Local Delivery of Gemcitabine Inhibits Pancreatic and Cholangiocarcinoma Tumor Growth by Promoting Epidermal Growth Factor Receptor Degradation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
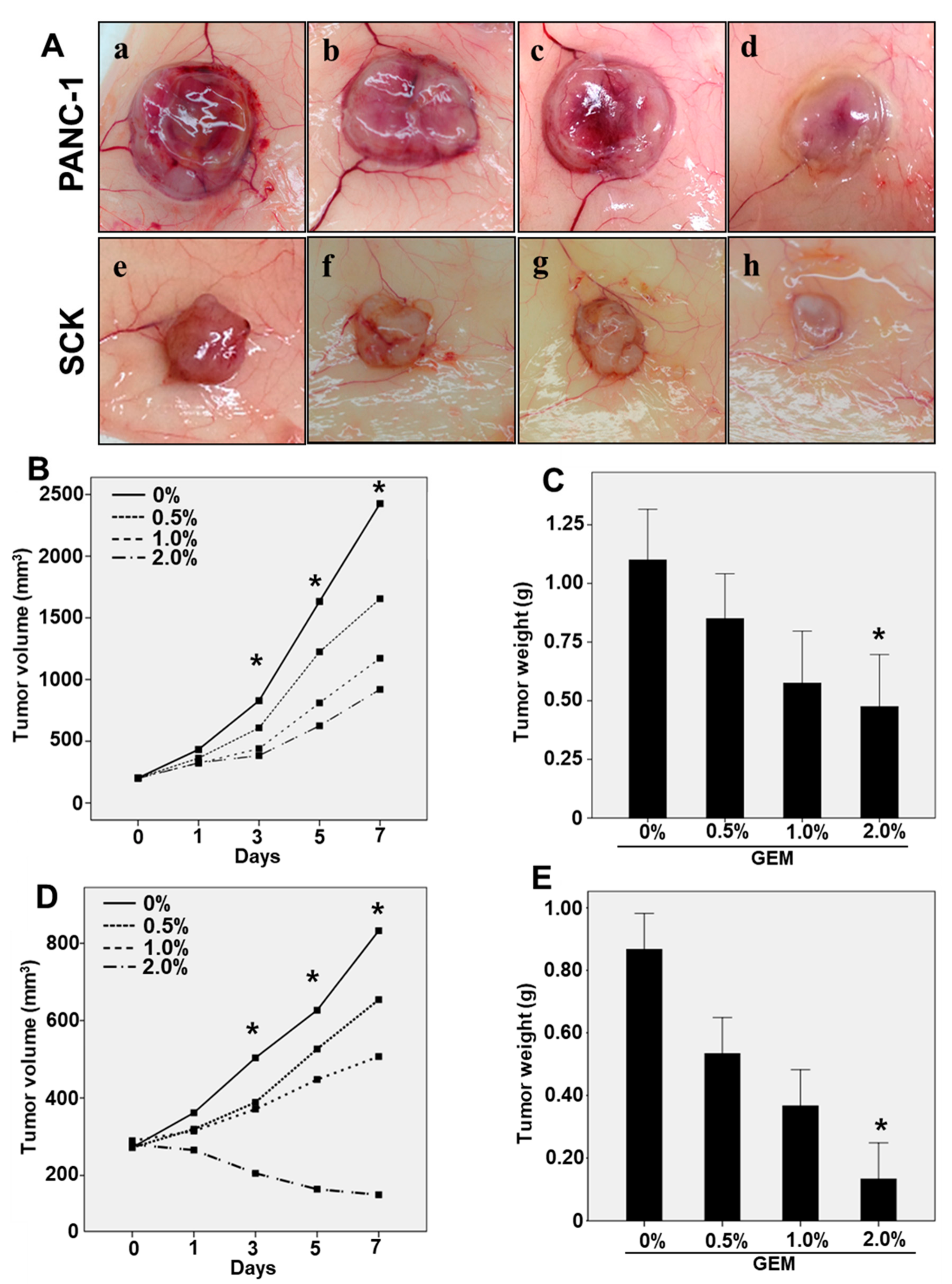
2.1. The GEM Inhibits the Growth of Pancreatic/Biliary Xenograft Tumors in Nude Mice
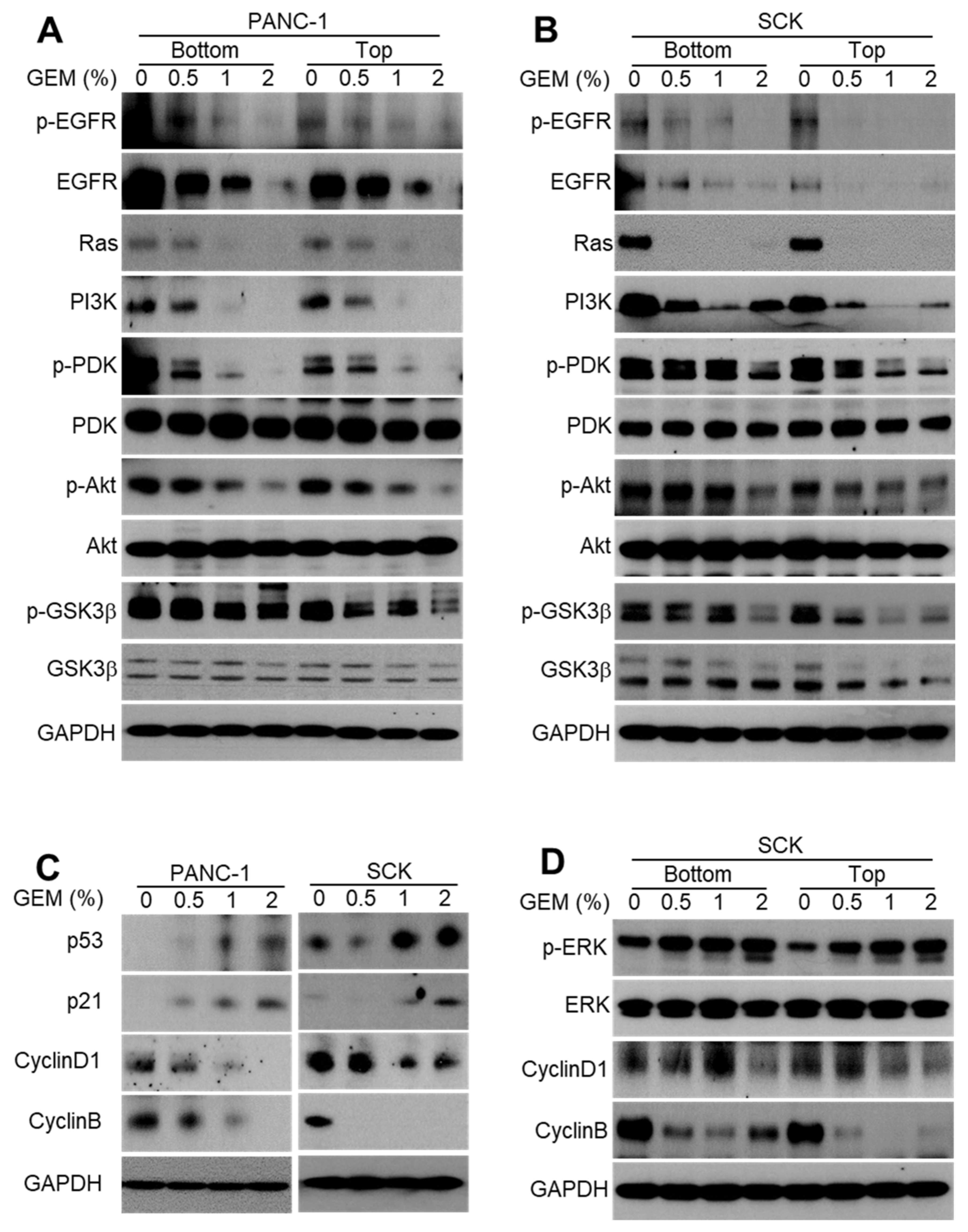
2.2. GEM Implantation Inhibits EGFR Signaling and the Cell Cycle in Xenograft Tumors
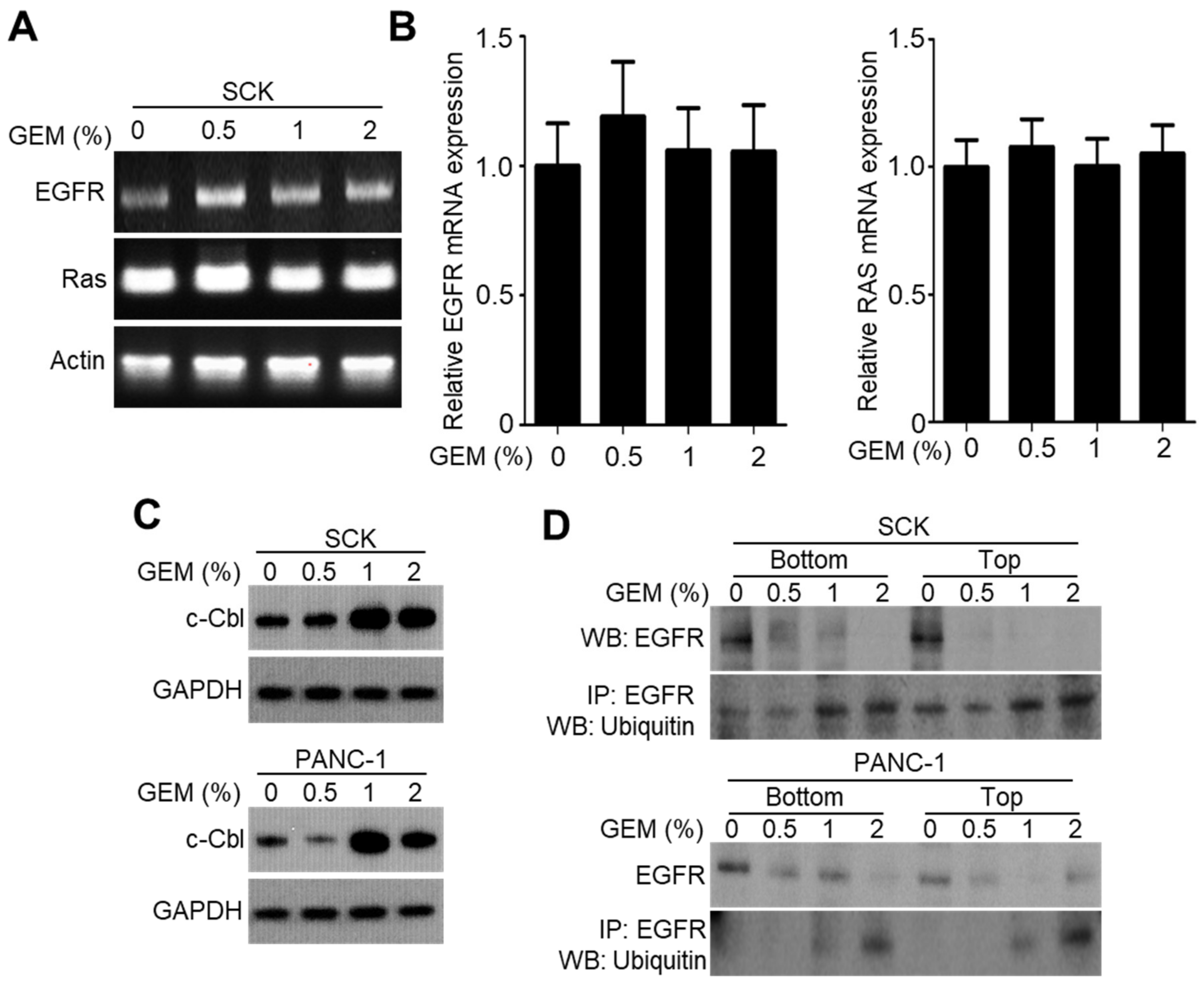
2.3. GEM Implantation Induces Ubiquitination and Degradation of EGFR
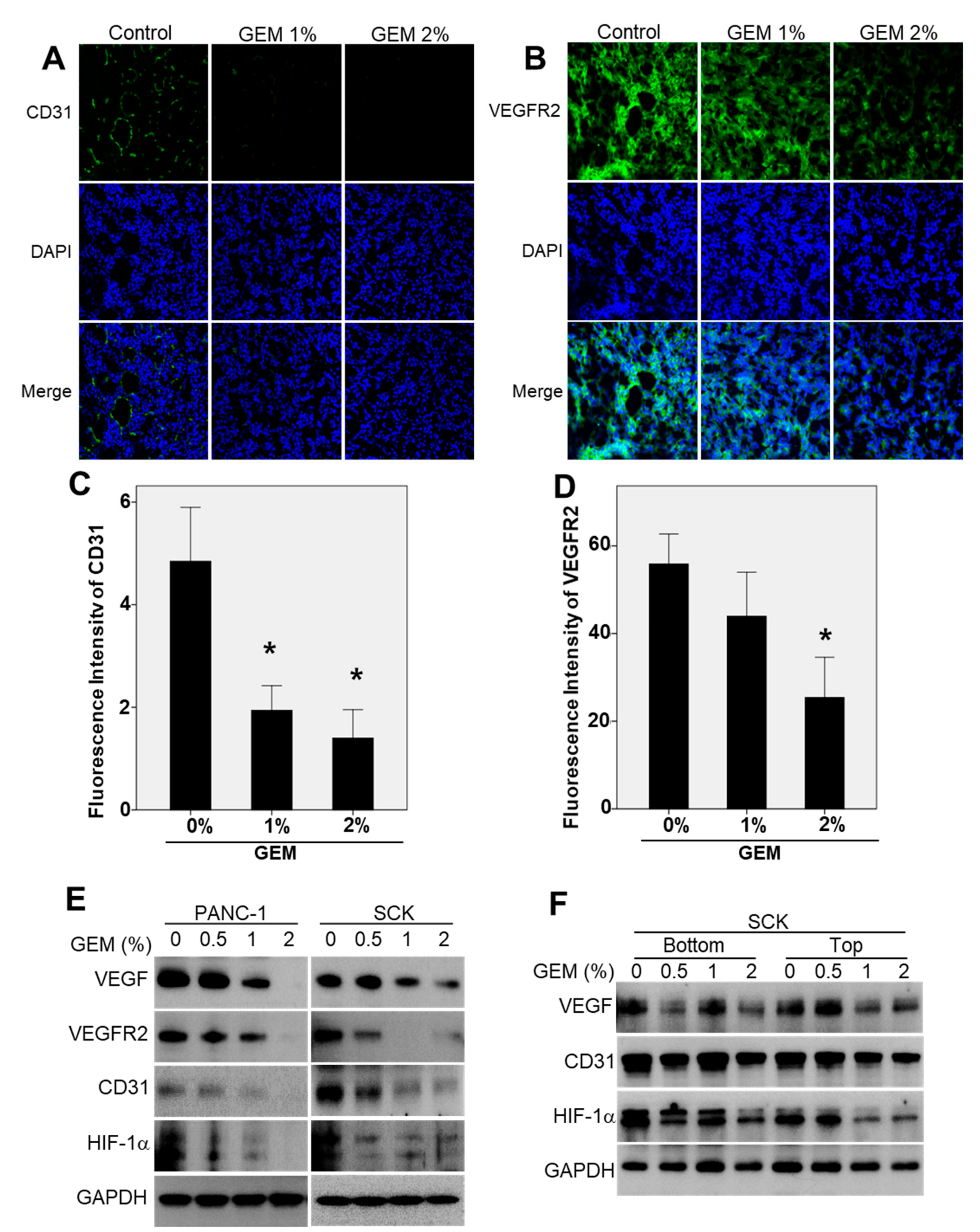
2.4. The GEM Reduces CD31 and Vascular Endothelial Growth Factor Receptor-2 (VEGFR2) Expression in Xenograft Tumors
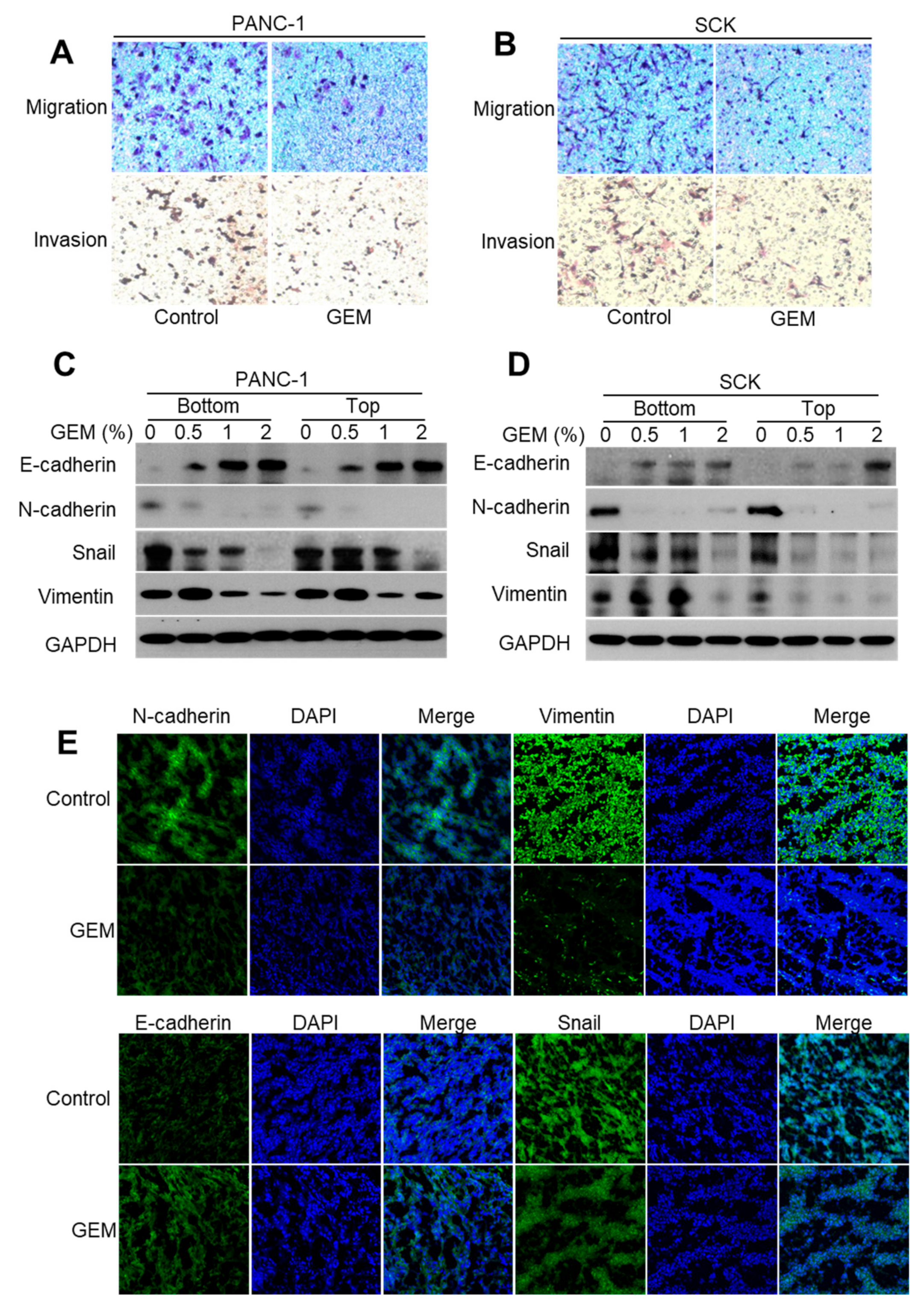
2.5. GEM Implantation Suppresses the EMT-like Features in Xenograft Tumors
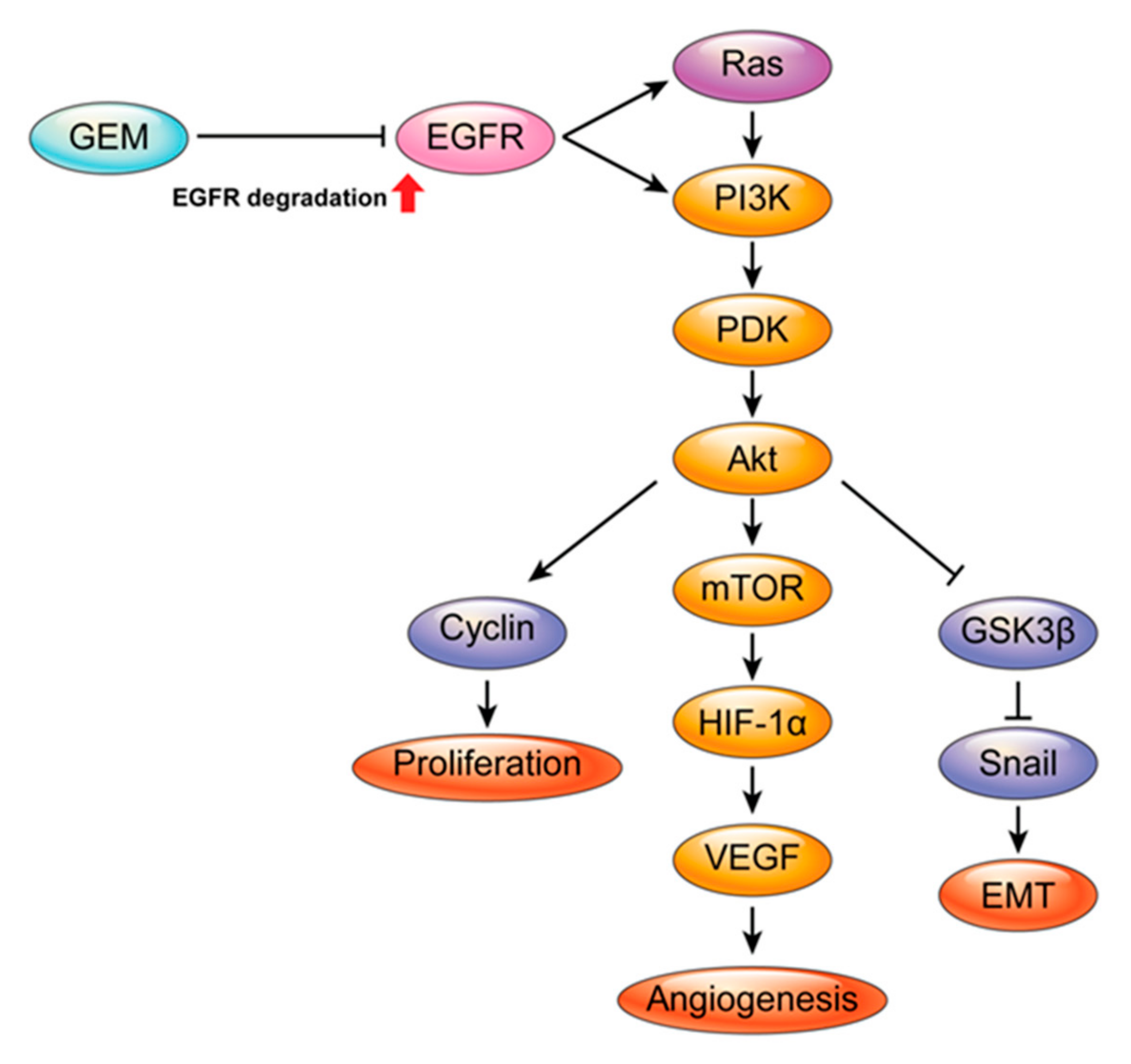
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Antibodies
4.2. Mouse Xenograft Experiment
4.3. Preparation of the GEM
4.4. Tumor Sample Immunofluorescence
4.5. Western Blot Analysis
4.6. Migration Assay
4.7. Invasion Assay
4.8. RNA Extraction and Reverse Transcription–Polymerase Chain Reaction
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Andersen, J.R.; Sørensen, S.M.; Kruse, A.; Rokkjaer, M.; Matzen, P. Randomised trial of endoscopic endoprosthesis versus operative bypass in malignant obstructive jaundice. Gut 1989, 30, 1132–1135. [Google Scholar] [CrossRef]
- Smith, A.C.; Dowsett, J.F.; Russell, R.C.G.; Hatfield, A.R.W.; Cotton, P.B. Randomised trial of endoscopic stenting versus surgical bypass in malignant low bileduct obstruction. Lancet 1994, 344, 1655–1660. [Google Scholar] [CrossRef]
- Wagner, H.J.; Knyrim, K.; Vakil, N.; Klose, K.J. Plastic endoprostheses versus metal stents in the palliative treatment of malignant hilar biliary obstruction. A prospective and randomized trial. Endoscopy 1993, 25, 213–218. [Google Scholar] [CrossRef]
- O’brien, S.; Hatfield, A.R.; Craig, P.I.; Williams, S.P. A three year follow up of self expanding metal stents in the endoscopic palliation of longterm survivors with malignant biliary obstruction. Gut 1995, 36, 618–621. [Google Scholar] [CrossRef]
- Hausegger, K.A.; Kleinert, R.; Lammer, J.E.; Klein, G.E.; Flückiger, F. Malignant biliary obstruction: Histologic findings after treatment with self-expandable stents. Radiology 1992, 185, 461–464. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, D.K.; Kim, H.G.; Park, J.J.; Park, S.H.; Kim, J.H.; Myung, S.J. Features of malignant biliary obstruction affecting the patency of metallic stents: A multicenter study. Gastrointest. Endosc. 2002, 55, 359–365. [Google Scholar] [CrossRef]
- Lee, D.K.; Kim, H.S.; Kim, K.S.; Lee, W.J.; Kim, H.K.; Won, Y.H.; Kwon, S.O. The effect on porcine bile duct of a metallic stent covered with a paclitaxel-incorporated membrane. Gastrointest. Endosc. 2005, 61, 296–301. [Google Scholar] [CrossRef]
- Suk, K.T.; Kim, J.W.; Kim, H.S.; Baik, S.K.; Oh, S.J.; Lee, S.J.; Lee, D.K. Human application of a metallic stent covered with a paclitaxel-incorporated membrane for malignant biliary obstruction: Multicenter pilot study. Gastrointest. Endosc. 2007, 66, 798–803. [Google Scholar] [CrossRef]
- Lee, S.S.; Shin, J.H.; Han, J.M.; Cho, C.H.; Kim, M.H.; Lee, S.K.; Song, H.Y. Histologic influence of paclitaxel-eluting covered metallic stents in a canine biliary model. Gastrointest. Endosc. 2009, 69, 1140–1147. [Google Scholar] [CrossRef]
- Song, T.J.; Lee, S.S.; Yun, S.C.; Park, D.H.; Seo, D.W.; Lee, S.K.; Kim, M.H. Paclitaxel-eluting covered metal stents versus covered metal stents for distal malignant biliary obstruction: A prospective comparative pilot study. Gastrointest. Endosc. 2011, 73, 727–733. [Google Scholar] [CrossRef]
- Jang, S.I.; Kim, J.H.; Kim, M.; Yang, S.; Jo, E.A.; Lee, J.W.; Lee, D. Porcine feasibility and safety study of a new paclitaxel-eluting biliary stent with a Pluronic-containing membrane. Endoscopy 2012, 44, 825–831. [Google Scholar] [CrossRef]
- Jang, S.I.; Kim, J.H.; You, J.W.; Rhee, K.; Lee, S.J.; Kim, H.G.; Lee, D.K. Efficacy of a metallic stent covered with a paclitaxel-incorporated membrane versus a covered metal stent for malignant biliary obstruction: A prospective comparative study. Dig. Dis. Sci. 2013, 58, 865–871. [Google Scholar] [CrossRef]
- Chung, M.J.; Kim, H.; Kim, K.S.; Park, S.; Chung, J.B.; Park, S.W. Safety evaluation of self-expanding metallic biliary stents eluting gemcitabine in a porcine model. J. Gastroenterol. Hepatol. 2012, 27, 261–267. [Google Scholar] [CrossRef]
- Bang, S.; Jang, S.I.; Lee, S.Y.; Baek, Y.Y.; Yun, J.; Oh, S.J.; Lee, D.H. Molecular mechanism of local drug delivery with Paclitaxel-eluting membranes in biliary and pancreatic cancer: New application for an old drug. Gastroenterol. Res. Pract. 2015, 2015, 568981. [Google Scholar] [CrossRef]
- Burris, H., III; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Tarassoff, P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Eckel, F.; Schmid, R.M. Chemotherapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Br. J. Cancer 2007, 96, 896–902. [Google Scholar] [CrossRef]
- Yokoi, K.; Sasaki, T.; Bucana, C.D.; Fan, D.; Baker, C.H.; Kitadai, Y.; Fidler, I.J. Simultaneous inhibition of EGFR, VEGFR, and platelet-derived growth factor receptor signaling combined with gemcitabine produces therapy of human pancreatic carcinoma and prolongs survival in an orthotopic nude mouse model. Cancer Res. 2005, 65, 10371–10380. [Google Scholar] [CrossRef]
- Cohenuram, M.; Saif, M.W. Epidermal growth factor receptor inhibition strategies in pancreatic cancer: Past, present and the future. JOP 2007, 8, 4–15. [Google Scholar]
- Feng, F.Y.; Varambally, S.; Tomlins, S.A.; Chun, P.Y.; Lopez, C.A.; Li, X.; Nyati, M.K. Role of epidermal growth factor receptor degradation in gemcitabine-mediated cytotoxicity. Oncogene 2007, 26, 3431–3439. [Google Scholar] [CrossRef][Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Yarden, Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Yu, W.D.; Ma, Y.; Flynn, G.; Muindi, J.R.; Kong, R.X.; Trump, D.L.; Johnson, C.S. Calcitriol enhances gemcitabine anti-tumor activity in vitro and in vivo by promoting apoptosis in a human pancreatic carcinoma model system. Cell Cycle 2010, 9, 3022–3029. [Google Scholar] [CrossRef]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Lee, J.; Park, S.H.; Chang, H.M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Shin, D.B. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Birhanu, G.; Javar, H.A.; Seyedjafari, E.; Zandi-Karimi, A. Nanotechnology for delivery of gemcitabine to treat pancreatic cancer. Biomed. Pharmacother. 2017, 88, 635–643. [Google Scholar] [CrossRef]
- Reddy, L.H.; Couvreur, P. Novel approaches to deliver gemcitabine to cancers. Curr. Pharm. Des. 2008, 14, 1124–1137. [Google Scholar] [CrossRef]
- Moon, S.; Yang, S.G.; Na, K. An acetylated polysaccharide-PTFE membrane-covered stent for the delivery of gemcitabine for treatment of gastrointestinal cancer and related stenosis. Biomaterials 2011, 32, 3603–3610. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003, 21, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef]
- Waterman, H.; Yarden, Y. Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases. FEBS Lett. 2001, 490, 142–152. [Google Scholar] [CrossRef]
- Ravid, T.; Heidinger, J.M.; Gee, P.; Khan, E.M.; Goldkorn, T. c-Cbl-mediated ubiquitinylation is required for epidermal growth factor receptor exit from the early endosomes. J. Biol. Chem. 2004, 279, 37153–37162. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Suyama, K.; Baba, H. Recent Advances in Targeting the EGFR Signaling Pathway for the Treatment of Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 752. [Google Scholar] [CrossRef]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Nurse, P. Cyclin dependent kinases and cell cycle control (nobel lecture). Chembiochem 2002, 3, 596–603. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Yuan, J.; Yan, R.; Krämer, A.; Eckerdt, F.; Roller, M.; Kaufmann, M.; Strebhardt, K. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 2004, 23, 5843–5852. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Strizzi, L.; Bianco, C.; Raafat, A.; Abdallah, W.; Chang, C.; Raafat, D.; Callahan, R. Netrin-1 regulates invasion and migration of mouse mammary epithelial cells overexpressing Cripto-1 in vitro and in vivo. J. Cell Sci. 2005, 118, 4633–4643. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cheng, L.; Xia, J.; Wang, Z.; Wu, Q.; Wang, Z. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1alpha in pancreatic cancer cells. Curr. Cancer Drug Targets. 2014, 14, 407–417. [Google Scholar] [CrossRef]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.I.; Fang, S.; Baek, Y.-Y.; Lee, D.H.; Na, K.; Lee, S.Y.; Lee, D.K. Local Delivery of Gemcitabine Inhibits Pancreatic and Cholangiocarcinoma Tumor Growth by Promoting Epidermal Growth Factor Receptor Degradation. Int. J. Mol. Sci. 2020, 21, 1605. https://doi.org/10.3390/ijms21051605
Jang SI, Fang S, Baek Y-Y, Lee DH, Na K, Lee SY, Lee DK. Local Delivery of Gemcitabine Inhibits Pancreatic and Cholangiocarcinoma Tumor Growth by Promoting Epidermal Growth Factor Receptor Degradation. International Journal of Molecular Sciences. 2020; 21(5):1605. https://doi.org/10.3390/ijms21051605
Chicago/Turabian StyleJang, Sung Ill, Sungsoon Fang, Yi-Yong Baek, Don Haeng Lee, Kun Na, Su Yeon Lee, and Dong Ki Lee. 2020. "Local Delivery of Gemcitabine Inhibits Pancreatic and Cholangiocarcinoma Tumor Growth by Promoting Epidermal Growth Factor Receptor Degradation" International Journal of Molecular Sciences 21, no. 5: 1605. https://doi.org/10.3390/ijms21051605
APA StyleJang, S. I., Fang, S., Baek, Y.-Y., Lee, D. H., Na, K., Lee, S. Y., & Lee, D. K. (2020). Local Delivery of Gemcitabine Inhibits Pancreatic and Cholangiocarcinoma Tumor Growth by Promoting Epidermal Growth Factor Receptor Degradation. International Journal of Molecular Sciences, 21(5), 1605. https://doi.org/10.3390/ijms21051605