Vitamin D as a Primer for Oncolytic Viral Therapy in Colon Cancer Models

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. VD Analogue Does Not Affect Viral Replication but Reduces Cellular Proliferation in Vd-Responsive Cells
2.2. The Differences Seen in Viral Replication in HT29 Cells with and without VD Was Attributed to the Baseline VD-Responsive Nature of HT29 Cells
2.3. VD Potentiates Oncolytic Viral Replication and Increases the Survival of Mice Bearing VD-Responsive Xenograft Tumors
2.4. VD Blunts Inflammation and Promotes Tumor Destruction After Viral Infection in VD-Responsive HT29 Xenografts
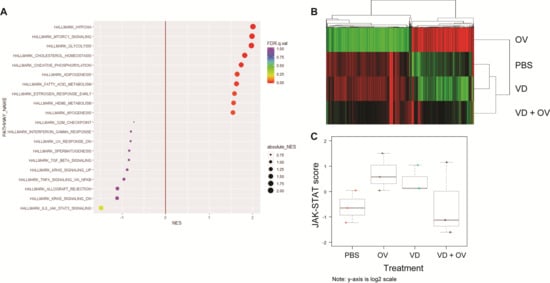
2.5. RNA-Sequencing Analysis of HT29 Treatment Groups Shows Downregulated Jak-STAT Pathway Signaling When Comparing Virus Alone to VD Combined with Virus
3. Discussion
4. Materials and Methods
4.1. CF33 Chimerization and hNIS, Anti-PD-L1 or ΔF14.5 Cloning
4.2. Cell Lines and Mice
4.3. Tumor Models
4.4. Viral Growth Assays
4.5. Cytotoxicity Assays
4.6. Immunohistochemistry (IHC)
4.7. RNA-Sequencing
4.8. nCounter
4.9. Colony Assay
4.10. Immunofluorescence
4.11. Statistical Analysis
4.12. RT-PCR
4.13. Western Blotting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | Colorectal Cancer |
| MSI | Microsatellite instability |
| OV | Oncolytic Virus |
| PFU | Plaque-forming unit |
| TME | Tumor microenvironment |
| VD | Vitamin D |
References
- Siegel, R.L.; Mph, K.D.M.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA: A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Tsikitis, V.L.; Larson, D.W.; Huebner, M.; Lohse, C.M.; A Thompson, P. Predictors of recurrence free survival for patients with stage II and III colon cancer. BMC Cancer 2014, 14, 336. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. Interpretation of the updates of NCCN 2017 version 1.0 guideline for colorectal cancer. Zhonghua wei chang wai ke za zhi Chin. J. Gastrointest. Surg. 2017, 20, 28–33. [Google Scholar]
- Sandhu, J.; Lavingia, V.; Fakih, M. Systemic treatment for metastatic colorectal cancer in the era of precision medicine. J. Surg. Oncol. 2019, 119, 564–582. [Google Scholar] [CrossRef] [PubMed]
- Creasy, J.M.; Sadot, E.; Koerkamp, B.G.; Chou, J.F.; Gonen, M.; Kemeny, N.E.; Balachandran, V.P.; Kingham, T.P.; DeMatteo, R.P.; Allen, P.J.; et al. Actual 10-year survival after hepatic resection of colorectal liver metastases: What factors preclude cure? Surgery 2018, 163, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.H.; O’Leary, M.P.; Fong, Y.; Chen, N.G. From Benchtop to Bedside: A Review of Oncolytic Virotherapy. Biomed. 2016, 4, 18. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. OncoImmunology 2018, 7, e1442169. [Google Scholar] [CrossRef]
- Choi, A.H.; O’Leary, M.P.; Lu, J.; Kim, S.-I.; Fong, Y.; Chen, N.G. Endogenous Akt Activity Promotes Virus Entry and Predicts Efficacy of Novel Chimeric Orthopoxvirus in Triple-Negative Breast Cancer. Mol. Ther. Oncolytics 2018, 9, 22–29. [Google Scholar] [CrossRef]
- O’Leary, M.P.; Choi, A.H.; Kim, S.-I.; Chaurasiya, S.; Lu, J.; Park, A.K.; Woo, Y.; Warner, S.G.; Fong, Y.; Chen, N.G. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J. Transl. Med. 2018, 16, 110. [Google Scholar] [CrossRef]
- O’Leary, M.P.; Warner, S.G.; Kim, S.-I.; Chaurasiya, S.; Lu, J.; Choi, A.H.; Park, A.K.; Woo, Y.; Fong, Y.; Chen, N.G. A Novel Oncolytic Chimeric Orthopoxvirus Encoding Luciferase Enables Real-Time View of Colorectal Cancer Cell Infection. Mol. Ther. Oncolytics 2018, 9, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.G.; Kim, S.-I.; Chaurasiya, S.; O’Leary, M.P.; Lu, J.; Sivanandam, V.; Woo, Y.; Chen, N.G.; Fong, Y. A Novel Chimeric Poxvirus Encoding hNIS Is Tumor-Tropic, Imageable, and Synergistic with Radioiodine to Sustain Colon Cancer Regression. Mol. Ther. Oncolytics 2019, 13, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Yang, A.; Kang, S.; Lu, J.; Kim, S.-I.; Park, A.K.; Sivanandam, V.; Zhang, Z.; Woo, Y.; Warner, S.G.; et al. Oncolytic poxvirus CF33-hNIS-ΔF14.5 favorably modulates tumor immune microenvironment and works synergistically with anti-PD-L1 antibody in a triple-negative breast cancer model. OncoImmunology 2020, 9, 1729300. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.-I.; Warner, S.G.; Von Hoff, D.; Fong, Y. Novel Chimeric Immuno-Oncolytic Virus CF33-hNIS-antiPDL1 for the Treatment of Pancreatic Cancer. J. Am. Coll. Surg. 2020, 230, 709–717. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Boil. 2014, 25, 198–213. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, P.; Wang, F.; Yang, J.; Liu, Z.; Qin, H. Association Between Vitamin D and Risk of Colorectal Cancer: A Systematic Review of Prospective Studies. J. Clin. Oncol. 2011, 29, 3775–3782. [Google Scholar] [CrossRef]
- Ng, K.; Nimeiri, H.S.; McCleary, N.J.; Abrams, T.A.; Yurgelun, M.B.; Cleary, J.M.; Rubinson, D.A.; Schrag, D.; Miksad, R.; Bullock, A.J.; et al. Effect of High-Dose vs Standard-Dose Vitamin D3 Supplementation on Progression-Free Survival Among Patients With Advanced or Metastatic Colorectal Cancer: The SUNSHINE Randomized Clinical Trial. JAMA 2019, 321, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Leyssens, C.; Verlinden, L.; Verstuyf, A. The future of vitamin D analogs. Front. Physiol. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, C.J.; Warner, S.G. A New Role for Vitamin D: The Enhancement of Oncolytic Viral Therapy in Pancreatic Cancer. Biomedicines 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Feldman, D. Regulation of vitamin D receptor abundance and responsiveness during differentiation of HT-29 human colon cancer cells. Endocrinology 1993, 132, 1808–1814. [Google Scholar] [CrossRef]
- Milczarek, M.; Filip-Psurska, B.; Świętnicki, W.; Kutner, A.; Wietrzyk, J. Vitamin D analogs combined with 5-fluorouracil in human HT-29 colon cancer treatment. Oncol. Rep. 2014, 32, 491–504. [Google Scholar] [CrossRef]
- Huang, J.-F.; Ko, Y.; Huang, C.-F.; Yeh, M.-L.; Dai, C.-Y.; Hsieh, M.; Huang, C.-I.; Yang, H.; Wang, S.; Lin, Z.; et al. 25-Hydroxy vitamin D suppresses hepatitis C virus replication and contributes to rapid virological response of treatment efficacy. Hepatol. Res. 2017, 47, 1383–1389. [Google Scholar] [CrossRef]
- Lange, C.M.; Gouttenoire, J.; Duong, F.H.T.; Morikawa, K.; Heim, M.H.; Moradpour, D. Vitamin D Receptor and Jak–STAT Signaling Crosstalk Results in Calcitriol-Mediated Increase of Hepatocellular Response to IFN-α. J. Immunol. 2014, 192, 6037–6044. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Huang, L.C.; Romanowski, E.G.; Yates, K.A.; Proske, R.J.; McDermott, A.M. Human Cathelicidin (LL-37), a Multifunctional Peptide, is Expressed by Ocular Surface Epithelia and has Potent Antibacterial and Antiviral Activity. Curr. Eye Res. 2005, 30, 385–394. [Google Scholar] [CrossRef]
- Horikawa, Y.; Wang, Y.; Nagano, S.; Kamizono, J.; Ikeda, M.; Komiya, S.; Kosai, K.-I. Assessment of an altered E1B promoter on the specificity and potency of triple-regulated conditionally replicating adenoviruses: Implications for the generation of ideal m-CRAs. Cancer Gene Ther. 2011, 18, 724–733. [Google Scholar] [CrossRef][Green Version]
- Chen, P.-T.; Hsieh, C.-C.; Wu, C.-T.; Yen, T.-C.; Lin, P.-Y.; Chen, W.-C.; Chen, M.-F. 1,25-Dihydroxyvitamin D3 Inhibits Esophageal Squamous Cell Carcinoma Progression by Reducing IL6 Signaling. Mol. Cancer Ther. 2015, 14, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Muthian, G.; Raikwar, H.P.; Rajasingh, J.; Bright, J.J. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci. Res. 2006, 83, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.C.; Kulling, P.M.; Olson, T.L.; Tan, S.-F.; Rainbow, R.J.; Feith, D.J.; Loughran, T.P. Vitamin D decreases STAT phosphorylation and inflammatory cytokine output in T-LGL leukemia. Cancer Boil. Ther. 2016, 18, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Zella, L.A.; Meyer, M.B.; Nerenz, R.D.; Lee, S.M.; Martowicz, M.L.; Pike, J.W. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol. Endocrinol. 2009, 24, 128–147. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Chen, N.G.; Lu, J.; Martin, N.; Shen, Y.; Kim, S.-I.; Warner, S.G.; Woo, Y.; Fong, Y. A chimeric poxvirus with J2R (thymidine kinase) deletion shows safety and anti-tumor activity in lung cancer models. Cancer Gene Ther. 2019, 27, 125–135. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathological Changes | Control | Treated | ||
|---|---|---|---|---|
| PBS | OV | VD | VD + OV | |
| Necrosis | + | ++ | ++ | +++ |
| Lymphocytes | - | ++ | ++ | + |
| Inflammatory cells | + | +++ | ++ | + |
| cellular differentiation | Moderate to poor | Moderate | Moderate | Moderate |
| Vasculature | + | +++ | ++ | + |
| Cancer-associated fibroblast | + | ++ | +++ | ++ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-I.; Chaurasiya, S.; Park, A.K.; Kang, S.; Lu, J.; Woo, Y.; Yin, H.H.; Yin, Z.; Fong, Y.; Warner, S.G. Vitamin D as a Primer for Oncolytic Viral Therapy in Colon Cancer Models. Int. J. Mol. Sci. 2020, 21, 7326. https://doi.org/10.3390/ijms21197326
Kim S-I, Chaurasiya S, Park AK, Kang S, Lu J, Woo Y, Yin HH, Yin Z, Fong Y, Warner SG. Vitamin D as a Primer for Oncolytic Viral Therapy in Colon Cancer Models. International Journal of Molecular Sciences. 2020; 21(19):7326. https://doi.org/10.3390/ijms21197326
Chicago/Turabian StyleKim, Sang-In, Shyambabu Chaurasiya, Anthony K. Park, Seonah Kang, Jianming Lu, Yanghee Woo, Hongwei Holly Yin, Zhirong Yin, Yuman Fong, and Susanne G. Warner. 2020. "Vitamin D as a Primer for Oncolytic Viral Therapy in Colon Cancer Models" International Journal of Molecular Sciences 21, no. 19: 7326. https://doi.org/10.3390/ijms21197326
APA StyleKim, S.-I., Chaurasiya, S., Park, A. K., Kang, S., Lu, J., Woo, Y., Yin, H. H., Yin, Z., Fong, Y., & Warner, S. G. (2020). Vitamin D as a Primer for Oncolytic Viral Therapy in Colon Cancer Models. International Journal of Molecular Sciences, 21(19), 7326. https://doi.org/10.3390/ijms21197326