Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters

Abstract

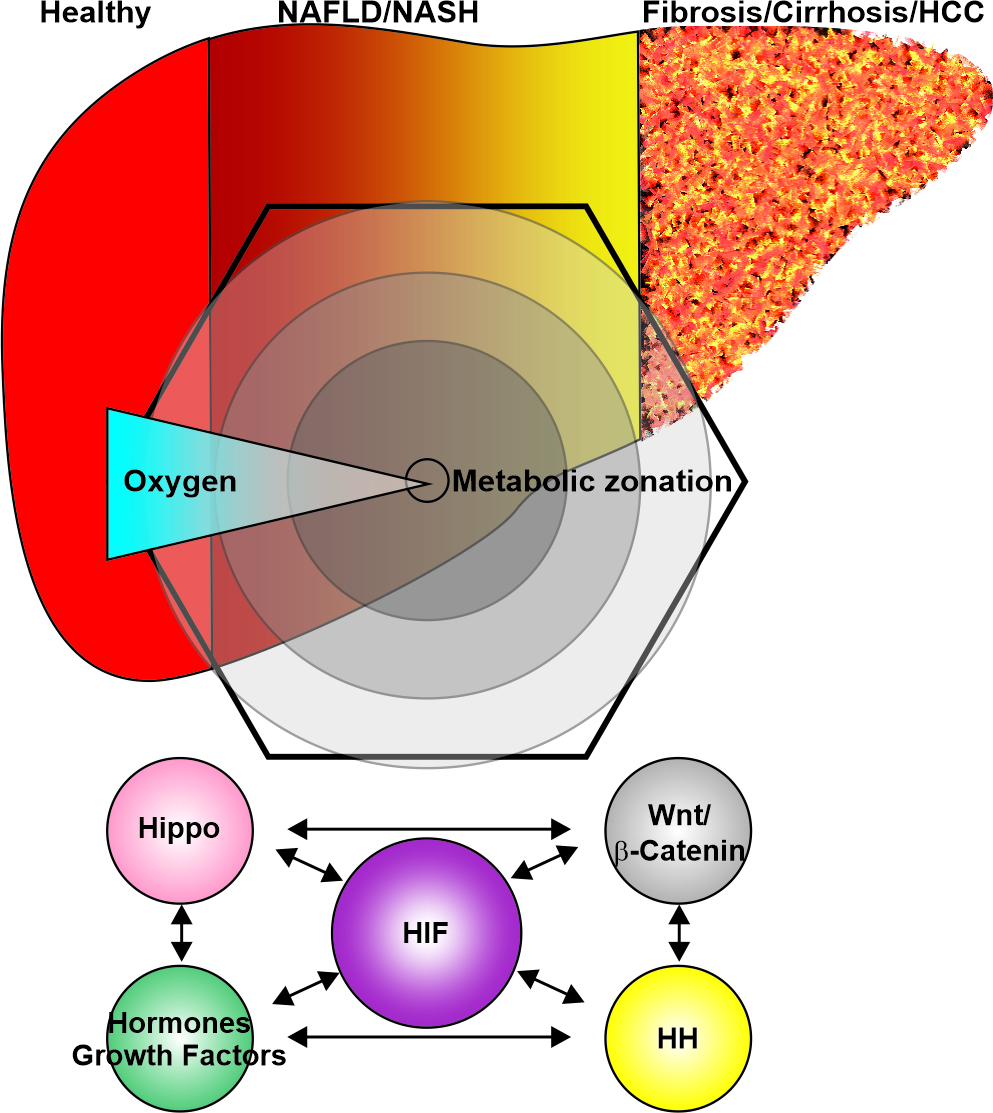{kind=link}
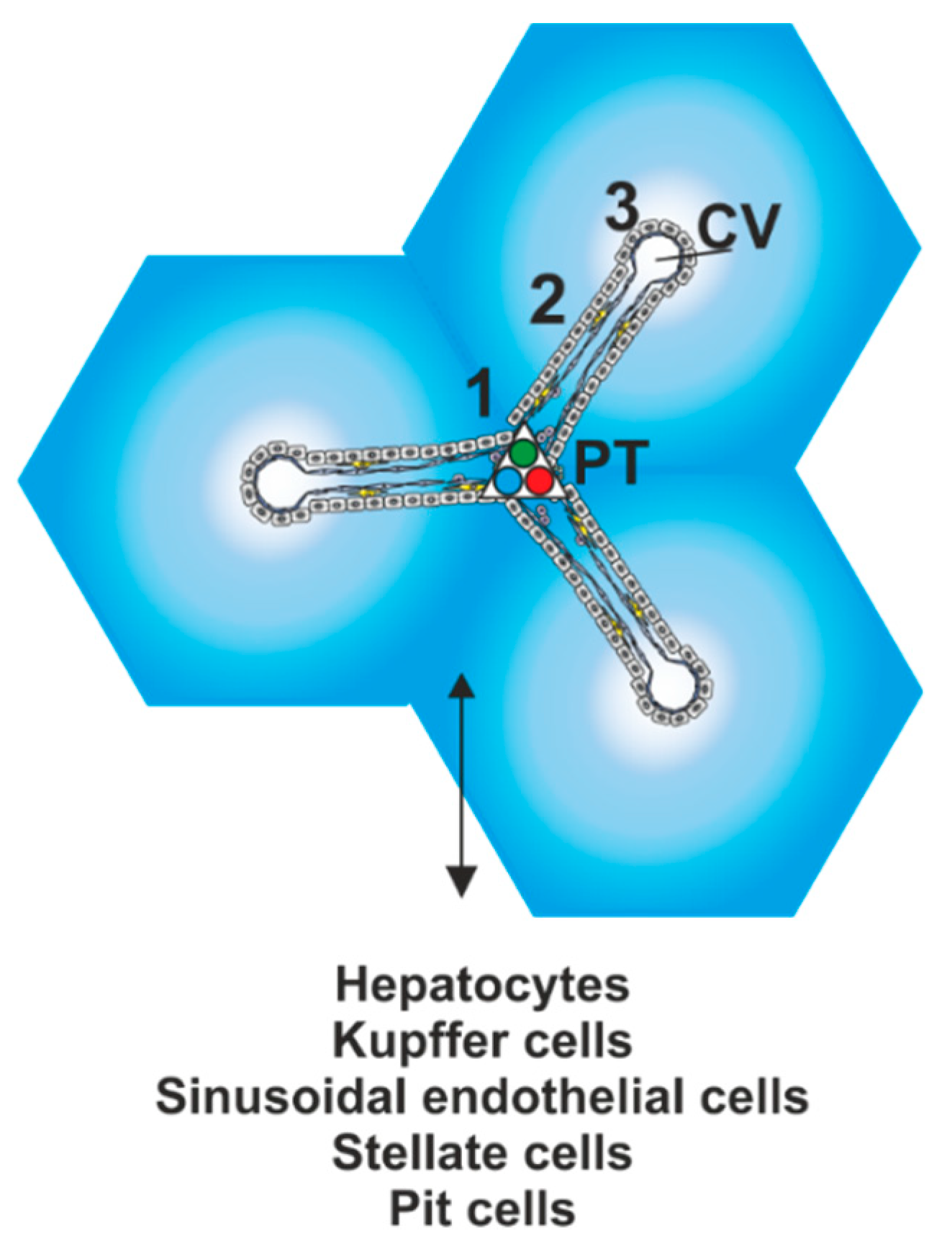{kind=link}
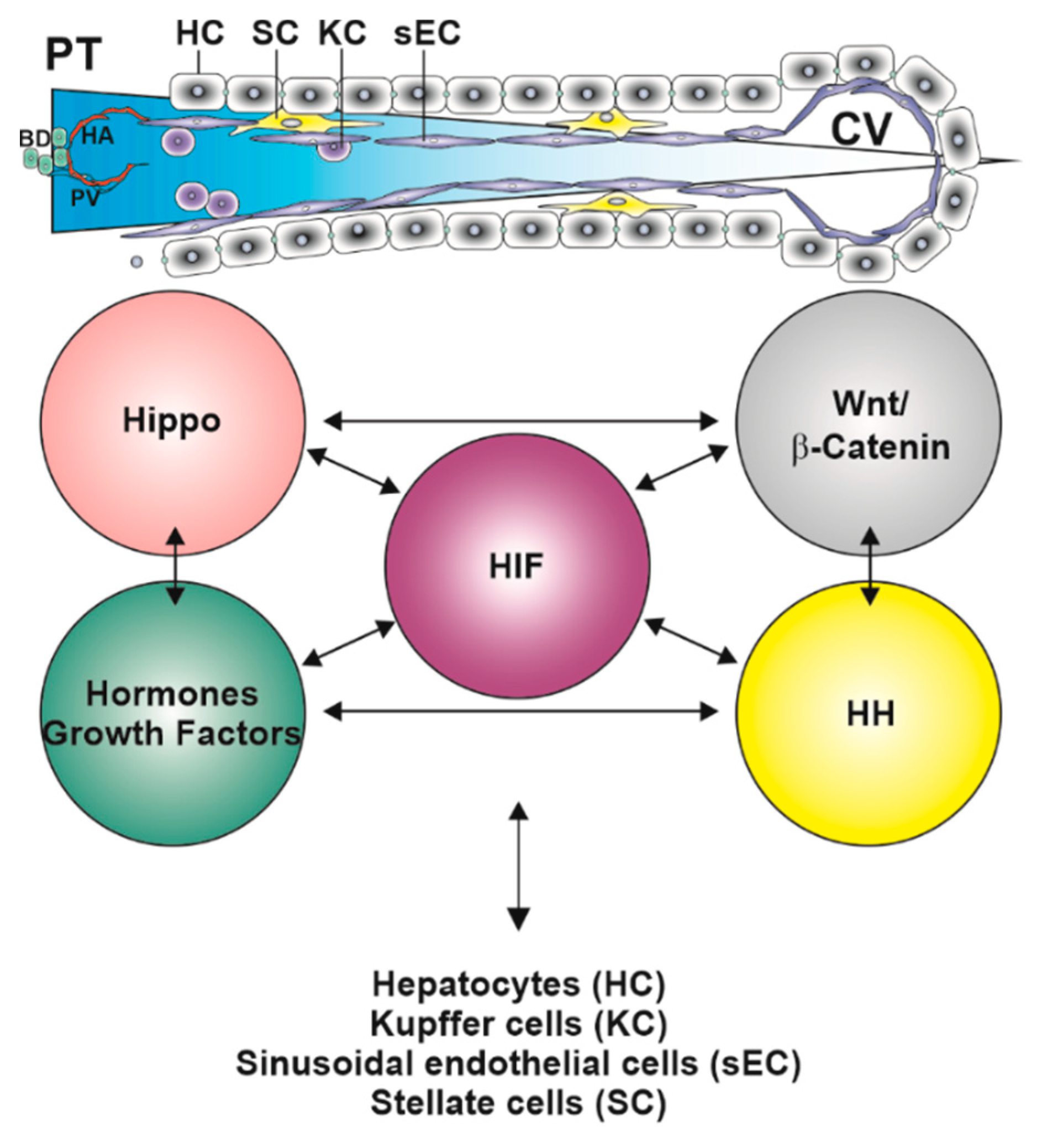{kind=link}
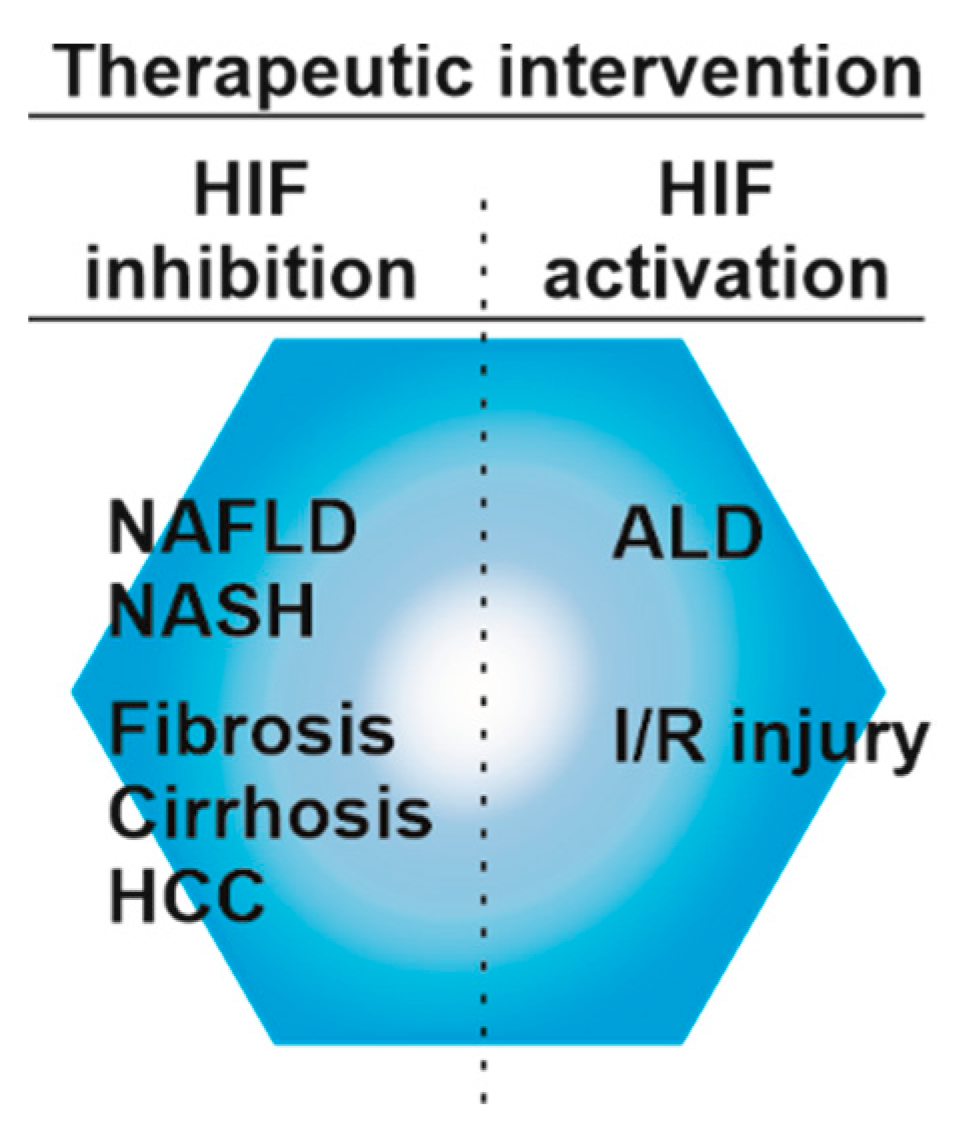{kind=link}
1. Introduction
2. Liver Lobule and Acinus—Structural and Functional Units Displaying Metabolic Zonation
3. Regulatory Pathways Involved in Liver Zonation
4. Oxygen Sensing and Hypoxia-Inducible Transcription Factors (HIFs)
5. HIFs, Redox, and HIF Prolyl 4-Hydroxylases in Liver
6. HIFs, Beta-Catenin, and Zonation
7. HIFs, Hedgehog, and Zonation
8. HIFs, Hippo, and Zonation
9. HIFs, Steatosis, and Fatty Liver Disease
9.1. HIFs and NAFLD
9.2. HIFs and ALD
10. HIFs, Liver Fibrosis, Cirrhosis, and HCC
11. HIFs in Ischemia-Reperfusion-Mediated Liver Injury
12. Hypoxia, HIF, and HIF-Prolyl Hydroxylase-Related Therapies to Treat Liver Diseases
13. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Sasse, D.; Spornitz, U.M.; Maly, I.P. Liver architecture. Enzyme 1992, 46, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Dimova, E.Y.; Flügel, D.; Scharf, J.G. Oxygen: Modulator of physiological and pathophysiological processes in the liver. Z. Gastroenterol. 2006, 44, 67–76. [Google Scholar] [CrossRef]
- Novikoff, A.B. Cell heterogeneity within the hepatic lobule of the rat: Staining reactions. J. Histochem. Cytochem. 1959, 7, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Loud, A.V. A quantitative stereological description of the ultrastructure of normal rat liver parenchymal cells. J. Cell Biol. 1968, 37, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Jungermann, K.; Sasse, D. Heterogeneity of liver parenchymal cells. Trends Biochem. Sci. 1978, 3, 198–202. [Google Scholar] [CrossRef]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Toth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E.; et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Matz-Soja, M. Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 2014, 20, 8491–8504. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, J.; Tokarski, C.; Marbach, E.; Matz-Soja, M.; Zellmer, S.; Gebhardt, R.; Schuster, S. Zonation of hepatic fatty acid metabolism—The diversity of its regulation and the benefit of modeling. Biochim. Biophys. Acta 2015, 1851, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Jungermann, K.; Kietzmann, T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu. Rev. Nutr. 1996, 16, 179–203. [Google Scholar] [CrossRef] [PubMed]
- Halpern, K.B.; Shenhav, R.; Massalha, H.; Toth, B.; Egozi, A.; Massasa, E.E.; Medgalia, C.; David, E.; Giladi, A.; Moor, A.E.; et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol. 2018, 36, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Benhamouche, S.; Decaens, T.; Godard, C.; Chambrey, R.; Rickman, D.S.; Moinard, C.; Vasseur-Cognet, M.; Kuo, C.J.; Kahn, A.; Perret, C.; et al. APC tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev. Cell 2006, 10, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Mart, P.; et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Braeuning, A.; Menzel, M.; Kleinschnitz, E.-M.; Harada, N.; Tamai, Y.; Köhle, C.; Buchmann, A.; Schwarz, M. Serum components and activated Ha-ras antagonize expression of perivenous marker genes stimulated by β-catenin signaling in mouse hepatocytes. FEBS J. 2007, 274, 4766–4777. [Google Scholar] [CrossRef]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef]
- Matz-Soja, M.; Hovhannisyan, A.; Gebhardt, R. Hedgehog signalling pathway in adult liver: A major new player in hepatocyte metabolism and zonation? Med. Hypotheses 2013, 80, 589–594. [Google Scholar] [CrossRef]
- Cheng, X.; Kim, S.Y.; Okamoto, H.; Xin, Y.; Yancopoulos, G.D.; Murphy, A.J.; Gromada, J. Glucagon contributes to liver zonation. Proc. Natl. Acad. Sci. USA 2018, 115, E4111–E4119. [Google Scholar] [CrossRef]
- Stanulovic, V.S.; Kyrmizi, I.; Kruithof-de Julio, M.; Hoogenkamp, M.; Vermeulen, J.L.; Ruijter, J.M.; Talianidis, I.; Hakvoort, T.B.; Lamers, W.H. Hepatic HNF4α deficiency induces periportal expression of glutamine synthetase and other pericentral enzymes. Hepatology 2007, 45, 433–444. [Google Scholar] [CrossRef]
- Sekine, S.; Ogawa, R.; Mcmanus, M.T.; Kanai, Y.; Hebrok, M. Dicer is required for proper liver zonation. J. Pathol. 2009, 219, 365–372. [Google Scholar] [CrossRef]
- Russell, J.O.; Monga, S.P. Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu. Rev. Pathol. 2018, 13, 351–378. [Google Scholar] [CrossRef]
- Blitzer, J.T.; Nusse, R. A critical role for endocytosis in Wnt signaling. BMC Cell Biol. 2006, 7, 28. [Google Scholar] [CrossRef]
- Rocha, A.S.; Vidal, V.; Mertz, M.; Kendall, T.J.; Charlet, A.; Okamoto, H.; Schedl, A. The Angiocrine Factor Rspondin3 Is a Key Determinant of Liver Zonation. Cell Rep. 2015, 13, 1757–1764. [Google Scholar] [CrossRef]
- Choi, S.S.; Omenetti, A.; Syn, W.K.; Diehl, A.M. The role of Hedgehog signaling in fibrogenic liver repair. Int. J. Biochem. Cell Biol. 2011, 43, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Leibing, T.; Geraud, C.; Augustin, I.; Boutros, M.; Augustin, H.G.; Okun, J.G.; Langhans, C.D.; Zierow, J.; Wohlfeil, S.A.; Olsavszky, V.; et al. Angiocrine Wnt signaling controls liver growth and metabolic maturation in mice. Hepatology 2018, 68, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Cornesse, Y.; Brechtel, K.; Modaressi, S.; Jungermann, K. Perivenous expression of the mRNA of the three hypoxia-inducible factor α-subunits, HIF1α, HIF2α and HIF3α, in rat liver. Biochem. J. 2001, 354, 531–537. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Löfstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Ovenberger, M.; Poellinger, L.; Påhlman, S. Hypoxia inducible factor-2a in cancer. Cell Cycle 2007, 6, 919–926. [Google Scholar] [CrossRef]
- Heikkila, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3α variants in the hypoxia response. Cell. Mol. Life Sci. 2011, 68, 3885–3901. [Google Scholar] [CrossRef]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and characterization of hypoxia-inducible factor (HIF)-3α in human kidney: Suppression of HIF-mediated gene expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3α locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, L.; Yao, Q.; Li, Y.; Zhou, J.; Liu, Y.; Duan, C. Molecular, functional, and gene expression analysis of zebrafish hypoxia-inducible factor-3α. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1165–R1174. [Google Scholar] [CrossRef]
- Scharf, J.G.; Unterman, T.G.; Kietzmann, T. Oxygen-dependent modulation of insulin-like growth factor binding protein biosynthesis in primary cultures of rat hepatocytes. Endocrinology 2005, 146, 5433–5443. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J.; Semenza, G.L. Pathways for oxygen regulation and homeostasis: The 2016 albert lasker basic medical research award. JAMA 2016, 316, 1252–1253. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Kietzmann, T. Hypoxia-Inducible Factor Prolyl 4-Hydroxylases and Metabolism. Trends Mol. Med. 2018, 24, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Ginouvès, A.; Ilc, K.; Macías, N.; Pouysségur, J.; Berra, E. PHDs overactivation during chronic hypoxia “desensitizes” HIFa and protects cells from necrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4745–4750. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Michalopoulos, G.K.; Stolz, D.B. Peroxisomal localization of hypoxia-inducible factors and hypoxia-inducible factor regulatory hydroxylases in primary rat hepatocytes exposed to hypoxia-reoxygenation. Am J. Pathol. 2006, 169, 1251–1269. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Tajima, T.; Hori, S.; Matsuura, T.; Johnson, R.S.; Goda, N.; Suematsu, M. Hypoxia-inducible factor-1 is a determinant of lobular structure and oxygen consumption in the liver. Microcirculation 2013, 20, 385–393. [Google Scholar] [CrossRef]
- Tajima, T.; Goda, N.; Fujiki, N.; Hishiki, T.; Nishiyama, Y.; Senoo-Matsuda, N.; Shimazu, M.; Soga, T.; Yoshimura, Y.; Johnson, R.S.; et al. HIF-1a is necessary to support gluconeogenesis during liver regeneration. Biochem. Biophys. Res. Commun. 2009, 387, 789–794. [Google Scholar] [CrossRef]
- Wei, K.; Piecewicz, S.M.; McGinnis, L.M.; Taniguchi, C.M.; Wiegand, S.J.; Anderson, K.; Chan, C.W.; Mulligan, K.X.; Kuo, D.; Yuan, J.; et al. A liver Hif-2α-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat. Med. 2013, 19, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell. Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Suzuki, R.; Lee, K.; Tran, T.; Gunton, J.E.; Saha, A.K.; Patti, M.-E.; Goldfine, A.; Ruderman, N.B.; Gonzalez, F.J.; et al. Ablation of ARNT/HIF1ß in Liver Alters Gluconeogenesis, Lipogenic Gene Expression, and Serum Ketones. Cell Metab. 2009, 9, 565. [Google Scholar] [CrossRef]
- Minamishima, Y.A.; Kaelin, W.G., Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science 2010, 329, 407. [Google Scholar] [CrossRef]
- Schmucker, D.L.; Mooney, J.S.; Jones, A.L. Stereological analysis of hepatic fine structure in the Fischer 344 rat. Influence of sublobular location and animal age. J. Cell Biol. 1978, 78, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Jungermann, K. Metabolic zonation of liver parenchyma. Semin. Liver Dis. 1988, 8, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Carrero, P.; Okamoto, K.; Coumailleau, P.; O’Brien, S.; Tanaka, H.; Poellinger, L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1α. Mol. Cell. Biol. 2000, 20, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1α. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Diebold, I.; Flügel, D.; Becht, S.; Belaiba, R.S.; Bonello, S.; Hess, J.; Kietzmann, T.; Görlach, A. The hypoxia-inducible factor-2α is stabilized by oxidative stress involving NOX4. Antioxid. Redox Signal. 2010, 13, 425–436. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef]
- Liu, Q.; Berchner-Pfannschmidt, U.; Moller, U.; Brecht, M.; Wotzlaw, C.; Acker, H.; Jungermann, K.; Kietzmann, T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 4302–4307. [Google Scholar] [CrossRef]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive oxygen species activate the HIF-1α promoter via a functional NF-κB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef]
- BelAiba, R.S.; Bonello, S.; Zahringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia Up-Regulates Hypoxia-Inducible Factor-1α Transcription by Involving Phosphatidylinositol 3-Kinase and Nuclear Factor κB in Pulmonary Artery Smooth Muscle Cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef]
- Görlach, A.; Bonello, S. The cross-talk between NF-κB and HIF-1: Further evidence for a significant liaison. Biochem. J. 2008, 412, e17–e19. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1αa by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef]
- Scholz, C.C.; Taylor, C.T. Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 2013, 13, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Al Taleb, Z.; Petry, A.; Chi, T.F.; Mennerich, D.; Görlach, A.; Dimova, E.Y.; Kietzmann, T. Differential transcriptional regulation of hypoxia-inducible factor-1a by arsenite under normoxia and hypoxia: Involvement of Nrf2. J. Mol. Med. 2016, 94, 1153–1166. [Google Scholar] [CrossRef]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef]
- Koong, A.C.; Chen, E.Y.; Mivechi, N.F.; Denko, N.C.; Stambrook, P.; Giaccia, A.J. Hypoxic Activation of Nuclear Factor-kB Is Mediated by a Ras and Raf Signaling Pathway and Does Not Involve MAP Kinase (ERK1 or ERK2). Cancer Res. 1994, 54, 5273–5279. [Google Scholar] [PubMed]
- Nilakantan, H.; Kuttippurathu, L.; Parrish, A.; Hoek, J.B.; Vadigepalli, R. In vivo zonal variation and liver cell-type specific NF-κB localization after chronic adaptation to ethanol and following partial hepatectomy. PLoS ONE 2015, 10, e0140236. [Google Scholar] [CrossRef]
- Skoko, J.J.; Wakabayashi, N.; Noda, K.; Kimura, S.; Tobita, K.; Shigemura, N.; Tsujita, T.; Yamamoto, M.; Kensler, T.W. Loss of Nrf2 in mice evokes a congenital intrahepatic shunt that alters hepatic oxygen and protein expression gradients and toxicity. Toxicol. Sci. 2014, 141, 112–119. [Google Scholar] [CrossRef]
- Varela-Nallar, L.; Rojas-Abalos, M.; Abbott, A.C.; Moya, E.A.; Iturriaga, R.; Inestrosa, N.C. Chronic hypoxia induces the activation of the Wnt/β-catenin signaling pathway and stimulates hippocampal neurogenesis in wild-type and APPswe-PS1ΔE9 transgenic mice in vivo. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Mazumdar, J.; O’Brien, W.T.; Johnson, R.S.; Lamanna, J.C.; Chavez, J.C.; Klein, P.S.; Simon, M.C. O2 regulates stem cells through Wnt/ß-catenin signalling. Nat. Cell Biol. 2010, 12, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Zhang, J.; Fu, Q.; Wei, T.; Sun, X.; Chen, Q.; Yang, J.; Bai, X.; Liang, T. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol. Cancer 2017, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Kaidi, A.; Williams, A.C.; Paraskeva, C. Interaction between ß-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 2007, 9, 210–217. [Google Scholar] [CrossRef]
- Verras, M.; Papandreou, I.; Lim, A.L.; Denko, N.C. Tumor hypoxia blocks Wnt processing and secretion through the induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2008, 28, 7212–7224. [Google Scholar] [CrossRef]
- Näthke, I.; Rocha, S. Antagonistic crosstalk between APC and HIF-1α. Cell Cycle 2011, 10, 1545–1547. [Google Scholar] [CrossRef] [PubMed]
- Newton, I.P.; Kenneth, N.S.; Appleton, P.L.; Näthke, I.; Rocha, S. Adenomatous polyposis coli and hypoxia-inducible factor-1α have an antagonistic connection. Mol. Biol. Cell 2010, 21, 3630–3638. [Google Scholar] [CrossRef]
- Brocardo, M.; Lei, Y.; Tighe, A.; Taylor, S.S.; Mok, M.T.S.; Henderson, B.R. Mitochondrial targeting of adenomatous polyposis coli protein is stimulated by truncating cancer mutations: Regulation of Bcl-2 and implications for cell survival. J. Biol. Chem. 2008, 283, 5950–5959. [Google Scholar] [CrossRef]
- Lui, C.; Mills, K.; Brocardo, M.G.; Sharma, M.; Henderson, B.R. APC as a mobile scaffold: Regulation and function at the nucleus, centrosomes, and mitochondria. IUBMB Life 2012, 64, 209–214. [Google Scholar] [CrossRef]
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC Min/+ mice. Am. J. Pathol. 2012, 180, 24–31. [Google Scholar] [CrossRef]
- Hoogeboom, D.; Burgering, B.M.T. Should I stay or should I go: β-catenin decides under stress. Biochim. Biophys. Acta Rev. Cancer 2009, 1796, 63–74. [Google Scholar] [CrossRef]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Lenart, J.; Dombrowski, F.; Görlach, A.; Kietzmann, T. Deficiency of manganese superoxide dismutase in hepatocytes disrupts zonated gene expression in mouse liver. Arch. Biochem. Biophys. 2007, 462, 238–244. [Google Scholar] [CrossRef]
- Konzack, A.; Jakupovic, M.; Kubaichuk, K.; Görlach, A.; Dombrowski, F.; Miinalainen, I.; Sormunen, R.; Kietzmann, T. Mitochondrial Dysfunction Due to Lack of Manganese Superoxide Dismutase Promotes Hepatocarcinogenesis. Antioxid. Redox Signal. 2015, 23, 1059–1075. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.R.; Athineos, D.; Meniel, V.S.; Wilkins, J.A.; Ridgway, R.A.; Burke, Z.D.; Muncan, V.; Clarke, A.R.; Sansom, O.J. B-catenin deficiency, but not Myc deletion, suppresses the immediate phenotypes of APC loss in the liver. Proc. Natl. Acad. Sci. USA 2008, 105, 18919–18923. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Chute, J. Underlying potential: Cellular and molecular determinants of adult liver repair. J. Clin. Investig. 2013, 123, 1858–1860. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.-X.; Choi, S.S.; Qi, Y.; Chen, W.; Bustamante, M.; Huang, J.; Zdanowicz, M.; Camp, T.; Torbenson, M.S.; et al. Role for Hedgehog signaling in hepatic stellate cell activation and viability. Lab. Investig. 2005, 85, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R. Metabolic zonation of the liver: Regulation and implications for liver function. Pharmacol. Ther. 1992, 53, 275–354. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008, 34, 643–648. [Google Scholar] [CrossRef]
- Matz-Soja, M.; Rennert, C.; Schonefeld, K.; Aleithe, S.; Boettger, J.; Schmidt-Heck, W.; Weiss, T.S.; Hovhannisyan, A.; Zellmer, S.; Kloting, N.; et al. Hedgehog signaling is a potent regulator of liver lipid metabolism and reveals a GLI-code associated with steatosis. Elife 2016, 5. [Google Scholar] [CrossRef]
- Hazel, S.J.; Sandberg Nordqvist, A.; Hall, K.; Nilsson, M.; Schalling, M. Differential expression of IGF-I and IGF-binding protein-1 and -2 in periportal and perivenous zones of rat liver. J. Endocrinol. 1998, 157, 285–294. [Google Scholar] [CrossRef]
- Matz-Soja, M.; Aleithe, S.; Marbach, E.; Böttger, J.; Arnold, K.; Schmidt-Heck, W.; Kratzsch, J.; Gebhardt, R. Hepatic Hedgehog signaling contributes to the regulation of IGF1 and IGFBP1 serum levels. Cell Commun. Signal. 2014, 12. [Google Scholar] [CrossRef]
- Bijlsma, M.F.; Groot, A.P.; Oduro, J.P.; Franken, R.J.; Schoenmakers, S.H.; Peppelenbosch, M.P.; Spek, C.A. Hypoxia induces a hedgehog response mediated by HIF-1α. J. Cell. Mol. Med. 2009, 13, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, K.; Gao, D.; Zhu, G.; Wu, D.; Wang, X.; Chen, Y.; Du, Y.; Song, W.; Ma, Z.; et al. Reciprocal regulation of hypoxia-inducible factor 2α and GLI1 expression associated with the radioresistance of renal cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Yamasaki, A.; Kawamoto, M.; Imaizumi, A.; Katano, M. Hypoxia but not normoxia promotes Smoothened transcription through upregulation of RBPJ and Mastermind-like 3 in pancreatic cancer. Cancer Lett. 2016, 371, 143–150. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, M.; Xing, L.; Wang, Y.; Xiao, Y.; Wu, Y. HIF-1a contributes to proliferation and invasiveness of neuroblastoma cells via SHH signaling. PLoS ONE 2015, 10, e0121115. [Google Scholar]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-Mediated Suppression of Acyl-CoA Dehydrogenases and Fatty Acid Oxidation Is Critical for Cancer Progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe-Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Fitamant, J.; Kottakis, F.; Benhamouche, S.; Tian, H.S.; Chuvin, N.; Parachoniak, C.A.; Nagle, J.M.; Perera, R.M.; Lapouge, M.; Deshpande, V.; et al. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Rep. 2015, 10, 1692–1707. [Google Scholar] [CrossRef]
- Font-Burgada, J.; Shalapour, S.; Ramaswamy, S.; Hsueh, B.; Rossell, D.; Umemura, A.; Taniguchi, K.; Nakagawa, H.; Valasek, M.A.; Ye, L.; et al. Hybrid Periportal Hepatocytes Regenerate the Injured Liver without Giving Rise to Cancer. Cell 2015, 162, 766–779. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef]
- Grijalva, J.L.; Huizenga, M.; Mueller, K.; Rodriguez, S.; Brazzo, J.; Camargo, F.; Sadri-Vakili, G.; Vakili, K. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G196–G204. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Takano, N.; Luo, W.; Lu, H.; Bullen, J.W.; Samanta, D.; Liang, H.; Semenza, G.L. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget 2014, 5, 12509–12527. [Google Scholar] [CrossRef] [PubMed]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Desiderio, M.A. Hypoxia inducible factor-1 is activated by transcriptional co-activator with PDZ-binding motif (TAZ) versus WWdomain-containing oxidoreductase (WWOX) in hypoxic microenvironment of bone metastasis from breast cancer. Eur. J. Cancer 2013, 49, 2608–2618. [Google Scholar] [CrossRef]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Luo, W.; Bullen, J.W.; Liang, H.; Semenza, G.L. HIF-1α and TAZ serve as reciprocal co-activators in human breast cancer cells. Oncotarget 2015, 6, 11768–11778. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-associated protein (YAP) binds to HIF-1α and sustains HIF-1α protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.O. Angiogenesis and liver fibrosis. World J. Hepatol. 2015, 7, 377–391. [Google Scholar] [CrossRef]
- Zhang, C.; Bian, M.; Chen, X.; Jin, H.; Zhao, S.; Yang, X.; Shao, J.; Chen, A.; Guo, Q.; Zhang, F.; et al. Oroxylin A prevents angiogenesis of LSECs in liver fibrosis via inhibition of YAP/HIF-1α signaling. J. Cell. Biochem. 2018, 119, 2258–2268. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.D.; Chen, W.D.; Wang, X.; Lou, G.; Liu, N.; Lin, M.; Forman, B.M.; Huang, W. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology 2012, 56, 2336–2343. [Google Scholar] [CrossRef]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef]
- Pear, W.S.; Simon, M.C. Lasting longer without oxygen: The influence of hypoxia on Notch signaling. Cancer Cell 2005, 8, 435–437. [Google Scholar] [CrossRef]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Systematic review: The diagnosis and staging of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2011, 33, 525–540. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Argo, C.K.; Caldwell, S.H. Epidemiology and Natural History of Non-Alcoholic Steatohepatitis. Clin. Liver Dis. 2009, 13, 511–531. [Google Scholar] [CrossRef]
- Hui, J.M.; Kench, J.G.; Chitturi, S.; Sud, A.; Farrell, G.C.; Byth, K.; Hall, P.; Khan, M.; George, J. Long-term outcomes of cirrhosis in nonalcoholic steatohepatitis compared with hepatitis C. Hepatology 2003, 38, 420–427. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St. Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Kondo, K.; Sugioka, T.; Tsukada, K.; Aizawa, M.; Takizawa, M.; Shimizu, K.; Morimoto, M.; Suematsu, M.; Goda, N. Fenofibrate, a peroxisome proliferator-activated receptor a agonist, improves hepatic microcirculatory patency and oxygen availability in a high-fat-diet-induced fatty liver in mice. In Oxygen Transport to Tissue XXXI; Springer: Boston, MA, USA, 2010. [Google Scholar]
- Piguet, A.-C.; Stroka, D.; Zimmermann, A.; Dufour, J.-F. Hypoxia aggravates non-alcoholic steatohepatitis in mice lacking hepatocellular PTEN. Clin. Sci. 2010, 118, 401–410. [Google Scholar] [CrossRef]
- Schleicher, J.; Guthke, R.; Dahmen, U.; Dirsch, O.; Holzhuetter, H.G.; Schuster, S. A theoretical study of lipid accumulation in the liver—Implications for nonalcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2014, 1841, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Quistorff, B.; Katz, N.; Witters, L.A. Hepatocyte heterogeneity in the metabolism of fatty acids: Discrepancies on zonation of acetyl-CoA carboxylase. Enzyme 1992, 46, 59–71. [Google Scholar] [CrossRef]
- Debois, D.; Bralet, M.-P.; Le Naour, F.; Brunelle, A.; Laprévote, O. In Situ lipidomic analysis of nonalcoholic fatty liver by cluster TOF-SIMS imaging. Anal. Chem. 2009, 81, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Wattacheril, J.; Seeley, E.H.; Angel, P.; Chen, H.; Bowen, B.P.; Lanciault, C.; Caprioli, R.M.; Abumrad, N.; Flynn, C.R. Differential Intrahepatic Phospholipid Zonation in Simple Steatosis and Nonalcoholic Steatohepatitis. PLoS ONE 2013, 8, e57165. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Safran, M.; Buckley, M.R.; Ebert, B.L.; Glickman, J.; Bosenberg, M.; Regan, M.; Kaelin, W.G., Jr. Failure to prolyl hydroxylate hypoxia-inducible factor α phenocopies VHL inactivation in vivo. EMBO J. 2006, 25, 4650–4662. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.M.; Schonenberger, M.J.; Trotzmuller, M.; Horn, M.; Elsasser, H.P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Farr, R.L.; Lismont, C.; Terlecky, S.R.; Fransen, M. Peroxisome biogenesis in mammalian cells: The impact of genes and environment. Biochim. Biophys. Acta 2016, 1863, 1049–1060. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Xue, J.; Avery, J.; Wu, J.; Lind, S.E.; Ding, W.Q. Activation of peroxisome proliferator-activated receptor α (PPARα) suppresses hypoxia-inducible factor-1α (HIF-1α) signaling in cancer cells. J. Biol. Chem. 2012, 287, 35161–35169. [Google Scholar] [CrossRef]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef]
- Morello, E.; Sutti, S.; Foglia, B.; Novo, E.; Cannito, S.; Bocca, C.; Rajsky, M.; Bruzzì, S.; Abate, M.L.; Rosso, C.; et al. Hypoxia-inducible factor 2a drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine-rich glycoprotein. Hepatology 2018, 67, 2196–2214. [Google Scholar] [CrossRef]
- Xie, C.; Yagai, T.; Luo, Y.; Liang, X.; Chen, T.; Wang, Q.; Sun, D.; Zhao, J.; Ramakrishnan, S.K.; Sun, L.; et al. Activation of intestinal hypoxia-inducible factor 2α during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Lu, C.-W.; Lin, S.-C.; Chen, K.-F.; Lai, Y.-Y.; Tsai, S.-J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, A.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting complex i activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 Controls Mitochondrial Metabolism during Hypoxia by Repressing the Iron-Sulfur Cluster Assembly Proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J. Excess weight risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Siu, L.; Foont, J.; Wands, J.R. Hepatitis C virus and alcohol. Semin. Liver Dis. 2009, 29, 188–199. [Google Scholar] [CrossRef]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J.; Shuhart, M.C.; Davis, G.L.; Franco, J.; Harrison, S.A.; Howell, C.D.; Ling, S.C.; Liu, L.U.; et al. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Mandayam, S.; Jamal, M.M.; Morgan, T.R. Epidemiology of alcoholic liver disease. Semin. Liver Dis. 2004, 24, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Israel, Y.; Videla, L.; MacDonald, A.; Bernstein, J. Metabolic alterations produced in the liver by chronic ethanol administration. Comparison between the effects produced by ethanol and by thyroid hormones. Biochem. J. 1973, 134, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Yuki, T.; Thurman, R.G. The swift increase in alcohol metabolism: Time course for the increase in hepatic oxygen uptake and the involvement of glycolysis. Biochem. J. 1980, 186, 119–126. [Google Scholar] [CrossRef] [PubMed]
- French, S.W. The role of hypoxia in the pathogenesis of alcoholic liver disease. Hepatol. Res. 2004, 29, 69–74. [Google Scholar] [CrossRef]
- Jungermann, K.; Kietzmann, T. Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology 2000, 31, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Raleigh, J.A.; Bradford, B.U.; Thurman, R.G.; Hara, H. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: Role of Kupffer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 1996, 271, G494–G500. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Iimuro, Y.; Yin, M.; Raleigh, J.A.; Thurman, R.G. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology 1997, 25, 920–926. [Google Scholar] [CrossRef]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1α is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef]
- Laitakari, A.; Ollonen, T.; Kietzmann, T.; Walkinshaw, G.; Mennerich, D.; Izzi, V.; Haapasaari, K.M.; Myllyharju, J.; Serpi, R.; Dimova, E.Y.; et al. Systemic inactivation of hypoxia-inducible factor prolyl 4-hydroxylase 2 in mice protects from alcohol-induced fatty liver disease. Redox Biol. 2019, 22, 101145. [Google Scholar] [CrossRef]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef]
- Novo, E.; Povero, D.; Busletta, C.; Paternostro, C.; Di Bonzo, L.V.; Cannito, S.; Compagnone, A.; Bandino, A.; Marra, F.; Colombatto, S.; et al. The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J. Pathol. 2012, 226, 588–597. [Google Scholar] [CrossRef]
- Moon, J.O.; Welch, T.P.; Gonzalez, F.J.; Copple, B.L. Reduced liver fibrosis in hypoxia-inducible factor-1α-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G582–G592. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Chiang, D.J.; McMullen, M.R.; Nagy, L.E. Moderate, chronic ethanol feeding exacerbates carbon tetrachloride–induced hepatic fibrosis via hepatocyte-specific hypoxia-inducible factor 1α. Pharmacol. Res. Perspect. 2014, 2, e00061. [Google Scholar] [CrossRef]
- Copple, B.L.; Kaska, S.; Wentling, C. Hypoxia-inducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J. Pharmacol. Exp. Ther. 2012, 341, 307–316. [Google Scholar] [CrossRef]
- Copple, B.L.; Bai, S.; Burgoon, L.D.; Moon, J.-O. Hypoxia-inducible factor-1α regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int. 2011, 31, 230–244. [Google Scholar] [CrossRef]
- Zagzag, D.; Krishnamachary, B.; Yee, H.; Okuyama, H.; Chiriboga, L.; Ali, M.A.; Melamed, J.; Semenza, G.L. Stromal cell-derived factor-1α and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: Von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 2005, 65, 6178–6188. [Google Scholar] [CrossRef]
- Hempel, M.; Schmitz, A.; Winkler, S.; Kucukoglu, O.; Bruckner, S.; Niessen, C.; Christ, B. Pathological implications of cadherin zonation in mouse liver. Cell. Mol. Life Sci. 2015, 72, 2599–2612. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Lin, Y.; Shimizu-Hirota, R.; Hanada, S.; Neilson, E.G.; Greenson, J.K.; Weiss, S.J. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol. Cell. Biol. 2011, 31, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, C.; Amicone, L.; Alonzi, T.; Marchetti, A.; Mancone, C.; Tripodi, M. Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte. Liver Int. 2015, 35, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Yan, X.; Xu, Y.; Luo, F.; Ye, J.; Yan, H.; Yang, X.; Huang, X.; Zhang, J.; et al. HIFs enhance the migratory and neoplastic capacities of hepatocellular carcinoma cells by promoting EMT. Tumour Biol. 2014, 35, 8103–8114. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, L.; Sun, C.; Ireland, N.; Shah, Y.M.; Liu, Y.; Rui, L. Insulin/Snail1 axis ameliorates fatty liver disease by epigenetically suppressing lipogenesis. Nat. Commun. 2018, 9, 2751. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Guo, X.; Li, D.; Chen, Y.; An, J.; Wang, K.; Xu, Z.; Chen, Z.; Xing, J. SNP rs2057482 in HIF1A gene predicts clinical outcome of aggressive hepatocellular carcinoma patients after surgery. Sci. Rep. 2015, 5, 11846. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, P.C.; Chen, M.K.; Su, S.C.; Ueng, K.C.; Chen, Y.C.; Hsieh, Y.H.; Liu, Y.F.; Tsai, H.T.; Yang, S.F. Hypoxia inducible factor-1α gene polymorphism G1790A and its interaction with tobacco and alcohol consumptions increase susceptibility to hepatocellular carcinoma. J. Surg. Oncol. 2010, 102, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yao, D.; Wang, L.; Wu, W.; Qiu, L.; Yao, M.; Yao, N.; Zhang, H.; Yu, D.; Ni, Q. Expression characteristics of hypoxia-inducible factor-1α and its clinical values in diagnosis and prognosis of hepatocellular carcinoma. Hepatitis Mon. 2011, 11, 36–43. [Google Scholar] [CrossRef]
- Bangoura, G.; Liu, Z.S.; Qian, Q.; Jiang, C.Q.; Yang, G.F.; Jing, S. Prognostic significance of HIF-2α/EPAS1 expression in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 3176–3182. [Google Scholar] [CrossRef]
- Xie, H.; Song, J.; Liu, K.; Ji, H.; Shen, H.; Hu, S.; Yang, G.; Du, Y.; Zou, X.; Jin, H.; et al. The expression of hypoxia-inducible factor-1α in hepatitis B virus-related hepatocellular carcinoma: Correlation with patients’ prognosis and hepatitis B virus X protein. Dig. Dis. Sci. 2008, 53, 3225–3233. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Bockhorn, M.; Praha, C.; Baba, H.A.; Broelsch, C.E.; Frilling, A.; Weber, F. Deregulation of HIF1-α and hypoxia-regulated pathways in hepatocellular carcinoma and corresponding non-malignant liver tissue-influence of a modulated host stroma on the prognosis of HCC. Langenbeck’s Arch. Surg. 2010, 395, 395–405. [Google Scholar] [CrossRef]
- Xiang, Z.L.; Zeng, Z.C.; Fan, J.; Tang, Z.Y.; He, J.; Zeng, H.Y.; Chang, J.Y. The expression of HIF-1α in primary hepatocellular carcinoma and its correlation with radiotherapy response and clinical outcome. Mol. Biol. Rep. 2012, 39, 2021–2029. [Google Scholar] [CrossRef]
- Yang, S.L.; Liu, L.P.; Jiang, J.X.; Xiong, Z.F.; He, Q.J.; Wu, C. The correlation of expression levels of HIF-1α and HIF-2α in hepatocellular carcinoma with capsular invasion, portal vein tumor thrombi and patients’ clinical outcome. Jpn. J. Clin. Oncol. 2014, 44, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.S.; Chen, X.H.; Yin, X.; Zhang, B.H. Prognostic Significance of HIF-1α Expression in Hepatocellular Carcinoma: A Meta-Analysis. PLoS ONE 2013, 8, e65753. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.K.; Brimacombe, C.L.; Rowe, I.A.; Reynolds, G.M.; Fletcher, N.F.; Stamataki, Z.; Bhogal, R.H.; Simões, M.L.; Ashcroft, M.; Afford, S.C.; et al. A dual role for hypoxia inducible factor-1α in the hepatitis C virus lifecycle and hepatoma migration. J. Hepatol. 2012, 56, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.X.; Xu, Y.; Yang, X.R.; Wang, W.M.; Bai, H.; Shi, R.Y.; Nayar, S.K.; Devbhandari, R.P.; He, Y.Z.; Zhu, Q.F.; et al. Hypoxia inducible factor 2 α inhibits hepatocellular carcinoma growth through the transcription factor dimerization partner 3/E2F transcription factor 1-dependent apoptotic pathway. Hepatology 2013, 57, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef]
- Zhen, L.; Shijie, N.; Shuijun, Z. Tumor PHD2 expression is correlated with clinical features and prognosis of patients with HCC receiving liver resection. Medicine 2014, 93, e179. [Google Scholar] [CrossRef]
- Tao, Y.; Lin, F.; Li, R.; Shen, J.; Wang, Z. Prolyl hydroxylase-2 inhibits liver tumor cell proliferation and cyclin D1 expression in a hydroxylase-dependent manner. Int. J. Biochem. Cell Biol. 2016, 77, 129–140. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Ma, C.; Li, B. MicroRNA-30a suppresses the proliferation, migration and invasion of human renal cell carcinoma cells by directly targeting ADAM9. Oncol. Lett. 2018, 16, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Hua, S.; Li, G.; Wang, S.; Cheng, X.; He, S.; Wu, P.; Chen, X. Prolyl hydroxylase domain protein 3 and asparaginyl hydroxylase factor inhibiting HIF-1 levels are predictive of tumoral behavior and prognosis in hepatocellular carcinoma. Oncotarget 2017, 8, 12983–13002. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Harnoss, J.M.; Platzer, L.K.; Burhenne, J.; Radhakrishnan, P.; Cai, J.; Strowitzki, M.J.; Weiss, J.; Ritter, A.S.; Mollenhauer, M.; Schmidt, T.; et al. Prolyl Hydroxylase Inhibition Enhances Liver Regeneration Without Induction of Tumor Growth. Ann. Surg. 2017, 265, 782–791. [Google Scholar] [CrossRef]
- Van de Groenendaal-Meent, D.; Adel, M.D.; Noukens, J.; Rijnders, S.; Krebs-Brown, A.; Mateva, L.; Alexiev, A.; Schaddelee, M. Effect of Moderate Hepatic Impairment on the Pharmacokinetics and Pharmacodynamics of Roxadustat, an Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor. Clin. Drug Investig. 2016, 36, 743–751. [Google Scholar] [CrossRef]
- Howard, T.K.; Klintmalm, G.B.G.; Cofer, J.B.; Husberg, B.S.; Goldstein, R.M.; Gonwa, T.A. The influence of preservation injury on rejection in the hepatic transplant recipient. Transplantation 1990, 49, 103–107. [Google Scholar] [CrossRef]
- Serracino-Inglott, F.; Habib, N.A.; Mathie, R.T. Hepatic ischemia-reperfusion injury. Am. J. Surg. 2001, 181, 160–166. [Google Scholar] [CrossRef]
- Guo, J.Y.; Yang, T.; Sun, X.G.; Zhou, N.Y.; Li, F.S.; Long, D.; Lin, T.; Li, P.Y.; Feng, L. Ischemic postconditioning attenuates liver warm ischemia-reperfusion injury through Akt-eNOS-NO-HIF pathway. J. Biomed. Sci. 2011, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Plock, J.; Frese, S.; Keogh, A.; Bisch-Knaden, S.; Ayuni, E.; Corazza, N.; Weikert, C.; Jakob, S.; Erni, D.; Dufour, J.-F.; et al. Activation of non-ischemic, hypoxia-inducible signalling pathways up-regulate cytoprotective genes in the murine liver. J. Hepatol. 2007, 47, 538–545. [Google Scholar] [CrossRef]
- Zhong, Z.; Ramshesh, V.K.; Rehman, H.; Currin, R.T.; Sridharan, V.; Theruvath, T.P.; Kim, I.; Wright, G.L.; Lemasters, J.J. Activation of the oxygen-sensing signal cascade prevents mitochondrial injury after mouse liver ischemia-reperfusion. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G823–G832. [Google Scholar] [CrossRef]
- Ben Mosbah, I.; Mouchel, Y.; Pajaud, J.; Ribault, C.; Lucas, C.; Laurent, A.; Boudjema, K.; Morel, F.; Corlu, A.; Compagnon, P. Pretreatment with Mangafodipir Improves Liver Graft Tolerance to Ischemia/Reperfusion Injury in Rat. PLoS ONE 2012, 7, e50235. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Lee, P.C.; Huang, Y.T.; Lee, W.P.; Kuo, Y.J.; Lee, K.C.; Hsieh, Y.C.; Lee, T.Y.; Lin, H.C. Involvement of the HIF-1α and wnt/β-catenin pathways in the protective effects of losartan on fatty liver graft with ischaemia/reperfusion injury. Clin. Sci. 2014, 126, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Van Geyte, K.; Fraisl, P.; Kiss, J.; Aragonés, J.; Mazzone, M.; Mairbäurl, H.; De Bock, K.; Jeoung, N.H.; Mollenhauer, M.; et al. Loss or Silencing of the PHD1 Prolyl Hydroxylase Protects Livers of Mice Against Ischemia/Reperfusion Injury. Gastroenterology 2010, 138, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, S.F.; Fabian, Z.; Schaible, B.; Lenihan, C.R.; Schwarzl, T.; Rodriguez, J.; Zheng, X.; Li, Z.; Tambuwala, M.M.; Higgins, D.G.; et al. Prolyl hydroxylase-1 regulates hepatocyte apoptosis in an NF-κB-dependent manner. Biochem. Biophys. Res. Commun. 2016, 474, 579–586. [Google Scholar] [CrossRef]
- Mollenhauer, M.; Kiss, J.; Dudda, J.; Kirchberg, J.; Rahbari, N.; Radhakrishnan, P.; Niemietz, T.; Rausch, V.; Weitz, J.; Schneider, M. Deficiency of the oxygen sensor PHD1 augments liver regeneration after partial hepatectomy. Langenbeck’s Arch. Surg. 2012, 397, 1313–1322. [Google Scholar] [CrossRef]
- Adluri, R.S.; Thirunavukkarasu, M.; Dunna, N.R.; Zhan, L.; Oriowo, B.; Takeda, K.; Sanchez, J.A.; Otani, H.; Maulik, G.; Fong, G.H.; et al. Disruption of hypoxia-inducible transcription factor-prolyl hydroxylase domain-1 (PHD-1-/-) attenuates ex vivo myocardial ischemia/reperfusion injury through hypoxia-inducible factor-1α transcription factor and its target genes in mice. Antioxid. Redox Signal. 2011, 15, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Hyvarinen, J.; Hassinen, I.E.; Sormunen, R.; Maki, J.M.; Kivirikko, K.I.; Koivunen, P.; Myllyharju, J. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J. Biol. Chem. 2010, 285, 13646–13657. [Google Scholar] [CrossRef] [PubMed]
- Karsikas, S.; Myllymäki, M.; Heikkilä, M.; Sormunen, R.; Kivirikko, K.I.; Myllyharju, J.; Serpi, R.; Koivunen, P. HIF-P4H-2 deficiency protects against skeletal muscle ischemia-reperfusion injury. J. Mol. Med. 2016, 94, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Pi, X.; Wang, Z.; He, J.; Willis, M.S.; Patterson, C. Depletion of PHD3 protects heart from ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis. J. Mol. Cell. Cardiol. 2015, 80, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Luo, W.; Zhan, H.; Semenza, G.L. Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc. Natl. Acad. Sci. USA 2013, 110, 17462–17467. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, K.; Cai, Z.; Gupta, R.; Parajuli, N.; Fox-Talbot, K.; Darshan, M.S.; Gonzalez, F.J.; Semenza, G.L. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2012, 109, 10504–10509. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-α-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat. Commun. 2018, 9, 816. [Google Scholar] [CrossRef] [PubMed]
- Olenchock, B.A.; Moslehi, J.; Baik, A.H.; Davidson, S.M.; Williams, J.; Gibson, W.J.; Chakraborty, A.A.; Pierce, K.A.; Miller, C.M.; Hanse, E.A.; et al. EGLN1 Inhibition and Rerouting of α-Ketoglutarate Suffice for Remote Ischemic Protection. Cell 2016, 164, 884–895. [Google Scholar] [CrossRef]
- FibroGen. FibroGen Announces Approval of Roxadustat in China for the Treatment of Anemia in Chronic Kidney Disease Patients on Dialysis [Media Release]. Available online: http://investor.fibrogen.com/phoenix.zhtml?c=253783&p=irol-newsArticle&ID=2380952 (accessed on 17 December 2018).
- European Association for the Study of the Liver. Electronic address: Easloffice@easloffice.eu, European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kietzmann, T. Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters. Int. J. Mol. Sci. 2019, 20, 2347. https://doi.org/10.3390/ijms20092347
Kietzmann T. Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters. International Journal of Molecular Sciences. 2019; 20(9):2347. https://doi.org/10.3390/ijms20092347
Chicago/Turabian StyleKietzmann, Thomas. 2019. "Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters" International Journal of Molecular Sciences 20, no. 9: 2347. https://doi.org/10.3390/ijms20092347
APA StyleKietzmann, T. (2019). Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters. International Journal of Molecular Sciences, 20(9), 2347. https://doi.org/10.3390/ijms20092347