Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections


Abstract
1. Introduction
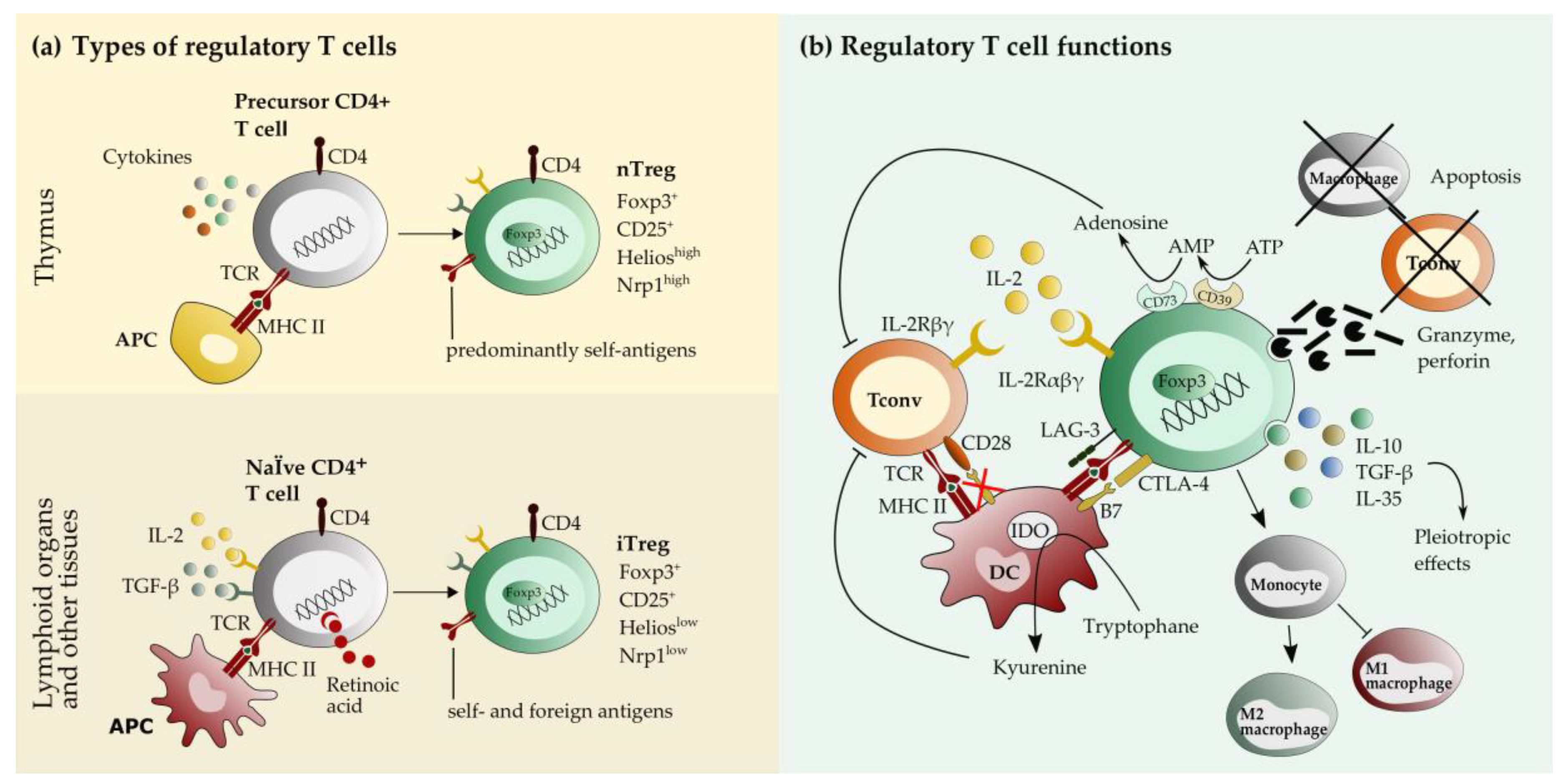
2. Biology of Regulatory T cells
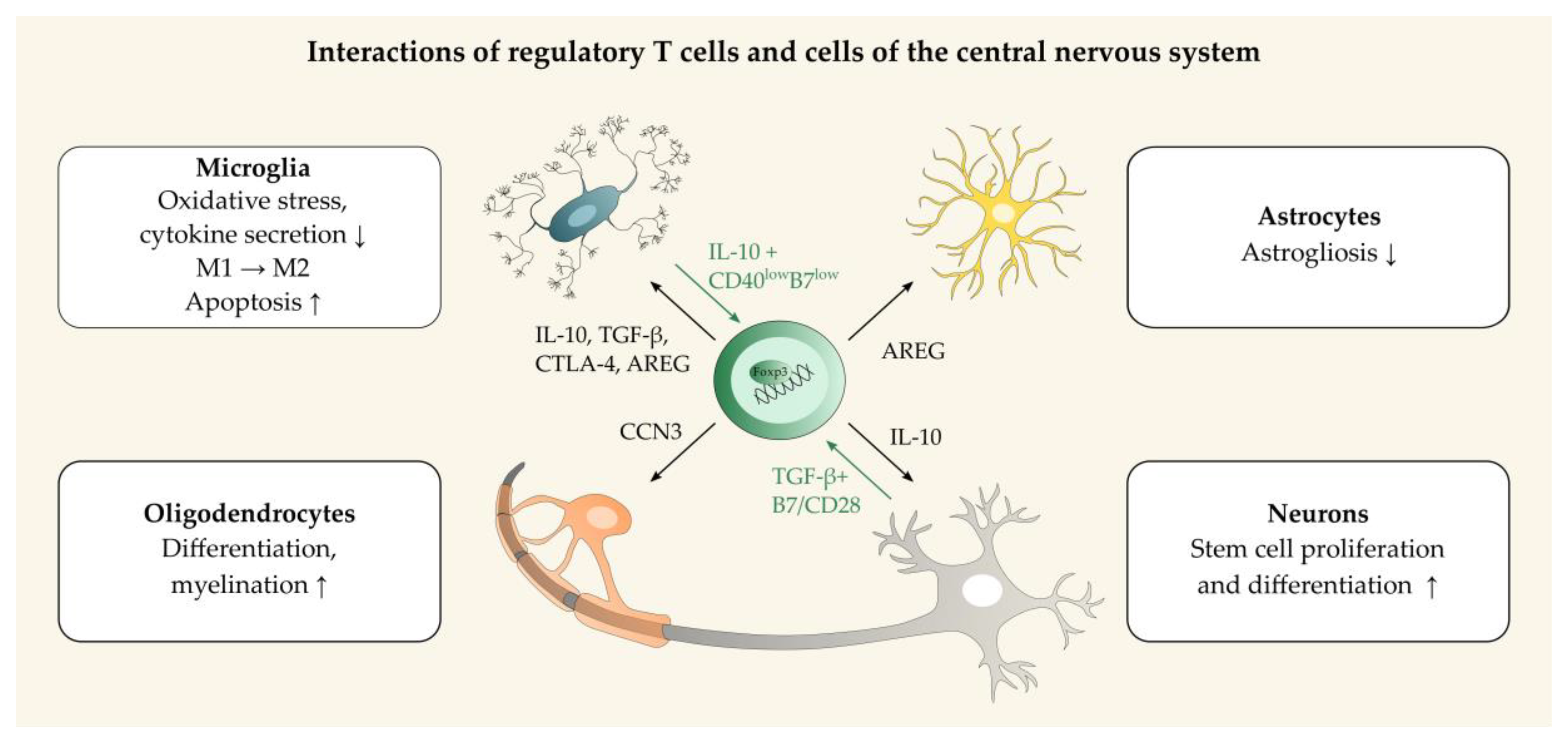
3. Regulatory T Cells in Neuroinflammation and Neuroprotection
4. Regulatory T Cells in Virus Infection of the Nervous System
4.1. Role of Regulatory T Cells in Viral Encephalitis of Humans and their Animal Models
4.1.1. Regulatory T Cells Show Neuroprotective and Antiviral Effects in Retroviral Encephalitis
4.1.2. Regulatory T Cells Inhibit Antiviral Immunity and Facilitate Virus Latency and Spread, but also Protect from Excessive Immunopathology in Herpesvirus Infection
Involvement of Regulatory T Cells in the Establishment and Reactivation of Latency
Involvement of Regulatory T Cells in Herpesvirus Spread to the Central Nervous System
Involvement of Regulatory T Cells in the Suppression of Immunopathology
4.1.3. Regulatory T Cells Protect from Excessive Immunopathology in Acute Flaviviral Encephalitis, but Might Be Involved in the Establishment of Neuroinvasion
Role of Regulatory T Cells in West Nile Virus Infection
Role of Regulatory T Cells in Japanese Encephalitis Virus Infection
Role of Regulatory T Cells in Zika virus Infection
4.1.4. Regulatory T Cells Inhibit Antiviral Immunity in Persistent Measles Virus Infection of the Central Nervous System
4.1.5. Other Viruses
4.2. Regulatory T Cells in Animal Models for Virus-Induced Demyelinating Disorders
4.2.1. Theiler’s murine encephalomyelitis virus model
4.2.2. Coronavirus Model of Demyelination
5. Conclusions and Outlook
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | adenosine monophosphate |
| AREG | amphiregulin |
| Asm | acid sphingomyelinase |
| ATP | adenosine triphosphate |
| BDNF | brain-derived neurotrophic factor |
| CC | collaborative cross |
| CCL, CXCL | chemokine ligand |
| CCN3 | cellular network communication factor 3 |
| CCR, CXCR | chemokine receptor |
| CD | cluster of differentiation |
| CLN | cervical lymph node |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| CTLA-4 | cytotoxic T-lymphocyte-associated Protein 4 |
| DC | dendritic cell |
| DEREG | depletion of regulatory T cells |
| DTR | diphtheria toxin receptor |
| EAE | experimental autoimmune encephalomyelitis |
| EBV | Epstein Barr virus |
| Foxp3 | forkhead box protein P3 |
| GDNF | glial cell line-derived neurotrophic factor |
| GFP | green fluorescent protein |
| GITR | glucocorticoid-induced TNFR-related protein |
| HAND | HIV-1 associated neurocognitive disorder |
| HIV | human immunodeficiency virus |
| HSV | herpes simplex virus |
| ICOS | inducible T cell costimulator |
| IDO-1 | indoleamine 2,3-dioxygenase 1 |
| IFN | interferon |
| IgM | immunglobulin M |
| IL | interleukin |
| IL-2C | IL-2-anti-IL-2-antibody complexes |
| iNOS | inducible nitric oxide synthase |
| IPEX | immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome |
| iTreg | induced regulatory T cell |
| JEV | Japanese encephalitis virus |
| KLRG1 | killer cell lectin-like receptor subfamily G member 1 |
| LAG3 | lymphocyte activation gene 3 |
| MCMV | murine cytomegalovirus encephalitis |
| MHC | major histocompatibility complex |
| MHV | mouse hepatitis virus |
| MS | multiple sclerosis |
| MV | measles virus |
| NMDA | N-methyl-D-aspartate |
| NPC | neural precursor cells |
| nTreg | natural regulatory T cell |
| PD-L1 | programmed death-ligand 1 |
| RAG | recombination-activating gene |
| SSPE | subacute sclerotizing panencephalitis |
| STAT | signal transducer and activator of transcription |
| Tconv | conventional T cell |
| TCR | T cell receptor |
| TGF | transforming growth factor |
| TLR | toll-like receptor |
| TMEV | Theiler’s murine encephalomyelitis virus |
| TMEV-IDD | Theiler’s murine encephalomyelitis virus-induced demyelinating disease |
| TNF | tumor necrosis factor |
| TNFR | tumor necrosis factor receptor |
| Tr1 | type 1 regulatory T cell |
| Treg | regulatory T cell |
| VZV | Varizella-zoster virus |
| WNV | West Nile virus |
References
- Broer, S.; Hage, E.; Kaufer, C.; Gerhauser, I.; Anjum, M.; Li, L.; Baumgärtner, W.; Schulz, T.F.; Loscher, W. Viral mouse models of multiple sclerosis and epilepsy: Marked differences in neuropathogenesis following infection with two naturally occurring variants of Theiler’s virus BeAn strain. Neurobiol. disease 2017, 99, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.M.; Blumcke, I.; Sander, J.W.; Loscher, W. Infections, inflammation and epilepsy. Acta. Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; Kortekaas, J.; Herden, C.; Hoffmann, B.; Tappe, D.; Trebst, C.; Griffin, D.E.; Brindle, H.E.; Solomon, T.; Brown, A.S.; et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta. Neuropathol. 2016, 131, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hua, S.; Chen, H.R.; Ouyang, Z.; Einkauf, K.; Tse, S.; Ard, K.; Ciaranello, A.; Yawetz, S.; Sax, P.; et al. Transcriptional Changes during Naturally Acquired Zika Virus Infection Render Dendritic Cells Highly Conducive to Viral Replication. Cell Rep. 2017, 21, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Martines, R.B.; Bhatnagar, J.; de Oliveira Ramos, A.M.; Davi, H.P.; Iglezias, S.D.; Kanamura, C.T.; Keating, M.K.; Hale, G.; Silva-Flannery, L.; Muehlenbachs, A.; et al. Pathology of congenital Zika syndrome in Brazil: A case series. Lancet 2016, 388, 898–904. [Google Scholar] [CrossRef]
- Martines, R.B.; Bhatnagar, J.; Keating, M.K.; Silva-Flannery, L.; Muehlenbachs, A.; Gary, J.; Goldsmith, C.; Hale, G.; Ritter, J.; Rollin, D.; et al. Notes from the Field: Evidence of Zika Virus Infection in Brain and Placental Tissues from Two Congenitally Infected Newborns and Two Fetal Losses--Brazil, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 159–160. [Google Scholar] [CrossRef]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N Engl J Med 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Hayes, E.B.; Sejvar, J.J.; Zaki, S.R.; Lanciotti, R.S.; Bode, A.V.; Campbell, G.L. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1174–1179. [Google Scholar] [CrossRef]
- Miyake, M. The Pathology of Japanese Encephalitis. A Review. Bull World Health Organ. 1964, 30, 153–160. [Google Scholar]
- Rhodes, R.H. Histopathology of the central nervous system in the acquired immunodeficiency syndrome. Hum. Pathol. 1987, 18, 636–643. [Google Scholar] [CrossRef]
- Morishima, T.; Togashi, T.; Yokota, S.; Okuno, Y.; Miyazaki, C.; Tashiro, M.; Okabe, N.; Collaborative Study Group on Influenza-Associated Encephalopathy in, J. Encephalitis and encephalopathy associated with an influenza epidemic in Japan. Clin Infect Dis 2002, 35, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.F.; Li, W.; Li, K. Acute encephalopathy and encephalitis caused by influenza virus infection. Curr. Opin. Neurol. 2010, 23, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Itoh, M.; Mizuguchi, M.; Ozawa, H.; Okazaki, E.; Kobayashi, Y.; Takahashi, M.; Ohtani, K.; Ogawa, A.; Narita, M.; et al. Apoptosis and microglial activation in influenza encephalopathy. Acta. Neuropathol. 2003, 105, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Bitnun, A.; Shannon, P.; Durward, A.; Rota, P.A.; Bellini, W.J.; Graham, C.; Wang, E.; Ford-Jones, E.L.; Cox, P.; Becker, L.; et al. Measles inclusion-body encephalitis caused by the vaccine strain of measles virus. Clin. Infect. Dis. 1999, 29, 855–861. [Google Scholar] [CrossRef]
- Budka, H.; Urbanits, S.; Liberski, P.P.; Eichinger, S.; Popow-Kraupp, T. Subacute measles virus encephalitis: A new and fatal opportunistic infection in a patient with AIDS. Neurology 1996, 46, 586–587. [Google Scholar] [CrossRef]
- ter Meulen, V.; Muller, D.; Kackell, Y.; Katz, M.; Meyermann, R. Isolation of infectious measles virus in measles encephalitis. Lancet 1972, 2, 1172–1175. [Google Scholar] [CrossRef]
- Ong, K.C.; Wong, K.T. Henipavirus Encephalitis: Recent Developments and Advances. Brain. Pathol. 2015, 25, 605–613. [Google Scholar] [CrossRef]
- Donohue, W.L.; Playfair, F.D.; Whitaker, L. Mumps encephalitis; pathology and pathogenesis. J. Pediatr. 1955, 47, 395–412. [Google Scholar] [CrossRef]
- Farahtaj, F.; Alizadeh, L.; Gholami, A.; Tahamtan, A.; Shirian, S.; Fazeli, M.; Nejad, A.S.M.; Gorji, A.; Niknam, H.M.; Ghaemi, A. Natural Infection with Rabies Virus: A Histopathological and Immunohistochemical Study of Human Brains. Osong. Public Health Res. Perspect 2019, 10, 6–11. [Google Scholar] [CrossRef]
- Tappe, D.; Schmidt-Chanasit, J.; Rauch, J.; Allartz, P.; Herden, C. Immunopathology of Fatal Human Variegated Squirrel Bornavirus 1 Encephalitis, Germany, 2011–2013. Emerg. Infect. Dis. 2019, 25, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Tappe, D.; Schmidt-Chanasit, J.; Beer, M. Bornavirus Associated with Fatal Human Encephalitis. N. Engl. J. Med. 2015, 373, 1880–1881. [Google Scholar] [PubMed]
- Lad, E.M.; Ong, S.S.; Proia, A.D. Ocular histopathology in Eastern equine encephalitis: A case report. Am. J. Ophthalmol. Case Rep. 2017, 5, 99–102. [Google Scholar] [CrossRef] [PubMed]
- McJunkin, J.E.; Khan, R.; de los Reyes, E.C.; Parsons, D.L.; Minnich, L.L.; Ashley, R.G.; Tsai, T.F. Treatment of severe La Crosse encephalitis with intravenous ribavirin following diagnosis by brain biopsy. Pediatrics 1997, 99, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Zarafonetis, C.J.; Smadel, J.E. Fatal Herpes Simplex Encephalitis in Man. Am. J. Pathol. 1944, 20, 429–445. [Google Scholar]
- Raman, L.; Nelson, M. Cerebral vasculitis and encephalitis due to Epstein-Barr virus in a patient with newly diagnosed HIV infection. J. Clin. Virol. 2014, 59, 264–267. [Google Scholar] [CrossRef]
- Sabat, S.; Agarwal, A.; Zacharia, T.; Labib, S.; Yousef, J. Epstein-Barr virus encephalitis presenting as cerebellar hemorrhage. Neuroradiol J. 2015, 28, 555–558. [Google Scholar] [CrossRef]
- Kano, K.; Katayama, T.; Takeguchi, S.; Asanome, A.; Takahashi, K.; Saito, T.; Sawada, J.; Saito, M.; Anei, R.; Kamada, K.; et al. Biopsy-proven case of Epstein-Barr virus (EBV)-associated vasculitis of the central nervous system. Neuropathol. 2017, 37, 259–264. [Google Scholar] [CrossRef]
- Amlie-Lefond, C.; Kleinschmidt-DeMasters, B.K.; Mahalingam, R.; Davis, L.E.; Gilden, D.H. The vasculopathy of varicella-zoster virus encephalitis. Ann. Neurol. 1995, 37, 784–790. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Amlie-Lefond, C.; Gilden, D.H. The patterns of varicella zoster virus encephalitis. Hum. Pathol. 1996, 27, 927–938. [Google Scholar] [CrossRef]
- Bar-Or, A.; Pender, M.P.; Khanna, R.; Steinman, L.; Hartung, H.P.; Maniar, T.; Croze, E.; Aftab, B.T.; Giovannoni, G.; Joshi, M.J. Epstein-Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol. Med. 2020, 26, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Leitzen, E.; Raddatz, B.B.; Jin, W.; Goebbels, S.; Nave, K.A.; Baumgärtner, W.; Hansmann, F. Virus-triggered spinal cord demyelination is followed by a peripheral neuropathy resembling features of Guillain-Barre Syndrome. Sci. Rep. 2019, 9, 4588. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef]
- Hasenkrug, K.J.; Chougnet, C.A.; Dittmer, U. Regulatory T cells in retroviral infections. PLoS Pathog. 2018, 14, e1006776. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Rouse, B.T. Natural regulatory T cells in infectious disease. Nat. Immunol. 2005, 6, 353–360. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Deng, G.; Song, X.; Fujimoto, S.; Piccirillo, C.A.; Nagai, Y.; Greene, M.I. Foxp3 Post-translational Modifications and Treg Suppressive Activity. Front. Immunol. 2019, 10, 2486. [Google Scholar] [CrossRef]
- Kempkes, R.W.M.; Joosten, I.; Koenen, H.; He, X. Metabolic Pathways Involved in Regulatory T Cell Functionality. Front. Immunol. 2019, 10, 2839. [Google Scholar] [CrossRef]
- Wing, J.B.; Tanaka, A.; Sakaguchi, S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 2019, 50, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Tarbell, K. Regulatory T cells in the control of host-microorganism interactions (*). Annu. Rev. Immunol. 2009, 27, 551–589. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Parga, T.; Sehrawat, S.; Rouse, B.T. Role of regulatory T cells during virus infection. Immunol. Rev. 2013, 255, 182–196. [Google Scholar] [CrossRef]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef]
- D’Cruz, L.M.; Klein, L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat. Immunol. 2005, 6, 1152–1159. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Rasmussen, J.P.; Gavin, M.A.; Rudensky, A.Y. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 2005, 6, 1142–1151. [Google Scholar] [CrossRef]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Gasteiger, G.; Subramanian, N.; Sparwasser, T.; Busch, D.H.; Belkaid, Y.; Drexler, I.; Germain, R.N. Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction. J. Immunol. 2011, 187, 3186–3197. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Wing, J.B.; Sakaguchi, S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin. Immunol. 2011, 23, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, P.; Charbonnier, L.M.; Chatila, T.A. Regulatory T Cells: The Many Faces of Foxp3. J. Clin. Immunol. 2019, 39, 623–640. [Google Scholar] [CrossRef]
- Huang, X.; Stone, D.K.; Yu, F.; Zeng, Y.; Gendelman, H.E. Functional proteomic analysis for regulatory T cell surveillance of the HIV-1-infected macrophage. J. Proteome. Res. 2010, 9, 6759–6773. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Muschaweckh, A. Stability and Maintenance of Foxp3(+) Treg Cells in Non-lymphoid Microenvironments. Front. Immunol. 2019, 10, 2634. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Komai, K.; Nakamura, T.; Srirat, T.; Yoshimura, A. Tissue regulatory T cells and neural repair. Int. Immunol. 2019, 31, 361–369. [Google Scholar] [CrossRef]
- Lahl, K.; Loddenkemper, C.; Drouin, C.; Freyer, J.; Arnason, J.; Eberl, G.; Hamann, A.; Wagner, H.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007, 204, 57–63. [Google Scholar] [CrossRef]
- Kim, J.M.; Rasmussen, J.P.; Rudensky, A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007, 8, 191–197. [Google Scholar] [CrossRef]
- Kohm, A.P.; McMahon, J.S.; Podojil, J.R.; Begolka, W.S.; DeGutes, M.; Kasprowicz, D.J.; Ziegler, S.F.; Miller, S.D. Cutting Edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J. Immunol. 2006, 176, 3301–3305. [Google Scholar] [CrossRef]
- Webster, K.E.; Walters, S.; Kohler, R.E.; Mrkvan, T.; Boyman, O.; Surh, C.D.; Grey, S.T.; Sprent, J. In vivo expansion of T reg cells with IL-2-mAb complexes: Induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009, 206, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.W.; Teo, T.H.; Lum, F.M.; Andiappan, A.K.; Amrun, S.N.; Renia, L.; Rotzschke, O.; Ng, L.F. Virus infection drives IL-2 antibody complexes into pro-inflammatory agonists in mice. Sci. Rep. 2016, 6, 37603. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.L.; Wiendl, H. The role of regulatory T cells in multiple sclerosis. Nat. Clin. Pract. Neurol. 2008, 4, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Negi, N.; Das, B.K. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int. Rev. Immunol. 2018, 37, 57–68. [Google Scholar] [CrossRef]
- Lowther, D.E.; Hafler, D.A. Regulatory T cells in the central nervous system. Immunol. Rev. 2012, 248, 156–169. [Google Scholar] [CrossRef]
- Sallusto, F.; Impellizzieri, D.; Basso, C.; Laroni, A.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B. T-cell trafficking in the central nervous system. Immunol. Rev. 2012, 248, 216–227. [Google Scholar] [CrossRef]
- Kipnis, J.; Mizrahi, T.; Hauben, E.; Shaked, I.; Shevach, E.; Schwartz, M. Neuroprotective autoimmunity: Naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc. Natl. Acad. Sci. USA 2002, 99, 15620–15625. [Google Scholar] [CrossRef]
- Liu, Y.; Teige, I.; Birnir, B.; Issazadeh-Navikas, S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat. Med. 2006, 12, 518–525. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Fett, C.; Trandem, K.; Fleming, E.; Perlman, S. IFN-gamma- and IL-10-expressing virus epitope-specific Foxp3(+) T reg cells in the central nervous system during encephalomyelitis. J. Exp. Med. 2011, 208, 1571–1577. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Stone, D.K.; Mosley, R.L.; Gendelman, H.E. Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+CD25+ T cells. J. Proteome Res. 2009, 8, 3497–3511. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Stone, D.K.; Mosley, R.L.; Gendelman, H.E. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J. Immunol. 2009, 182, 4137–4149. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur. J. Immunol. 2015, 45, 180–191. [Google Scholar] [CrossRef]
- Ebner, F.; Brandt, C.; Thiele, P.; Richter, D.; Schliesser, U.; Siffrin, V.; Schueler, J.; Stubbe, T.; Ellinghaus, A.; Meisel, C.; et al. Microglial activation milieu controls regulatory T cell responses. J. Immunol. 2013, 191, 5594–5602. [Google Scholar] [CrossRef]
- Beurel, E.; Harrington, L.E.; Buchser, W.; Lemmon, V.; Jope, R.S. Astrocytes modulate the polarization of CD4+ T cells to Th1 cells. PLoS ONE 2014, 9, e86257. [Google Scholar] [CrossRef]
- Trajkovic, V.; Vuckovic, O.; Stosic-Grujicic, S.; Miljkovic, D.; Popadic, D.; Markovic, M.; Bumbasirevic, V.; Backovic, A.; Cvetkovic, I.; Harhaji, L.; et al. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia 2004, 47, 168–179. [Google Scholar] [CrossRef]
- Korn, T.; Reddy, J.; Gao, W.; Bettelli, E.; Awasthi, A.; Petersen, T.R.; Backstrom, B.T.; Sobel, R.A.; Wucherpfennig, K.W.; Strom, T.B.; et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007, 13, 423–431. [Google Scholar] [CrossRef]
- O’Connor, R.A.; Malpass, K.H.; Anderton, S.M. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J. Immunol. 2007, 179, 958–966. [Google Scholar] [CrossRef]
- Gobel, K.; Bittner, S.; Melzer, N.; Pankratz, S.; Dreykluft, A.; Schuhmann, M.K.; Meuth, S.G.; Wiendl, H. CD4(+) CD25(+) FoxP3(+) regulatory T cells suppress cytotoxicity of CD8(+) effector T cells: Implications for their capacity to limit inflammatory central nervous system damage at the parenchymal level. J. Neuroinflamm. 2012, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, M.; Young, A.; O’Hagan, T.; Eleftheriadis, G.; Bankhead, P.; Dombrowski, Y.; Medina, R.J.; Fitzgerald, D.C. Characterization of a murine mixed neuron-glia model and cellular responses to regulatory T cell-derived factors. Mol. Brain. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- de la Vega Gallardo, N.; Dittmer, M.; Dombrowski, Y.; Fitzgerald, D.C. Regenerating CNS myelin: Emerging roles of regulatory T cells and CCN proteins. Neurochem. Int. 2019, 130, 104349. [Google Scholar] [CrossRef]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef]
- Wang, J.; Xie, L.; Yang, C.; Ren, C.; Zhou, K.; Wang, B.; Zhang, Z.; Wang, Y.; Jin, K.; Yang, G.Y. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front. Cell. Neurosci. 2015, 9, 361. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Dey, M.; Chang, A.; Lesniak, M.S. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front. Immunol. 2013, 4, 116. [Google Scholar] [CrossRef]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef]
- Walsh, J.T.; Kipnis, J. Regulatory T cells in CNS injury: The simple, the complex and the confused. Trends Mol. Med. 2011, 17, 541–547. [Google Scholar] [CrossRef]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E.; et al. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.; Liu, J.; Reynolds, A.D.; Gorantla, S.; Mosley, R.L.; Gendelman, H.E. Brain ingress of regulatory T cells in a murine model of HIV-1 encephalitis. J. Neuroimmunol. 2011, 230, 33–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, J.; Gong, N.; Huang, X.; Reynolds, A.D.; Mosley, R.L.; Gendelman, H.E. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J. Immunol. 2009, 182, 3855–3865. [Google Scholar] [CrossRef] [PubMed]
- Zelinskyy, G.; Dietze, K.; Sparwasser, T.; Dittmer, U. Regulatory T cells suppress antiviral immune responses and increase viral loads during acute infection with a lymphotropic retrovirus. PLoS Pathog. 2009, 5, e1000406. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef]
- Yu, W.; Geng, S.; Suo, Y.; Wei, X.; Cai, Q.; Wu, B.; Zhou, X.; Shi, Y.; Wang, B. Critical Role of Regulatory T Cells in the Latency and Stress-Induced Reactivation of HSV-1. Cell Rep. 2018, 25, 2379–2389.e3. [Google Scholar] [CrossRef]
- Fernandez, M.A.; Yu, U.; Zhang, G.; White, R.; Sparwasser, T.; Alexander, S.I.; Jones, C.A. Treg depletion attenuates the severity of skin disease from ganglionic spread after HSV-2 flank infection. Virology 2013, 447, 9–20. [Google Scholar] [CrossRef]
- Fernandez, M.A.; Puttur, F.K.; Wang, Y.M.; Howden, W.; Alexander, S.I.; Jones, C.A. T regulatory cells contribute to the attenuated primary CD8+ and CD4+ T cell responses to herpes simplex virus type 2 in neonatal mice. J. Immunol. 2008, 180, 1556–1564. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Beck, M.A. The dendritic and T cell responses to herpes simplex virus-1 are modulated by dietary vitamin E. Free Radic. Biol. Med. 2009, 46, 1581–1588. [Google Scholar] [CrossRef][Green Version]
- Lund, J.M.; Hsing, L.; Pham, T.T.; Rudensky, A.Y. Coordination of early protective immunity to viral infection by regulatory T cells. Science 2008, 320, 1220–1224. [Google Scholar] [CrossRef]
- Lokensgard, J.R.; Schachtele, S.J.; Mutnal, M.B.; Sheng, W.S.; Prasad, S.; Hu, S. Chronic reactive gliosis following regulatory T cell depletion during acute MCMV encephalitis. Glia 2015, 63, 1982–1996. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Hu, S.; Sheng, W.S.; Singh, A.; Lokensgard, J.R. Tregs Modulate Lymphocyte Proliferation, Activation, and Resident-Memory T-Cell Accumulation within the Brain during MCMV Infection. PLoS ONE 2015, 10, e0145457. [Google Scholar] [CrossRef] [PubMed]
- Lanteri, M.C.; O’Brien, K.M.; Purtha, W.E.; Cameron, M.J.; Lund, J.M.; Owen, R.E.; Heitman, J.W.; Custer, B.; Hirschkorn, D.F.; Tobler, L.H.; et al. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. clin. Invest. 2009, 119, 3266–3277. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.B.; Da Costa, A.; Lund, J.M. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J. Immunol. 2014, 192, 683–690. [Google Scholar] [CrossRef]
- Kim, J.H.; Patil, A.M.; Choi, J.Y.; Kim, S.B.; Uyangaa, E.; Hossain, F.M.; Park, S.Y.; Lee, J.H.; Eo, S.K. CCR5 ameliorates Japanese encephalitis via dictating the equilibrium of regulatory CD4(+)Foxp3(+) T and IL-17(+)CD4(+) Th17 cells. J. Neuroinflamm. 2016, 13, 223. [Google Scholar] [CrossRef]
- Reuter, D.; Sparwasser, T.; Hunig, T.; Schneider-Schaulies, J. Foxp3+ regulatory T cells control persistence of viral CNS infection. PLoS ONE 2012, 7, e33989. [Google Scholar] [CrossRef]
- Hollmann, C.; Werner, S.; Avota, E.; Reuter, D.; Japtok, L.; Kleuser, B.; Gulbins, E.; Becker, K.A.; Schneider-Schaulies, J.; Beyersdorf, N. Inhibition of Acid Sphingomyelinase Allows for Selective Targeting of CD4+ Conventional versus Foxp3+ Regulatory T Cells. J. Immunol. 2016, 197, 3130–3141. [Google Scholar] [CrossRef]
- Derfuss, T.; Segerer, S.; Herberger, S.; Sinicina, I.; Hufner, K.; Ebelt, K.; Knaus, H.G.; Steiner, I.; Meinl, E.; Dornmair, K.; et al. Presence of HSV-1 immediate early genes and clonally expanded T-cells with a memory effector phenotype in human trigeminal ganglia. Brain Pathol. 2007, 17, 389–398. [Google Scholar] [CrossRef]
- Varanasi, S.K.; Donohoe, D.; Jaggi, U.; Rouse, B.T. Manipulating Glucose Metabolism during Different Stages of Viral Pathogenesis Can Have either Detrimental or Beneficial Effects. J. Immunol. 2017, 199, 1748–1761. [Google Scholar] [CrossRef]
- Ramakrishna, C.; Kujawski, M.; Chu, H.; Li, L.; Mazmanian, S.K.; Cantin, E.M. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat. Commun. 2019, 10, 2153. [Google Scholar] [CrossRef]
- Mutnal, M.B.; Hu, S.; Schachtele, S.J.; Lokensgard, J.R. Infiltrating regulatory B cells control neuroinflammation following viral brain infection. J. Immunol. 2014, 193, 6070–6080. [Google Scholar] [CrossRef] [PubMed]
- Salimi, H.; Cain, M.D.; Klein, R.S. Encephalitic Arboviruses: Emergence, Clinical Presentation, and Neuropathogenesis. Neurotherapeutics 2016, 13, 514–534. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Beckham, J.D. West Nile Virus Encephalitis 16 Years Later. Brain Pathol. 2015, 25, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.; Tesh, R.B.; Fisher-Hoch, S. Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 2010, 201, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, H.; Signs, K.; Somsel, P.; Downes, F.P.; Clark, P.A.; Massey, J.P. Persistence of West Nile Virus (WNV) IgM antibodies in cerebrospinal fluid from patients with CNS disease. J. Clin. Virol. 2004, 31, 289–291. [Google Scholar] [CrossRef] [PubMed]
- James, E.A.; Gates, T.J.; LaFond, R.E.; Yamamoto, S.; Ni, C.; Mai, D.; Gersuk, V.H.; O’Brien, K.; Nguyen, Q.A.; Zeitner, B.; et al. Neuroinvasive West Nile Infection Elicits Elevated and Atypically Polarized T Cell Responses That Promote a Pathogenic Outcome. PLoS Pathog. 2016, 12, e1005375. [Google Scholar] [CrossRef] [PubMed]
- Netland, J.; Bevan, M.J. CD8 and CD4 T cells in west nile virus immunity and pathogenesis. Viruses 2013, 5, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, A.; Das, S.; Ghosh, S.; Mishra, M.K.; Sharma, V.; Koli, P.; Sen, E.; Basu, A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 2007, 55, 483–496. [Google Scholar] [CrossRef]
- Chen, C.J.; Ou, Y.C.; Lin, S.Y.; Raung, S.L.; Liao, S.L.; Lai, C.Y.; Chen, S.Y.; Chen, J.H. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J. Gen. Virol. 2010, 91, 1028–1037. [Google Scholar] [CrossRef]
- Graham, J.B.; Swarts, J.L.; Thomas, S.; Voss, K.M.; Sekine, A.; Green, R.; Ireton, R.C.; Gale, M.; Lund, J.M. Immune Correlates of Protection From West Nile Virus Neuroinvasion and Disease. J. Infect. Dis. 2019, 219, 1162–1171. [Google Scholar] [CrossRef]
- Graham, J.B.; Thomas, S.; Swarts, J.; McMillan, A.A.; Ferris, M.T.; Suthar, M.S.; Treuting, P.M.; Ireton, R.; Gale, M., Jr.; Lund, J.M. Genetic diversity in the collaborative cross model recapitulates human West Nile virus disease outcomes. mBio 2015, 6, e00493–15. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.B.; Swarts, J.L.; Wilkins, C.; Thomas, S.; Green, R.; Sekine, A.; Voss, K.M.; Ireton, R.C.; Mooney, M.; Choonoo, G.; et al. A Mouse Model of Chronic West Nile Virus Disease. PLoS Pathog. 2016, 12, e1005996. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.S.; Demarest, V.L.; Wong, S.J.; Green, S.; Bernard, K.A. Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 2011, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Luhn, K.; Simmons, C.P.; Moran, E.; Dung, N.T.; Chau, T.N.; Quyen, N.T.; Thao le, T.T.; Van Ngoc, T.; Dung, N.M.; Wills, B.; et al. Increased frequencies of CD4+ CD25(high) regulatory T cells in acute dengue infection. J. Exp. Med. 2007, 204, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.; McDermott, D.H.; Lisco, A.; Foster, G.A.; Krysztof, D.; Follmann, D.; Stramer, S.L.; Murphy, P.M. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 2010, 201, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.; Louie, C.Y.; Glaser, C.; Jean, C.; Johnson, B.; Johnson, H.; McDermott, D.H.; Murphy, P.M. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: A meta-analysis of 4 cohorts in the US epidemic. J. Infect. Dis. 2008, 197, 262–265. [Google Scholar] [CrossRef]
- Nazmi, A.; Mohamed Arif, I.; Dutta, K.; Kundu, K.; Basu, A. Neural stem/progenitor cells induce conversion of encephalitogenic T cells into CD4+-CD25+- FOXP3+ regulatory T cells. Viral. Immunol. 2014, 27, 48–59. [Google Scholar] [CrossRef]
- Cao, S.; Li, Y.; Ye, J.; Yang, X.; Chen, L.; Liu, X.; Chen, H. Japanese encephalitis Virus wild strain infection suppresses dendritic cells maturation and function, and causes the expansion of regulatory T cells. Virol. J. 2011, 8, 39. [Google Scholar] [CrossRef]
- Gupta, N.; Hegde, P.; Lecerf, M.; Nain, M.; Kaur, M.; Kalia, M.; Vrati, S.; Bayry, J.; Lacroix-Desmazes, S.; Kaveri, S.V. Japanese encephalitis virus expands regulatory T cells by increasing the expression of PD-L1 on dendritic cells. Eur. J. Immunol. 2014, 44, 1363–1374. [Google Scholar] [CrossRef]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef]
- Li, Y.; Ye, J.; Yang, X.; Xu, M.; Chen, L.; Mei, L.; Zhu, J.; Liu, X.; Chen, H.; Cao, S. Infection of mouse bone marrow-derived dendritic cells by live attenuated Japanese encephalitis virus induces cells maturation and triggers T cells activation. Vaccine 2011, 29, 855–862. [Google Scholar] [CrossRef]
- Han, Y.W.; Choi, J.Y.; Uyangaa, E.; Kim, S.B.; Kim, J.H.; Kim, B.S.; Kim, K.; Eo, S.K. Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog. 2014, 10, e1004319. [Google Scholar] [CrossRef]
- Elong Ngono, A.; Young, M.P.; Bunz, M.; Xu, Z.; Hattakam, S.; Vizcarra, E.; Regla-Nava, J.A.; Tang, W.W.; Yamabhai, M.; Wen, J.; et al. CD4+ T cells promote humoral immunity and viral control during Zika virus infection. PLoS Pathog. 2019, 15, e1007474. [Google Scholar]
- Moss, W.J. Measles. Lancet 2017, 390, 2490–2502. [Google Scholar] [CrossRef]
- Laksono, B.M.; de Vries, R.D.; Verburgh, R.J.; Visser, E.G.; de Jong, A.; Fraaij, P.L.A.; Ruijs, W.L.M.; Nieuwenhuijse, D.F.; van den Ham, H.J.; Koopmans, M.P.G.; et al. Studies into the mechanism of measles-associated immune suppression during a measles outbreak in the Netherlands. Nat. Commun. 2018, 9, 4944. [Google Scholar] [CrossRef]
- Yu, X.L.; Cheng, Y.M.; Shi, B.S.; Qian, F.X.; Wang, F.B.; Liu, X.N.; Yang, H.Y.; Xu, Q.N.; Qi, T.K.; Zha, L.J.; et al. Measles virus infection in adults induces production of IL-10 and is associated with increased CD4+ CD25+ regulatory T cells. J. Immunol. 2008, 181, 7356–7366. [Google Scholar] [CrossRef]
- Lin, W.H.; Kouyos, R.D.; Adams, R.J.; Grenfell, B.T.; Griffin, D.E. Prolonged persistence of measles virus RNA is characteristic of primary infection dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 14989–14994. [Google Scholar] [CrossRef]
- Sellin, C.I.; Jegou, J.F.; Renneson, J.; Druelle, J.; Wild, T.F.; Marie, J.C.; Horvat, B. Interplay between virus-specific effector response and Foxp3 regulatory T cells in measles virus immunopathogenesis. PLoS ONE 2009, 4, e4948. [Google Scholar] [CrossRef]
- Schubert, S.; Moller-Ehrlich, K.; Singethan, K.; Wiese, S.; Duprex, W.P.; Rima, B.K.; Niewiesk, S.; Schneider-Schaulies, J. A mouse model of persistent brain infection with recombinant Measles virus. J. Gen. Virol. 2006, 87, 2011–2019. [Google Scholar] [CrossRef]
- Yentur, S.P.; Gurses, C.; Demirbilek, V.; Adin-Cinar, S.; Kuru, U.; Uysal, S.; Yapici, Z.; Yilmaz, G.; Cokar, O.; Onal, E.; et al. A decrease of regulatory T cells and altered expression of NK receptors are observed in subacute sclerosing panencephalitis. Viral. Immunol. 2014, 27, 506–511. [Google Scholar] [CrossRef]
- Frisullo, G.; Iorio, R.; Plantone, D.; Nociti, V.; Patanella, A.K.; Marti, A.; Palermo, C.; Valentini, P.; Mariotti, P.; Batocchi, A.P. Involvement of type I immune responses in swine-origin H1N1 influenza virus infection. Hum. Immunol. 2011, 72, 632–635. [Google Scholar] [CrossRef]
- Ireland, D.D.; Tami, C.; Pedras-Vasconcelos, J.; Verthelyi, D. CD4 and CD8 T cells mediate distinct lethal meningoencephalitis in mice challenged with Tacaribe arenavirus. Cell. Mol. Immunol. 2017, 14, 90–107. [Google Scholar] [CrossRef]
- Gerhauser, I.; Hansmann, F.; Ciurkiewicz, M.; Loscher, W.; Beineke, A. Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Perlman, S.; Zhao, J. Roles of regulatory T cells and IL-10 in virus-induced demyelination. J. Neuroimmunol. 2017, 308, 6–11. [Google Scholar] [CrossRef]
- Herder, V.; Gerhauser, I.; Klein, S.K.; Almeida, P.; Kummerfeld, M.; Ulrich, R.; Seehusen, F.; Rohn, K.; Schaudien, D.; Baumgärtner, W.; et al. Interleukin-10 expression during the acute phase is a putative prerequisite for delayed viral elimination in a murine model for multiple sclerosis. J. Neuroimmunol. 2012, 249, 27–39. [Google Scholar] [CrossRef]
- Richards, M.H.; Getts, M.T.; Podojil, J.R.; Jin, Y.H.; Kim, B.S.; Miller, S.D. Virus expanded regulatory T cells control disease severity in the Theiler’s virus mouse model of MS. J. autoimmun. 2011, 36, 142–154. [Google Scholar] [CrossRef]
- Martinez, N.E.; Karlsson, F.; Sato, F.; Kawai, E.; Omura, S.; Minagar, A.; Grisham, M.B.; Tsunoda, I. Protective and detrimental roles for regulatory T cells in a viral model for multiple sclerosis. Brain Pathol. 2014, 24, 436–451. [Google Scholar] [CrossRef]
- Mestre, L.; Carrillo-Salinas, F.J.; Mecha, M.; Feliu, A.; Espejo, C.; Alvarez-Cermeno, J.C.; Villar, L.M.; Guaza, C. Manipulation of Gut Microbiota Influences Immune Responses, Axon Preservation, and Motor Disability in a Model of Progressive Multiple Sclerosis. Front. Immunol. 2019, 10, 1374. [Google Scholar] [CrossRef]
- Omura, S.; Sato, F.; Martinez, N.E.; Range, T.; Ekshyyan, L.; Minagar, A.; Alexander, J.S.; Tsunoda, I. Immunoregulation of Theiler’s virus-induced demyelinating disease by glatiramer acetate without suppression of antiviral immune responses. Arch. Virol. 2018, 163, 1279–1284. [Google Scholar] [CrossRef]
- Prajeeth, C.K.; Beineke, A.; Iskandar, C.D.; Gudi, V.; Herder, V.; Gerhauser, I.; Haist, V.; Teich, R.; Huehn, J.; Baumgärtner, W.; et al. Limited role of regulatory T cells during acute Theiler virus-induced encephalitis in resistant C57BL/6 mice. J. Neuroinflamm. 2014, 11, 180. [Google Scholar] [CrossRef]
- Ciurkiewicz, M.; Herder, V.; Khan, M.A.; Uhde, A.K.; Teich, R.; Floess, S.; Baumgärtner, W.; Huehn, J.; Beineke, A. Cytotoxic CD8(+) T cell ablation enhances the capacity of regulatory T cells to delay viral elimination in Theiler’s murine encephalomyelitis. Brain Pathol. 2018, 28, 349–368. [Google Scholar] [CrossRef]
- Uhde, A.K.; Ciurkiewicz, M.; Herder, V.; Khan, M.A.; Hensel, N.; Claus, P.; Beckstette, M.; Teich, R.; Floess, S.; Baumgärtner, W.; et al. Intact interleukin-10 receptor signaling protects from hippocampal damage elicited by experimental neurotropic virus infection of SJL mice. Sci. Rep. 2018, 8, 6106. [Google Scholar] [CrossRef]
- Uhde, A.K.; Herder, V.; Akram Khan, M.; Ciurkiewicz, M.; Schaudien, D.; Teich, R.; Floess, S.; Baumgärtner, W.; Huehn, J.; Beineke, A. Viral Infection of the Central Nervous System Exacerbates Interleukin-10 Receptor Deficiency-Mediated Colitis in SJL Mice. PLoS ONE 2016, 11, e0161883. [Google Scholar] [CrossRef]
- Anghelina, D.; Zhao, J.; Trandem, K.; Perlman, S. Role of regulatory T cells in coronavirus-induced acute encephalitis. Virology 2009, 385, 358–367. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Firner, S.; Bechmann, I.; Waisman, A.; Lahl, K.; Sparwasser, T.; Thiel, V.; Ludewig, B. Regulatory T cells selectively preserve immune privilege of self-antigens during viral central nervous system infection. J. Immunol. 2012, 188, 3678–3685. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Perlman, S. Virus-specific regulatory T cells ameliorate encephalitis by repressing effector T cell functions from priming to effector stages. PLoS Pathog. 2014, 10, e1004279. [Google Scholar] [CrossRef]
- de Aquino, M.T.; Puntambekar, S.S.; Savarin, C.; Bergmann, C.C.; Phares, T.W.; Hinton, D.R.; Stohlman, S.A. Role of CD25(+) CD4(+) T cells in acute and persistent coronavirus infection of the central nervous system. Virology 2013, 447, 112–120. [Google Scholar] [CrossRef]
- Trandem, K.; Zhao, J.; Fleming, E.; Perlman, S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J. Immunol. 2011, 186, 3642–3652. [Google Scholar] [CrossRef]
- Trandem, K.; Anghelina, D.; Zhao, J.; Perlman, S. Regulatory T cells inhibit T cell proliferation and decrease demyelination in mice chronically infected with a coronavirus. J. Immunol. 2010, 184, 4391–4400. [Google Scholar] [CrossRef]
- Savarin, C.; Bergmann, C.C.; Hinton, D.R.; Stohlman, S.A. Differential Regulation of Self-reactive CD4(+) T Cells in Cervical Lymph Nodes and Central Nervous System during Viral Encephalomyelitis. Front. Immunol. 2016, 7, 370. [Google Scholar] [CrossRef]
- Chen, L.; Coleman, R.; Leang, R.; Tran, H.; Kopf, A.; Walsh, C.M.; Sears-Kraxberger, I.; Steward, O.; Macklin, W.B.; Loring, J.F.; et al. Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem. Cell Rep. 2014, 2, 825–837. [Google Scholar] [CrossRef]
- Plaisted, W.C.; Zavala, A.; Hingco, E.; Tran, H.; Coleman, R.; Lane, T.E.; Loring, J.F.; Walsh, C.M. Remyelination Is Correlated with Regulatory T Cell Induction Following Human Embryoid Body-Derived Neural Precursor Cell Transplantation in a Viral Model of Multiple Sclerosis. PLoS ONE 2016, 11, e0157620. [Google Scholar] [CrossRef]
- Brinkmeyer-Langford, C.L.; Rech, R.; Amstalden, K.; Kochan, K.J.; Hillhouse, A.E.; Young, C.; Welsh, C.J.; Threadgill, D.W. Host genetic background influences diverse neurological responses to viral infection in mice. Sci. Rep. 2017, 7, 12194. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X. Foxp3 Instability Helps tTregs Distinguish Self and Non-self. Front. Immunol. 2019, 10, 2226. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Group | Virus Family | Virus Name | Disease Name | Pathologic Findings in the Central Nervous System (Gross Findings; Histologic Findings) | References |
|---|---|---|---|---|---|
| RNA viruses | Flaviviridae | Zika virus | Congenital Zika syndrome | Microcephaly, ventriculomegaly; mononuclear infiltrates, gliosis, calcification, neuronal necrosis | [4,5,6,7,8] |
| West Nile virus | West Nile encephalitis | Mononuclear infiltrates, gliosis, neuronal necrosis, neuronophagia, occasionally demyelination | [9] | ||
| Japanese encephalitis virus | Japanese encephalitis | Cerebral congestion and edema; mononuclear infiltrates, gliosis, necrosis, hemorrhages, neuronophagia | [10] | ||
| Retroviridae | Human immuno-deficiency virus (HIV) | HIV encephalitis | Mononuclear infiltrates, multinucleated giant cells, gliosis, neuronal loss, spongy myelinopathy, demyelination, vascular damage | [11] | |
| Orthomyxoviridae | Influenza virus | Influenza-associated acute encephalopathy | Cerebral edema and hemorrhage; neuronal apoptosis, necrosis, gliosis, occasionally mononuclear infiltrates | [12,13,14] | |
| Paramyxoviridae | Measles virus | Measles inclusion body encephalitis | Gliosis, intranuclear and cytoplasmic inclusion bodies in neurons and glial cells, mild mononuclear infiltration | [15,16,17] | |
| Subacute sclerosing panencephalitis | Cortical atrophy; mononuclear infiltrates, gliosis, neuronal necrosis and neuronophagia, intranuclear inclusion bodies (Cowdry type A) in neurons and oligodendrocytes, demyelination | [15,16,17] | |||
| Nipah virus, Hendra virus | Henipavirus encephalitis | Mononuclear infiltrates, vasculitis and thrombosis with infarctions, neuronophagia, gliosis, endothelial syncytia with inclusion bodies | [18] | ||
| Mumps virus | Mumps encephalitis | Mononuclear infiltrates, demyelination, gliosis, neuronal degeneration, hemorrhage, hyaline thrombi | [19] | ||
| Rhabdoviridae | Rabies virus | Rabies | Mononuclear infiltrates, gliosis, neuronal necrosis, neuronophagia, cytoplasmic inclusion bodies (Negri bodies) in neurons | [20] | |
| Bornaviridae | Variegated squirrel 1 bornavirus | Borna virus-associated encephalitis | Mononuclear infiltrates, gliosis, edema, necrosis, neuronal necrosis, neuronophagia | [21,22] | |
| Togaviridae | Eastern equine encephalitis virus | Eastern equine encephalitis | Mononuclear infiltrates, gliosis, infarcts with hemorrhages, myelin pallor and Purkinje cell loss in cerebellum | [23] | |
| Bunyaviridae | La Crosse virus | La Crosse encephalitis | Mononuclear infiltrates, gliosis | [24] | |
| DNA viruses | Herpesviridae | Herpes simplex virus | Herpes simplex encephalitis | Mononuclear infiltrates, hemorrhages, necrosis, intranuclear inclusion bodies (Cowdry type A) in neurons and glial cells | [25] |
| Epstein–Barr virus (EBV) | EBV-associated encephalitis/vasculitis | Mononuclear infiltrates, vascular fibrinoid necrosis, hemorrhage, occasionally demyelination | [26,27,28] | ||
| Varicella-zoster virus (VZV) | VZV encephalitis | Vasculopathy, vascular fibrinoid necrosis and thrombosis, necrosis, hemorrhagic infarcts, demyelination, intranuclear inclusion bodies (Cowdry type A) in glial and ependymal cells | [29,30] |
| Model | Genetic Background of Mice | Method of Regulatory T Cell Manipulation | Timeframe | Effects on Antiviral Immunity | Effects on Immunopathology | References |
|---|---|---|---|---|---|---|
| Retroviridae | ||||||
| HIV-1 encephalitis model | C57BL/6 | Adoptive Treg transfer; Treg co-culture in vitro | Acute infection (Treg transfer: 1 dpi, analysis: 7 dpi) | Beneficial:Treg reduce viral replication and release, and destroy HIV-1-infected macrophages via caspase-3 and granzyme/perforin pathways | Beneficial: In vivo: Treg protect from neuronal loss, increase neurotrophic factor production, and reduce neuroinflammation In vitro: Treg induce proteomic changes in HIV-infected macrophages and transform them from M1 to M2 phenotype | [55,92,93] |
| Herpesviridae | ||||||
| Ocular HSV-1 infection | BalB/c | DT-mediated Foxp3 ablation with or w/o adoptive Treg transfer | Acute infection (depletion: 4–6 dpi, analysis: 28 dpi) and latent infection (depletion: 26–27 dpi, analysis: 36 dpi) | Detrimental: Acute phase: Treg facilitate establishment of latency in trigeminl ganglia Latent phase: Treg are involved in stress-induced reactivation of latent infection | n.d. | [96] |
| Subcutaneous HSV-2 infection | C57BL/6 | Antibody (CD25)-mediated Treg depletion or DT-mediated Foxp3 ablation | Acute infection(Treg depletion: -3 dpi, analysis: until 4 dpi) | Detrimental: Treg inhibit virus-specific CD4+ and CD8+ T cell responses, leading to increased viral load in the CNS of neonatal mice | n.d. | [97,98] |
| Intranasal HSV-1 infection | BALB/c | Vitamin E-deficient diet; antibody (CD25) mediated Treg depletion | Acute infection(diet: 4 weeks prior to infection; Treg depletion: -2 and 6 dpi, analysis: until 9 dpi) | Detrimental: Increased peripheral and CNS Treg numbers in vitamin E-deficient mice are associated with reduced trafficking of virus-specific CD8+T cells and increased viral load in the CNS | n.d. | [99] |
| Genital HSV-2 infection | C57BL/6 | DT-mediated Foxp3 ablation with or w/o Treg transfer | Acute infection (Treg depletion: -2, 0, 3 dpi, analysis: úntil 12 dpi) | Beneficial: Treg limit initial replication and virus spread into the CNS by promoting entry of immune cells into the infection site | n.d. | [100] |
| Intracerebro-ventricular MCMV infection | C57BL/6 | DT-mediated Foxp3 ablation | Acute-chronic infection (Treg depletion: -1, 1, 4 dpi, analysis: until 30 or 40 dpi) | Beneficial: Treg promote long-term immunity by supporting transition of effector T cells to tissue resident memory T cells | Beneficial: Treg reduce T cell numbers in acute encephalitis and supress microgliosis, astrogliosis, MHC class II expression, hippocampal neurotoxicity, and cognitive impairment in post-encephalitic phase | [101,102] |
| Flaviviridae | ||||||
| Subcutaneous WNV infection | C57BL/6 | DT-mediated Foxp3 ablation | Acute infection (Treg depletion: -1, 0 dpi, analysis: until 20 or 60 dpi) | No effect on viral load in acute infection; Treg limit effector T cell and inflammatory cytokine responses in acute encephalitis, but increase numbers of potentially protective memory T cells at later stages | Beneficial: Treg reduce morbidity and mortality in acute WNV encephalitis, presumably by reducing immunopathology | [103,104] |
| Intraperitoneal JEV infection | C57BL/6 | CCR5-/- mice with or w/o CCR5+ Treg or CCR+ Treg transfer | Acute infection (Treg tranfer: 3 dpi, analysis: until 15 dpi) | No effect | CCR5-mediated CNS homing of IL-10- and TGF-β-producing Treg reduces neuro-inflammation | [105] |
| Paramyxoviridae | ||||||
| Intracerebral infection with recombinant MV | C57BL/6 | Treg expansion by superagonistic CD28-antibodies; DT-mediated Foxp3 ablation | Persistent infection (Treg expansion: 14, 21 dpi, Treg depletion: 17–20 dpi, analysis: 28 dpi) | Detrimental: Treg inhibit virus-specific CD8+ T cell responses leading to increased virus replication in the persistently infected CNS | n.d. | [106] |
| Intracerebral infection with recombinant MV | C57BL/6, B6.129 | Asm deficiency/blockade with or w/o concurrent DT-mediated Foxp3 ablation | Persistent infection (Asm blockade with or w/o Treg depletion: 21–26 dpi, analysis: 28 dpi) | Detrimental: Deficiency or inhibition of Asm leads to an elevated Treg to T effector ratio and results in increased virus replication (effect is Treg-dependent); no effect on viral load of Treg-depletion alone | n.d. | [107] |
| Virus Strain | Mouse Strain | Treg ⇩ or ⇧ | Time of Treg Manipulation* | Method | Viral Load | Disease | Reference |
|---|---|---|---|---|---|---|---|
| BeAn | SJL/J | ⇩ | Pre-infection | Antibody–mediated depletion (anti-CD25 or -GITR) | ⇩ | Delayed chronic demyelination | [146] |
| DA | SJL/J | ⇧ | Pre-infection | Adoptive transfer of ex vivo generated iTreg | ⇧ | Acute disease ⇧ | [147] |
| DA | SJL/J | ⇧ | Chronic infection | Adoptive transfer of ex vivo generated iTreg | No effect | Chronic demyelination ⇩ | [147] |
| DA | SJL/J | ⇧** | Acute-chronic infection | Glatiramer acetate** | No effect | Chronic demyelination ⇩ | [149] |
| DA | SJL/J | ⇧** | Chronic infection | Oral antibiotics** | Not investigated | Chronic demyelination ⇩ | [148] |
| BeAn | C57BL/6 | ⇩ | Pre-infection | Antibody-mediated depletion (anti CD25) | No effect | No effect | [146] |
| BeAn | C57BL/6 | ⇩ | Pre-infection | DT-mediated depletion in DEREG mice | No effect | No effect | [150] |
| DA | C57BL/6 | ⇧ | Pre-infection | Adoptive transfer of ex vivo generated iTreg | No effect | No effect | [147] |
| BeAn | C57BL/6 | ⇧ | Pre-infection | Expansion by IL-2C | No effect | No effect | [151] |
| BeAn | C57BL/6 | Treg⇧ and CD8⇩ | Pre-infection | Expansion by IL-2C, antibody-mediated CD8-depletion | ⇧ | Chronic demyelination ⇩ | [151] |
| Virus Strain | Mouse Strain | Treg ⇩ or ⇧ | Time of Treg Manipulation* | Method | Viral Load | Disease Severity | Reference |
|---|---|---|---|---|---|---|---|
| Effects on acute encephalitis | |||||||
| A59 (low virulence) | C57BL/6 | ⇩ | Pre-infection-acute infection | DT-mediated depletion in DEREG mice | No effect | Encephalitis ⇧ | [155] |
| rJ.MY135Q (low virulence) | C57BL/6 | ⇩ | Pre-infection | Antibody–mediated depletion (anti-CD25) | No effect | Encephalitis ⇧ mortality ⇧ | [154] |
| rJ (high virulence) | C57BL/6 | ⇧ | Acute infection | Adoptive transfer of bulk Treg | No effect | Encephalitis ⇩ mortality ⇩ | [154] |
| rJ.2.2 (low virulence) | C57BL/6 | ⇧ | Pre-infection | Adoptive transfer of bulk or virus-specific Treg | No effect | Encephalitis ⇩ mortality ⇩ | [156] |
| Effects on chronic demyelinating disease | |||||||
| J2.2v-1 (low virulence) | C57BL/6 | ⇩ | Pre-infection -acute infection | Antibody–mediated depletion (anti-CD25) | No effect | Demyelination ⇧ | [157] |
| J2.2v-1, rJ2.2 (low virulence) | C57BL/6RAG1-/- | ⇧ | Acute infection | Adoptive transfer of bulk Treg | No effect | Demyelination ⇩ | [159] |
| J2.2v-1 (low virulence) | C57BL/6 | ⇩ | Transition from acute to chronic infection | DT-mediated depletion in DEREG mice | Virus RNA⇩ in CLN, no effect in CNS | No effect | [160] |
| J2.2v-1 (low virulence) | C57BL/6 | ⇧** | Transition from acute to chronic infection | Stem cell transfer**, antibody–mediated Treg depletion (anti-CD25) | No effect | Demyelination ⇩ remyelination ⇧ | [161,162] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciurkiewicz, M.; Herder, V.; Beineke, A. Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections. Int. J. Mol. Sci. 2020, 21, 1705. https://doi.org/10.3390/ijms21051705
Ciurkiewicz M, Herder V, Beineke A. Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections. International Journal of Molecular Sciences. 2020; 21(5):1705. https://doi.org/10.3390/ijms21051705
Chicago/Turabian StyleCiurkiewicz, Malgorzata, Vanessa Herder, and Andreas Beineke. 2020. "Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections" International Journal of Molecular Sciences 21, no. 5: 1705. https://doi.org/10.3390/ijms21051705
APA StyleCiurkiewicz, M., Herder, V., & Beineke, A. (2020). Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections. International Journal of Molecular Sciences, 21(5), 1705. https://doi.org/10.3390/ijms21051705