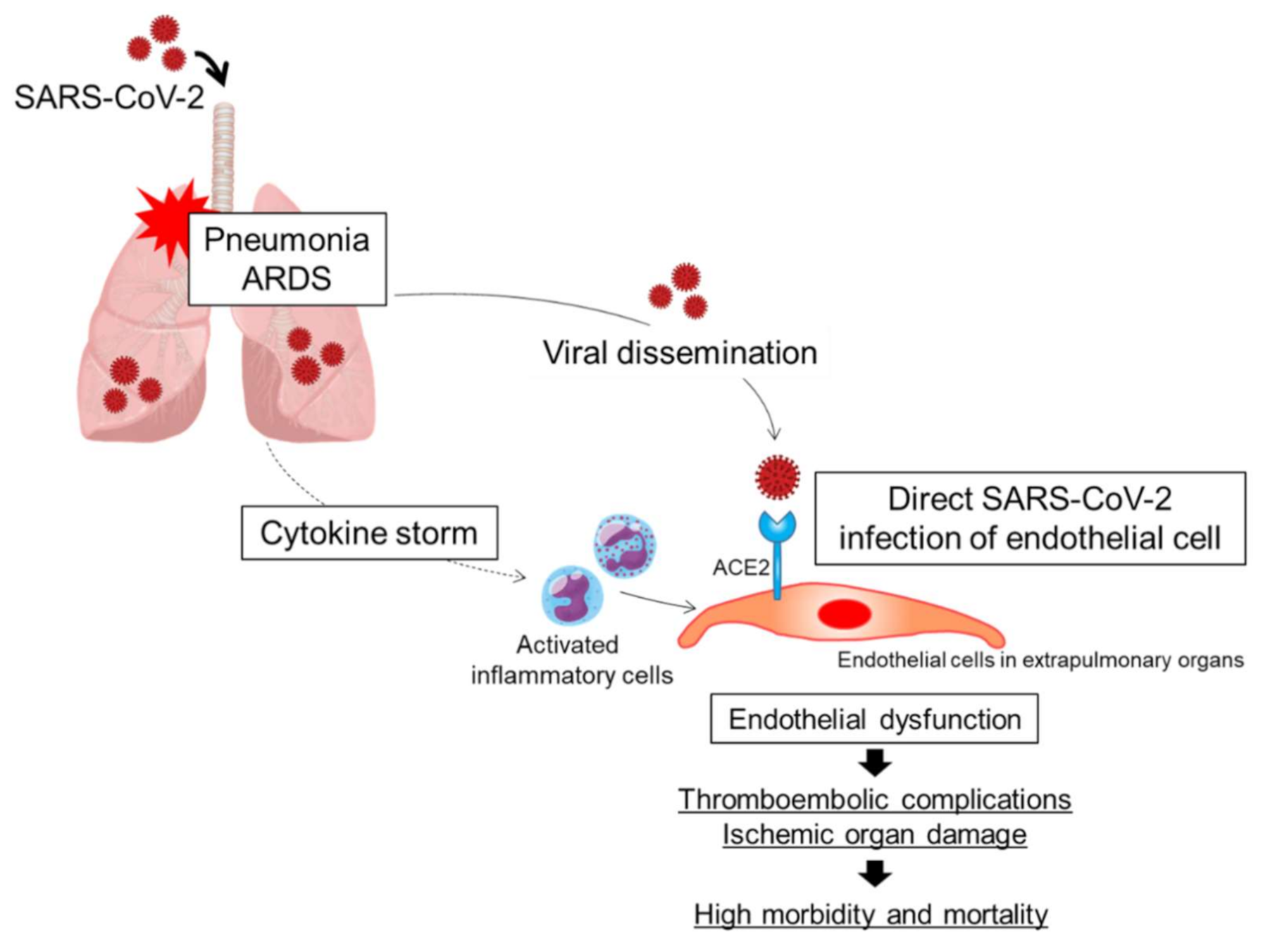
3. Possibility of Direct SARS-Cov-2 Infection of Endothelial Cells
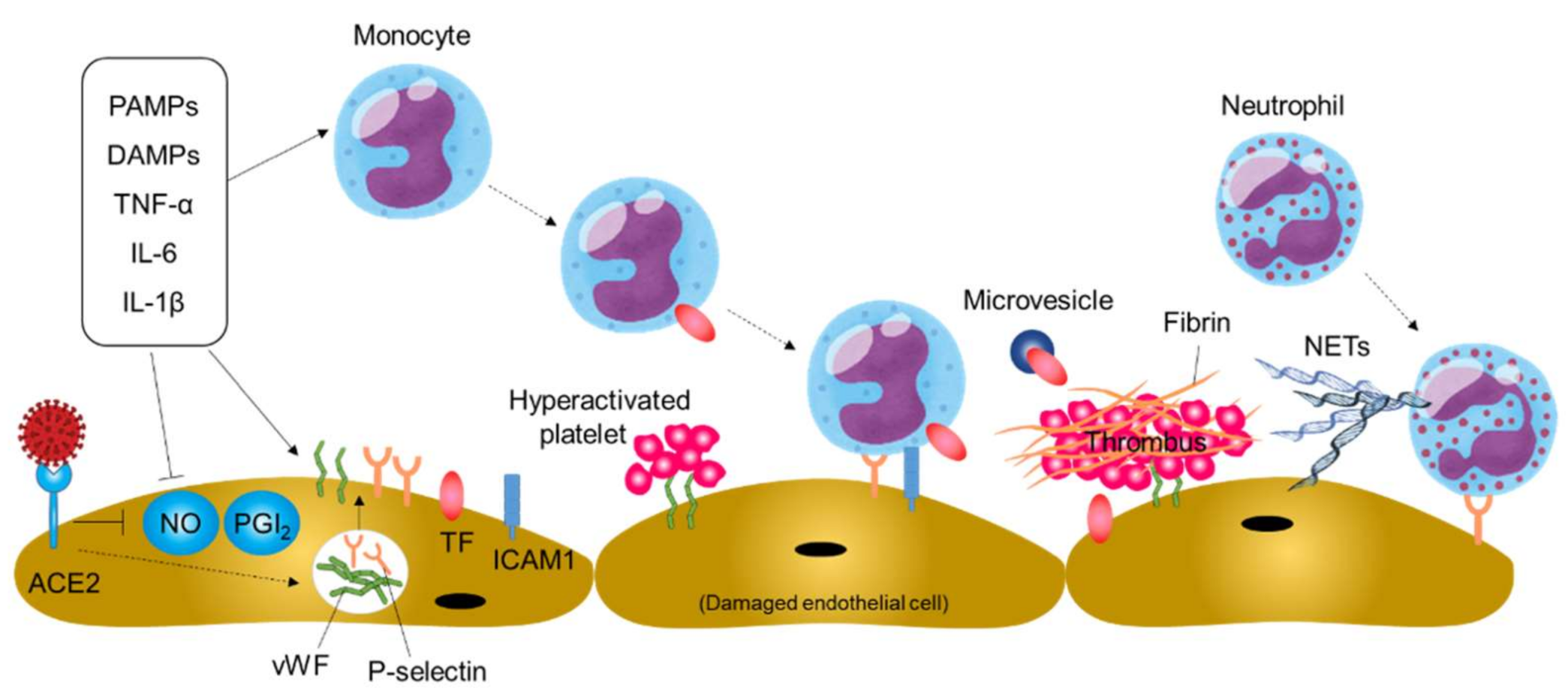
4. Downregulation of ACE2 Expression and Endothelial Dysfunction in COVID-19
5. Thromboinflammation and Endothelial Dysfunction In COVID-19
6. Endothelial Dysfunction in the Lung
7. Endothelial Dysfunction in Extrapulmonary Blood Vessels
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Wichmann, D. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann. Intern. Med. 2020, 173, 1030. [Google Scholar] [CrossRef]
- Thomas, W.; Varley, J.; Johnston, A.; Symington, E.; Robinson, M.; Sheares, K.; Lavinio, A.; Besser, M. Thrombotic complications of patients admitted to intensive care with COVID-19 at a teaching hospital in the United Kingdom. Thromb. Res. 2020, 191, 76–77. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 8, 1421–1424. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results From a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020, 173, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Vane, J.R.; Anggard, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar] [PubMed]
- Vallance, P.; Collier, J.; Moncada, S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989, 2, 997–1000. [Google Scholar] [CrossRef]
- Vanhoutte, P.M. Endothelium and control of vascular function. State of the Art lecture. Hypertension 1989, 13 Pt 2, 658–667. [Google Scholar] [CrossRef]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. Off. J. Jpn. Circ. Soc. 2009, 73, 411–418. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Arnout, J.; Hoylaerts, M.F.; Lijnen, H.R. Haemostasis. Handb. Exp. Pharmacol. 2006, 176, 1–41. [Google Scholar]
- Hoffman, M. Remodeling the blood coagulation cascade. J. Thromb. Thrombolysis 2003, 16, 17–20. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Reutershan, J. Leucocyte-endothelial interactions in health and disease. Handb. Exp. Pharmacol. 2006, 176 Pt 2, 97–133. [Google Scholar]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Morrell, C.N.; Cambien, B.; Yang, S.X.; Yamakuchi, M.; Bao, C.; Hara, M.R.; Quick, R.A.; Cao, W.; O’Rourke, B.; et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 2003, 115, 139–150. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, J.; Montezano, A.C.; Touyz, R.M. ACE2/Ang-(1-7)/Mas1 axis and the vascular system: Vasoprotection to COVID-19-associated vascular disease. Clin. Sci. 2021, 135, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Matarese, A.; Gambardella, J.; Sardu, C.; Santulli, G. miR-98 Regulates TMPRSS2 Expression in Human Endothelial Cells: Key Implications for COVID-19. Biomedicines 2020, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Ahmetaj-Shala, B.; Vaja, R.; Atanur, S.S.; George, P.M.; Kirkby, N.S.; Mitchell, J.A. Cardiorenal Tissues Express SARS-CoV-2 Entry Genes and Basigin (BSG/CD147) Increases With Age in Endothelial Cells. JACC Basic Transl. Sci. 2020, 5, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Dittmayer, C.; Meinhardt, J.; Radbruch, H.; Radke, J.; Heppner, B.I.; Heppner, F.L.; Stenzel, W.; Holland, G.; Laue, M. Why misinterpretation of electron micrographs in SARS-CoV-2-infected tissue goes viral. Lancet 2020, 396, e64–e65. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Colmenero, I.; Santonja, C.; Alonso-Riano, M.; Noguera-Morel, L.; Hernandez-Martin, A.; Andina, D.; Wiesner, T.; Rodriguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020, 183, 729–737. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, H.C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffre, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Miller, S.E.; Martines, R.B.; Bullock, H.A.; Zaki, S.R. Electron microscopy of SARS-CoV-2: A challenging task. Lancet 2020, 395, e99. [Google Scholar] [CrossRef]
- Baeck, M.; Hoton, D.; Marot, L.; Herman, A. Chilblains and COVID-19: Why SARS-CoV-2 endothelial infection is questioned. Br. J. Dermatol. 2020, 183, 1152–1153. [Google Scholar] [CrossRef]
- Brealey, J.K.; Miller, S.E. SARS-CoV-2 has not been detected directly by electron microscopy in the endothelium of chilblain lesions. Br. J. Dermatol. 2021, 184, 186. [Google Scholar] [CrossRef] [PubMed]
- Dittmayer, C.; Meinhardt, J.; Radbruch, H.; Radke, J.; Heppner, B.I.; Heppner, F.L.; Stenzel, W.; Holland, G.; Laue, M. Using EM data to understand COVID-19 pathophysiology—Authors’ reply. Lancet 2021, 397, 197–198. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod. Pathol. Off. J.U.S. Can. Acad. Pathol. Inc 2020, 33, 2104–2114. [Google Scholar] [CrossRef]
- Bhatnagar, J.; Gary, J.; Reagan-Steiner, S.; Estetter, L.B.; Tong, S.; Tao, Y.; Denison, A.M.; Lee, E.; DeLeon-Carnes, M.; Li, Y.; et al. Evidence of SARS-CoV-2 Replication and Tropism in the Lungs, Airways and Vascular Endothelium of Patients with Fatal COVID-19: An Autopsy Case-Series. J. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 receptors: Regulation, signalling and function. Blood Press. 2003, 12, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Sampaio, W.; Alzamora, A.; Motta-Santos, D.; Alenina, N.; Bader, M. The ACE2/Angiotensin-(1-7)/MAS1 axis of the Renin-angiotensin System: Focus on angiotensin-(1-7). Physiol. Rev 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Touyz, R.M.; Montezano, A.C. Angiotensin-(1-7) and Vascular Function: The Clinical Context. Hypertension 2018, 71, 68–69. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Dijkman, R.; Jebbink, M.F.; Deijs, M.; Milewska, A.; Pyrc, K.; Buelow, E.; van der Bijl, A.; van der Hoek, L. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. J. Gen. Virol. 2012, 93 Pt 9, 1924–1929. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, B.; Philips, M.I.; Mehta, J.L. Proapoptotic effects of ANG II in human coronary artery endothelial cells: Role of AT1 receptor and PKC activation. Am. J. Physiol. 1999, 276, H786–H792. [Google Scholar] [CrossRef]
- Lopez-Farre, A.; de Miguel, S.L.; Monton, M.; Jimenez, A.; Lopez-Bloya, A.; Gomez, J.; Nunez, A.; Rico, L.; Casado, S. Angiotensin II AT(1) receptor antagonists and platelet activation. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2001, 16 (Suppl. 1), 45–49. [Google Scholar]
- Keidar, S.; Kaplan, M.; Gamliel-Lazarovich, A. ACE2 of the heart: From angiotensin I to angiotensin (1-7). Cardiovasc. Res. 2007, 73, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.K.; Fujii, S.; Sawa, H.; Goto, D.; Ishimori, N.; Watano, K.; Kaneko, T.; Furumoto, T.; Sugawara, T.; Sakuma, I.; et al. Angiotensin-converting enzyme inhibition attenuates hypofibrinolysis and reduces cardiac perivascular fibrosis in genetically obese diabetic mice. Circulation 2001, 103, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef]
- Scholz, A.; Plate, K.H.; Reiss, Y. Angiopoietin-2: A multifaceted cytokine that functions in both angiogenesis and inflammation. Ann. N.Y. Acad. Sci. 2015, 1347, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [PubMed]
- De Mello, W.C. Chemical Communication between Heart Cells is Disrupted by Intracellular Renin and Angiotensin II: Implications for Heart Development and Disease. Front. Endocrinol. 2015, 6, 72. [Google Scholar] [CrossRef]
- Roche, J.A.; Roche, R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 7265–7269. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Figueroa, C.D.; Matus, C.E.; Pavicic, F.; Sarmiento, J.; Hidalgo, M.A.; Burgos, R.A.; Gonzalez, C.B.; Bhoola, K.D.; Ehrenfeld, P. Kinin B1 receptor regulates interactions between neutrophils and endothelial cells by modulating the levels of Mac-1, LFA-1 and intercellular adhesion molecule-1. Innate Immun. 2015, 21, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Lopatko Fagerstrom, I.; Stahl, A.L.; Mossberg, M.; Tati, R.; Kristoffersson, A.C.; Kahn, R.; Bascands, J.L.; Klein, J.; Schanstra, J.P.; Segelmark, M.; et al. Blockade of the kallikrein-kinin system reduces endothelial complement activation in vascular inflammation. EBioMedicine 2019, 47, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Alon, R.; Sportiello, M.; Kozlovski, S.; Kumar, A.; Reilly, E.C.; Zarbock, A.; Garbi, N.; Topham, D.J. Leukocyte trafficking to the lungs and beyond: Lessons from influenza for COVID-19. Nat. Rev. Immunol. 2021, 21, 49–64. [Google Scholar] [CrossRef]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Raman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bormikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifeatations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Masi, P.; Hekimian, G.; Lejeune, M.; Chommeloux, J.; Desnos, C.; De Chambrun, M.P.; Martin-Toutain, I.; Nieszkowska, A.; Lebreton, G.; Brechot, N.; et al. Systemic Inflammatory Response Syndrome Is a Major Contributor to COVID-19–Associated Coagulopathy: Insights from a Prospective, Single-Center Cohort Study. Circulation 2020, 142, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y.J. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1580. [Google Scholar] [CrossRef]
- Holt, P.G.; Strickland, D.H.; Wikstrom, M.E.; Jahnsen, F.L. Regulation of immunological homeostasis in the respiratory tract. Nat. Rev. Immunol. 2008, 8, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, T. COVID-19: Towards understanding of pathogenesis. Cell Res. 2020, 30, 367–369. [Google Scholar] [CrossRef]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M. Vascular Endothelial Glycocalyx Damage in COVID-19. Int. J. Mol. Sci. 2020, 21, 9712. [Google Scholar] [CrossRef] [PubMed]
- Polidoro, R.B.; Hagan, R.S.; de Santis, S.R.; Schmidt, N.W. Overview: Systemic Inflammatory Response Derived From Lung Injury Caused by SARS-CoV-2 Infection Explains Severe Outcomes in COVID-19. Front. Immunol. 2020, 11, 1626. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maruhashi, T.; Higashi, Y. Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19. Int. J. Mol. Sci. 2021, 22, 5131. https://doi.org/10.3390/ijms22105131
Maruhashi T, Higashi Y. Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19. International Journal of Molecular Sciences. 2021; 22(10):5131. https://doi.org/10.3390/ijms22105131
Chicago/Turabian StyleMaruhashi, Tatsuya, and Yukihito Higashi. 2021. "Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19" International Journal of Molecular Sciences 22, no. 10: 5131. https://doi.org/10.3390/ijms22105131
APA StyleMaruhashi, T., & Higashi, Y. (2021). Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19. International Journal of Molecular Sciences, 22(10), 5131. https://doi.org/10.3390/ijms22105131