The Regulation of Bone Metabolism and Disorders by Wnt Signaling

 , , ,
, , ,
Abstract
1. Introduction
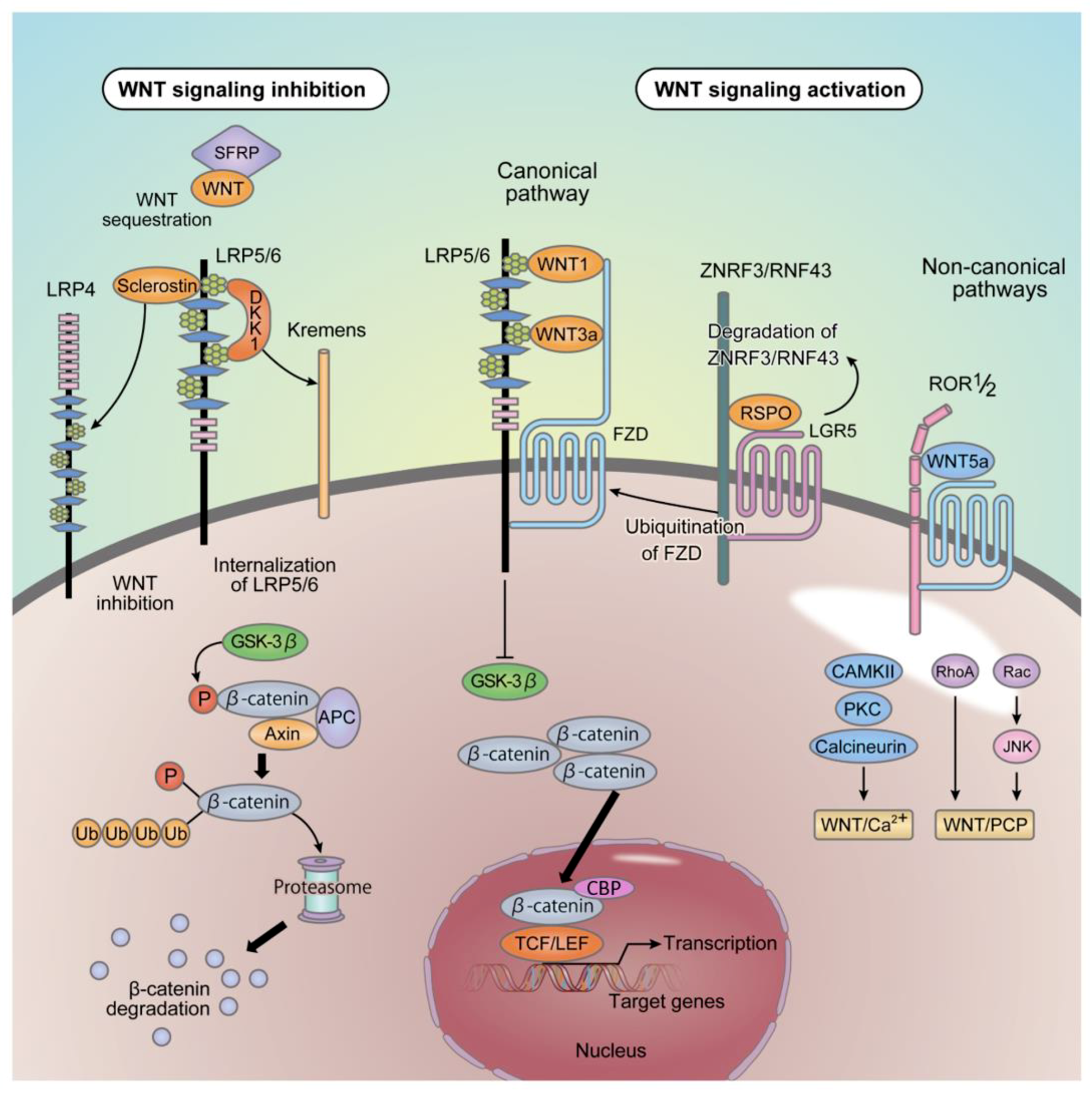
2. Outline of Wnt Signaling
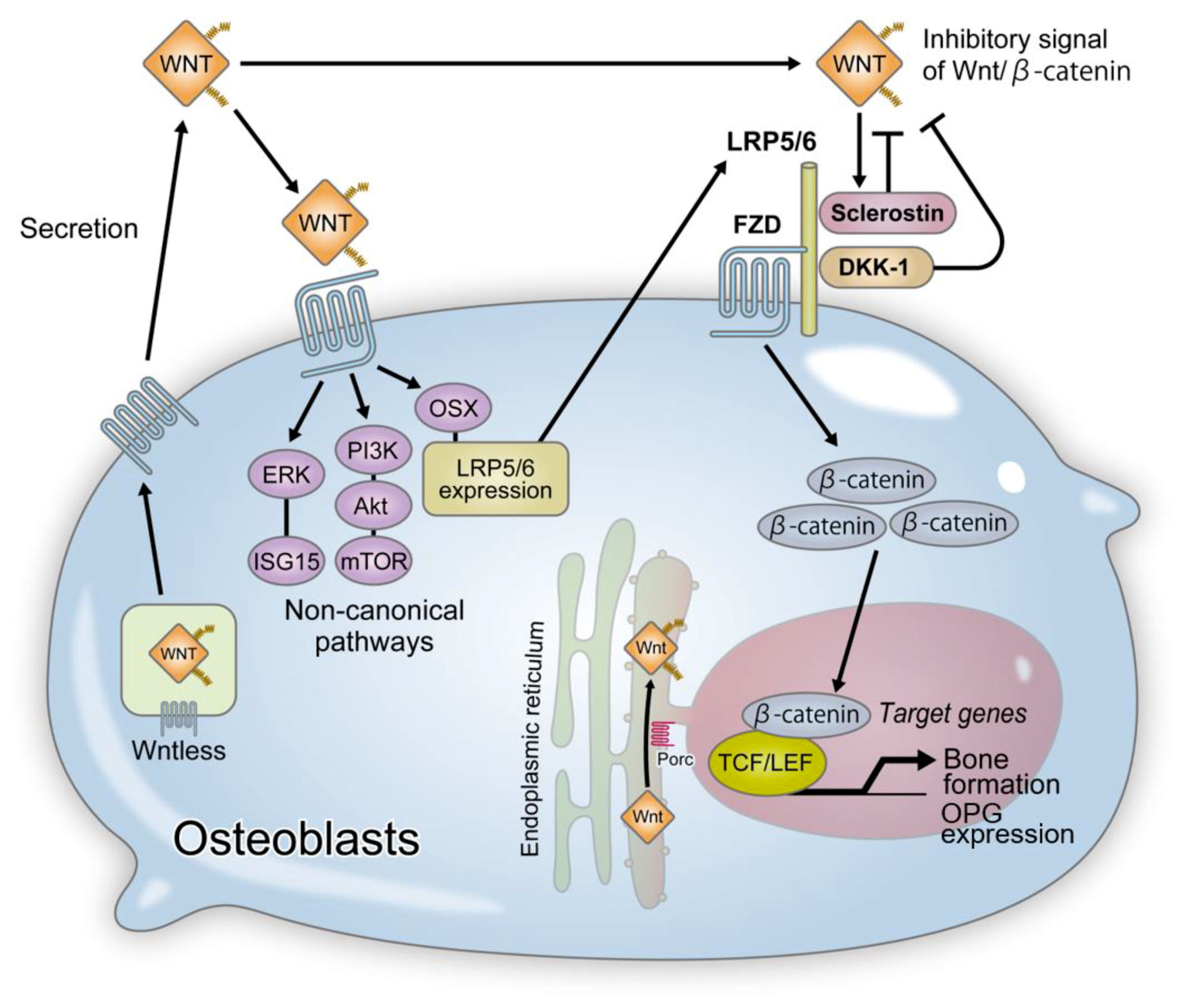
2.1. Porcupine (Porc) and Wntless (Wls)
2.2. Canonical and Non-Canonical Pathways
2.3. Inhibitors of Wnt Signaling
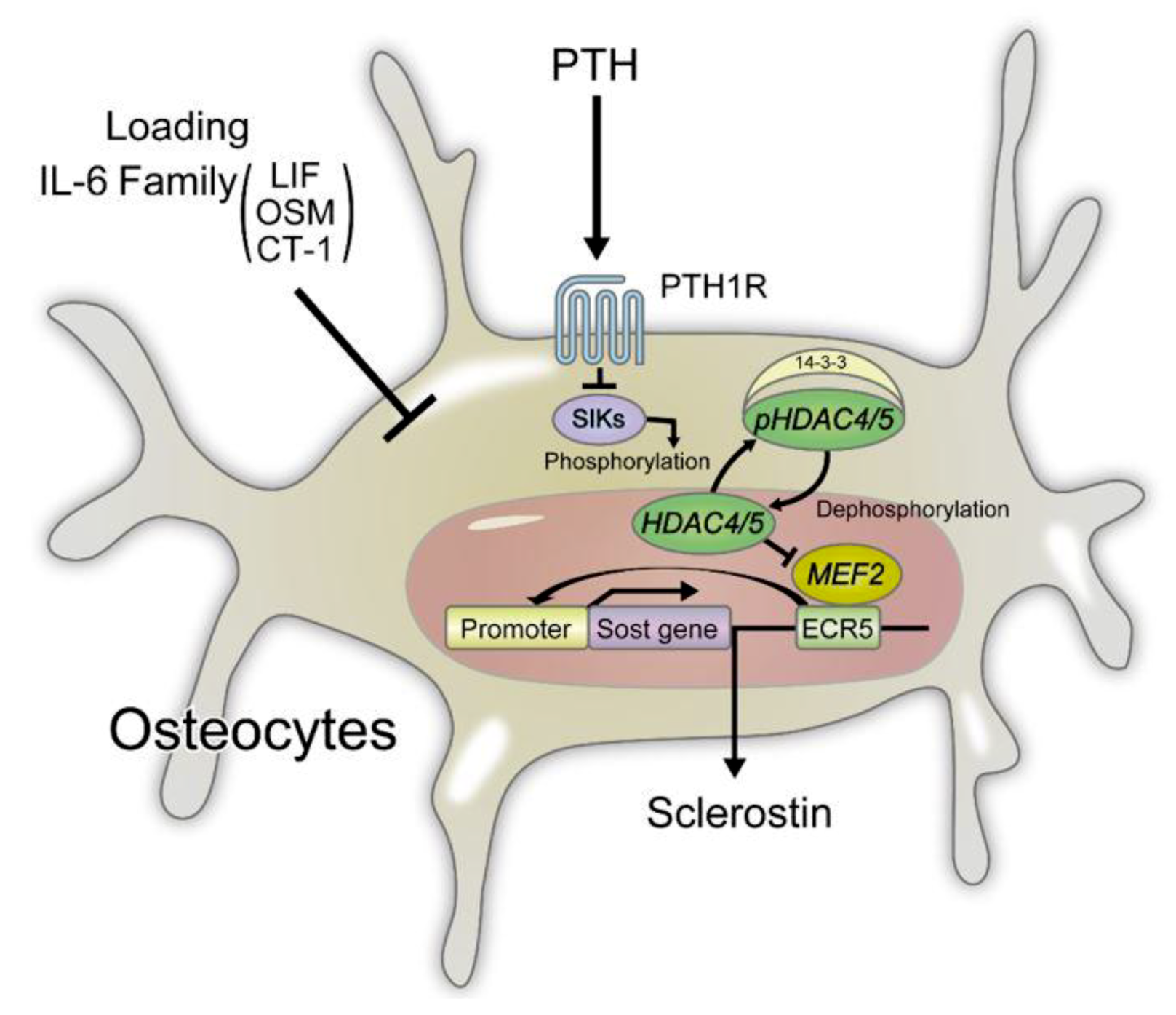
2.4. Sclerostin
2.5. ZNRF3 and RNF43
3. The Roles of Wnt Signaling in Bone Turnover
4. Wnt Signaling and Bone Formation
4.1. LRP5 and Osteoporosis-Pseudoglioma Syndrome
4.2. Sclerostin and Sclerosteosis/Endosteal Hyperostosis
4.3. Regulation of Sclerostin Expression in Osteocytes
4.4. Functional Regulation of Sclerostin by LRP4
4.5. Other Wnt Inhibitors and Bone Formation
4.6. Wnt Promotion and Bone Formation by RSPO and LGR
4.7. Porcupine/Wntless and Focal Dermal Hypoplasia
4.8. Wnt1 and Osteogenesis Imperfecta
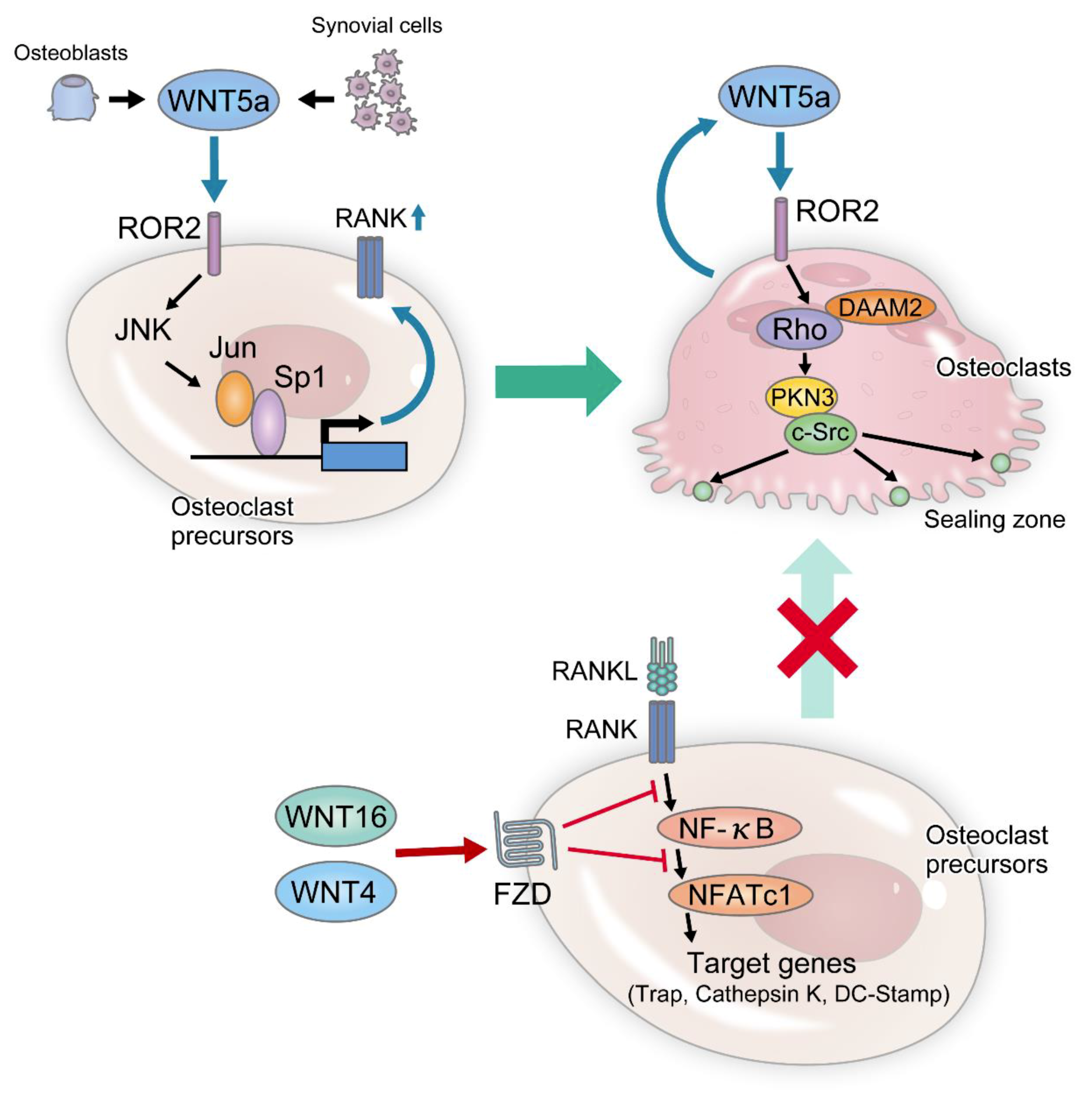
4.9. Non-Canonical Wnt Signaling and Bone Formation
5. Wnt Signaling and Bone Resorption
5.1. Canonical Wnt Signaling Pathway and Bone Resorption
5.2. Non-Canonical Wnt Signaling Pathway and Bone Resorption
6. Wnt Signaling and Musculoskeletal Disorders: Recent Findings and Clinical Application
6.1. Osteoporosis
6.1.1. Wnt-Related Molecules Involved in Osteoporosis
6.1.2. Existing Therapies for Osteoporosis
6.1.3. Novel Therapies for Osteoporosis
6.1.4. Clinical Issues
6.2. Osteoarthritis
6.2.1. Wnt-Related Molecules Involved in Osteoarthritis
6.2.2. Existing Therapies for Osteoarthritis
6.2.3. Preclinical Findings and Potential Novel Therapies for Osteoarthritis
6.2.4. Clinical Issues
6.3. Rheumatoid Arthritis
6.3.1. Wnt-Related Molecules Involved in Rheumatoid Arthritis
6.3.2. Existing Therapies for Rheumatoid Arthritis
6.3.3. Preclinical Findings and Potential Novel Therapies for Rheumatoid Arthritis
6.3.4. Clinical Issues
6.4. Neoplasm
6.4.1. Primary Bone Tumors
Wnt-Related Molecules Involved in Primary Bone Tumors
Existing Therapies for Primary Bone Tumors
Preclinical Findings and Potential Novel Therapies for Primary Bone Tumors
Clinical Issues
6.4.2. Multiple Myeloma
Wnt-Related Molecules involved in the Developing Multiple Myeloma
Existing Therapies for Multiple Myeloma
Preclinical Findings and Potential Novel Therapies for Multiple Myeloma
Clinical Issues
6.4.3. Bone Metastasis
Wnt-Related Molecules Involved in Bone Metastasis
Existing Therapies for Bone Metastasis
Preclinical Findings and Potential Novel Therapies for Bone Metastasis
Clinical Issues
7. Future Direction and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALP | alkaline phosphatase |
| APC | adenomatous polyposis coli |
| BMP | bone morphogenetic protein |
| LD | linear dichroism |
| BP domains | β-propeller domains |
| CaMKII | calmodulin-dependent protein kinase II |
| cAMP | cyclic adenosine monophosphate |
| CBP | CREB-binding protein |
| CRD | cysteine-rich domain |
| CT-1 | cardiotropin-1 |
| Ctsk | cathepsin K |
| Daam | dishevelled associated activator of morphogenesis |
| DKK | dickkopf |
| DMARDs | disease-modifying antirheumatic drugs |
| DMP 1 | dentin matrix protein 1 |
| ECR | evolutionarily conserved region |
| FZD | frizzled |
| GSK-3 β | glycogen synthase kinase-3 β |
| GWAS | genome-wide association study |
| HDAC | histone deacetylase |
| IL | interleukin |
| JNK | Jun N-terminal kinase |
| KO | knockout |
| LEF 1 | lymphocyte enhancer factor 1 |
| LGR | leucine-rich repeat-containing G protein-coupled receptor |
| LIF | leukemia inhibitory factor |
| LRP | low-density lipoprotein-related receptor |
| M-CSF | macrophage colony-stimulating factor |
| Mef | myocyte enhancer factor |
| mAb | monoclonal antibody |
| MMPs | matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NFATc1 | nuclear factor of activated T cell c1 |
| OA | osteoarthritis |
| OCN | osteocalcin |
| OSM | oncostatin M |
| OMIM® | Online Mendelian Inheritance in Man® |
| OPG | osteoprotegerin |
| OPPG | osteoporosis-pseudoglioma syndrome |
| OSX | osterix |
| PCP | planar cell polarity |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PKN3 | protein kinase N3 |
| Porc | porcupine |
| Postn | periostin |
| PTH | parathyroid hormone |
| PTHrP | parathyroid hormone -related protein |
| RA | rheumatoid arthritis |
| RANK | receptor activator NF-kB |
| RANKL | receptor activator NF-kB ligand |
| RNF43 | ring finger 43 |
| Ror1/2 | receptor tyrosine kinase-like orphan receptor 1/2 |
| RSPO | roof-plate specific spondin |
| Runx | runt-related transcription factor |
| S1P | sphingosine-1-phosphate |
| SERM | selective estrogen receptor modulators |
| sFRP | secreted frizzled-related protein |
| SIK | salt-inducible kinase |
| TCF | T-cell factor |
| TGF-β | transforming growth factor-beta |
| TNF | tumor necrosis factor |
| Wls | wntless |
| Wnt | wingless-related MMTV integration site |
| ZNRF3 | zinc and ring finger 3 |
References
- Nusse, R.; van Ooyen, A.; Cox, D.; Fung, Y.K.; Varmus, H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature 1984, 307, 131–136. [Google Scholar] [CrossRef]
- Sharma, R.P.; Chopra, V.L. Effect of the Wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev. Biol. 1976, 48, 461–465. [Google Scholar] [CrossRef]
- Rijsewijk, F.; Schuermann, M.; Wagenaar, E.; Parren, P.; Weigel, D.; Nusse, R. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987, 50, 649–657. [Google Scholar] [CrossRef]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Takahashi, N.; Kobayashi, Y. Roles of Wnt signals in bone resorption during physiological and pathological states. J. Mol. Med. 2013, 91, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Xu, H.E.; Williams, B.O. Lipid modification in Wnt structure and function. Curr. Opin. Lipidol. 2013, 24, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Bartscherer, K.; Pelte, N.; Ingelfinger, D.; Boutros, M. Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 2006, 125, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Banziger, C.; Soldini, D.; Schutt, C.; Zipperlen, P.; Hausmann, G.; Basler, K. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 2006, 125, 509–522. [Google Scholar] [CrossRef]
- Karner, C.M.; Long, F. Wnt signaling and cellular metabolism in osteoblasts. Cell. Mol. Life Sci. 2017, 74, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Multilayered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/betacatenin signaling activation (Review). Int. J. Mol. Med. 2018, 42, 713–725. [Google Scholar] [PubMed]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Harnish, K.; Nusse, R.; Van Hul, W. LRP5 and Wnt signaling: A union made for bone. J. Bone Miner. Res. 2004, 19, 1749–1757. [Google Scholar] [CrossRef]
- Endo, M.; Nishita, M.; Fujii, M.; Minami, Y. Insight into the role of Wnt5a-induced signaling in normal and cancer cells. Int. Rev. Cell Mol. Biol. 2015, 314, 117–148. [Google Scholar]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef]
- Van Lierop, A.H.; Appelman-Dijkstra, N.M.; Papapoulos, S.E. Sclerostin deficiency in humans. Bone 2017, 96, 51–62. [Google Scholar] [CrossRef]
- Sapir-Koren, R.; Livshits, G. Osteocyte control of bone remodeling: Is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos. Int. 2014, 25, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell... and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Nakashima, T.; Takayanagi, H. Osteocyte control of osteoclastogenesis. Bone 2013, 54, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R. The osteocyte: Key player in regulating bone turnover. RMD Open 2015, 1, e000049. [Google Scholar] [CrossRef]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef]
- Lara-Castillo, N.; Johnson, M.L. LRP receptor family member associated bone disease. Rev. Endocr. Metab. Disord. 2015, 16, 141–148. [Google Scholar] [CrossRef]
- Van Wesenbeeck, L.; Cleiren, E.; Gram, J.; Beals, R.K.; Benichou, O.; Scopelliti, D.; Key, L.; Renton, T.; Bartels, C.; Gong, Y.; et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 2003, 72, 763–771. [Google Scholar] [CrossRef]
- Koide, M.; Kobayashi, Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J. Bone Miner. Metab. 2019, 37, 9–17. [Google Scholar] [CrossRef]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nurnberg, G.; Cenani, A.; et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.; Zaman, Q.; Kornak, U.; Mundlos, S.; Malik, S.; Flottmann, R. Novel splice mutation in LRP4 causes severe type of Cenani-Lenz syndactyly syndrome with oro-facial and skeletal symptoms. Eur. J. Med. Genet. 2017, 60, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Kiper, P.O.S.; Saito, H.; Gori, F.; Unger, S.; Hesse, E.; Yamana, K.; Kiviranta, R.; Solban, N.; Liu, J.; Brommage, R.; et al. Cortical-Bone Fragility—Insights from sFRP4 Deficiency in Pyle’s Disease. N. Engl. J. Med. 2016, 374, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Szenker-Ravi, E.; Altunoglu, U.; Leushacke, M.; Bosso-Lefevre, C.; Khatoo, M.; Tran, H.T.; Naert, T.; Noelanders, R.; Hajamohideen, A.; Beneteau, C.; et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature 2018, 557, 564–569. [Google Scholar] [CrossRef]
- Grzeschik, K.H.; Bornholdt, D.; Oeffner, F.; Konig, A.; del Carmen Boente, M.; Enders, H.; Fritz, B.; Hertl, M.; Grasshoff, U.; Hofling, K.; et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat. Genet. 2007, 39, 833–835. [Google Scholar] [CrossRef]
- Laine, C.M.; Joeng, K.S.; Campeau, P.M.; Kiviranta, R.; Tarkkonen, K.; Grover, M.; Lu, J.T.; Pekkinen, M.; Wessman, M.; Heino, T.J.; et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 2013, 368, 1809–1816. [Google Scholar] [CrossRef]
- Fahiminiya, S.; Majewski, J.; Mort, J.; Moffatt, P.; Glorieux, F.H.; Rauch, F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J. Med. Genet. 2013, 50, 345–348. [Google Scholar] [CrossRef]
- Keupp, K.; Beleggia, F.; Kayserili, H.; Barnes, A.M.; Steiner, M.; Semler, O.; Fischer, B.; Yigit, G.; Janda, C.Y.; Becker, J.; et al. Mutations in WNT1 cause different forms of bone fragility. Am. J. Hum. Genet. 2013, 92, 565–574. [Google Scholar] [CrossRef]
- Pyott, S.M.; Tran, T.T.; Leistritz, D.F.; Pepin, M.G.; Mendelsohn, N.J.; Temme, R.T.; Fernandez, B.A.; Elsayed, S.M.; Elsobky, E.; Verma, I.; et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2013, 92, 590–597. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Mendelsohn, N.J.; Chung, B.H.; Zhang, W.; Cohn, D.H.; Fernandez, B.; Alkuraya, F.S.; Dobyns, W.B.; Curry, C.J. Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations. J. Med. Genet. 2016, 53, 427–430. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Sirirungruangsarn, Y.; Visrutaratna, P.; Petcharunpaisan, S.; Carlson, B.M.; Intachai, W.; Sudasna, J.; Kampuansai, J.; Dejkhamron, P. WNT1-associated osteogenesis imperfecta with atrophic frontal lobes and arachnoid cysts. J. Hum. Genet. 2019, 64, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Patton, M.A.; Afzal, A.R. Robinow syndrome. J. Med. Genet. 2002, 39, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Roifman, M.; Marcelis, C.L.; Paton, T.; Marshall, C.; Silver, R.; Lohr, J.L.; Yntema, H.G.; Venselaar, H.; Kayserili, H.; van Bon, B.; et al. De novo WNT5A-associated autosomal dominant Robinow syndrome suggests specificity of genotype and phenotype. Clin. Genet. 2015, 87, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lintern, K.B.; Guidato, S.; Rowe, A.; Saldanha, J.W.; Itasaki, N. Characterization of wise protein and its molecular mechanism to interact with both Wnt and BMP signals. J. Biol. Chem. 2009, 284, 23159–23168. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef]
- Bell, S.M.; Schreiner, C.M.; Wert, S.E.; Mucenski, M.L.; Scott, W.J.; Whitsett, J.A. R-spondin 2 is required for normal laryngeal-tracheal, lung and limb morphogenesis. Development 2008, 135, 1049–1058. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson KDMacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef]
- Bennett, C.N.; Ouyang, H.; Ma, Y.L.; Zeng, Q.; Gerin, I.; Sousa, K.M.; Lane, T.F.; Krishnan, V.; Hankenson, K.D.; MacDougald, O.A. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J. Bone Miner. Res. 2007, 22, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Visweswaran, M.; Pohl, S.; Arfuso, F.; Newsholme, P.; Dilley, R.; Pervaiz, S.; Dharmarajan, A. Multi-lineage differentiation of mesenchymal stem cells—To Wnt, or not Wnt. Int. J. Biochem. Cell Biol. 2015, 68, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, R.; Akiyama, H.; Kimura, H.; Otsuki, B.; Shimizu, M.; Tsuboyama, T.; Nakamura, T. Osteoblast-targeted expression of Sfrp4 in mice results in low bone mass. J. Bone Miner. Res. 2008, 23, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Kramer, I.; Huber, T.; Kinzel, B.; Guth-Gundel, S.; Leupin, O.; Kneissel, M. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc. Natl. Acad. Sci. USA 2014, 111, E5187–E5195. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Jung, J.U.; Wu, H.; Xia, W.F.; Pan, J.X.; Shen, C.; Mei, L.; Xiong, W.C. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc. Natl. Acad. Sci. USA 2015, 112, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xiong, W.C.; Mei, L. LRP4 in neuromuscular junction and bone development and diseases. Bone 2015, 80, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Patel, M.S.; Levasseur, R.; Lobov, I.; Chang, B.H.; Glass, D.A., II; Hartmann, C.; Li, L.; Hwang, T.H.; Brayton, C.F.; et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 2002, 157, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.K.; Ryu, J.H.; Suda, N.; Tanaka, K.F.; Gingrich, J.A.; Schutz, G.; Glorieux, F.H.; Chiang, C.Y.; Zajac, J.D.; Insogna, K.L.; et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 2008, 135, 825–837. [Google Scholar] [CrossRef]
- Cui, Y.; Niziolek, P.J.; MacDonald, B.T.; Zylstra, C.R.; Alenina, N.; Robinson, D.R.; Zhong, Z.; Matthes, S.; Jacobsen, C.M.; Conlon, R.A.; et al. Lrp5 functions in bone to regulate bone mass. Nat. Med. 2011, 17, 684–691. [Google Scholar] [CrossRef]
- Riddle, R.C.; Diegel, C.R.; Leslie, J.M.; Van Koevering, K.K.; Faugere, M.C.; Clemens, T.L.; Williams, B.O. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS ONE 2013, 8, e63323. [Google Scholar] [CrossRef]
- Weivoda, M.M.; Ruan, M.; Hachfeld, C.M.; Pederson, L.; Howe, A.; Davey, R.A.; Zajac, J.D.; Kobayashi, Y.; Williams, B.O.; Westendorf, J.J.; et al. Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways. J. Bone Miner. Res. 2016, 31, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.; Keller, J.; Baranowsky, A.; Beil, F.T.; Catala-Lehnen, P.; Schulze, J.; Amling, M.; Schinke, T. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J. Cell Biol. 2013, 200, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.; Schulze, J.; Beil, F.T.; Gebauer, M.; Baranowsky, A.; Keller, J.; Marshall, R.P.; Wintges, K.; Friedrich, F.W.; Priemel, M.; et al. Control of bone formation by the serpentine receptor Frizzled-9. J. Cell Biol. 2011, 192, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Kobayashi, Y.; Udagawa, N.; Uehara, S.; Ishihara, A.; Mizoguchi, T.; Kikuchi, Y.; Takada, I.; Kato, S.; Kani, S.; et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 2012, 18, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Udagawa, N.; Mukai, H.; Ishihara, A.; Maeda, K.; Yamashita, T.; Murakami, K.; Nishita, M.; Nakamura, T.; Kato, S.; et al. Protein kinase N3 promotes bone resorption by osteoclasts in response to Wnt5a-Ror2 signaling. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Haraguchi, R.; Kitazawa, R.; Mori, K.; Tachibana, R.; Kiyonari, H.; Imai, Y.; Abe, T.; Kitazawa, S. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci. Rep. 2016, 6, 25198. [Google Scholar] [CrossRef]
- Morvan, F.; Boulukos, K.; Clement-Lacroix, P.; Roman-Roman, S.; Suc-Royer, I.; Vayssiere, B.; Ammann, P.; Martin, P.; Pinho, S.; Pognonec, P.; et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J. Bone Miner. Res. 2006, 21, 934–945. [Google Scholar] [CrossRef]
- Colditz, J.; Thiele, S.; Baschant, U.; Niehrs, C.; Bonewald, L.F.; Hofbauer, L.C.; Rauner, M. Postnatal Skeletal Deletion of Dickkopf-1 Increases Bone Formation and Bone Volume in Male and Female Mice, Despite Increased Sclerostin Expression. J. Bone Miner. Res. 2018, 33, 1698–1707. [Google Scholar] [CrossRef]
- Knight, M.N.; Karuppaiah, K.; Lowe, M.; Mohanty, S.; Zondervan, R.L.; Bell, S.; Ahn, J.; Hankenson, K.D. R-spondin-2 is a Wnt agonist that regulates osteoblast activity and bone mass. Bone Res. 2018, 6, 24. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, W.; Zhou, X.; Li, D.; Weng, J.; Yi, Z.; Cho, S.G.; Li, C.; Yi, T.; Wu, X.; et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 2009, 136, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jia, K.; Zheng, C.; Zhu, X.; Li, J.; He, L.; Siwko, S.; Xue, F.; Liu, M.; Luo, J. Loss of Lgr4 inhibits differentiation, migration and apoptosis, and promotes proliferation in bone mesenchymal stem cells. J. Cell Physiol. 2019, 234, 10855–10867. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zylstra-Diegel, C.R.; Schumacher, C.A.; Baker, J.J.; Carpenter, A.C.; Rao, S.; Yao, W.; Guan, M.; Helms, J.A.; Lane, N.E.; et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc. Natl. Acad. Sci. USA 2012, 109, E2197–E2204. [Google Scholar] [CrossRef] [PubMed]
- Joeng, K.S.; Lee, Y.C.; Lim, J.; Chen, Y.; Jiang, M.M.; Munivez, E.; Ambrose, C.; Lee, B.H. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J. Clin. Investig. 2017, 127, 2678–2688. [Google Scholar] [CrossRef]
- Luther, J.; Yorgan, T.A.; Rolvien, T.; Ulsamer, L.; Koehne, T.; Liao, N.; Keller, D.; Vollersen, N.; Teufel, S.; Neven, M.; et al. Wnt1 is an Lrp5-independent bone-anabolic Wnt ligand. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Yu, B.; Chang, J.; Liu, Y.; Li, J.; Kevork, K.; Al-Hezaimi, K.; Graves, D.T.; Park, N.H.; Wang, C.Y. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat. Med. 2014, 20, 1009–1017. [Google Scholar] [CrossRef]
- Okamoto, M.; Udagawa, N.; Uehara, S.; Maeda, K.; Yamashita, T.; Nakamichi, Y.; Kato, H.; Saito, N.; Minami, Y.; Takahashi, N.; et al. Noncanonical Wnt5a enhances Wnt/beta-catenin signaling during osteoblastogenesis. Sci. Rep. 2014, 4, 4493. [Google Scholar] [CrossRef]
- Chen, J.; Tu, X.; Esen, E.; Joeng, K.S.; Lin, C.; Arbeit, J.M.; Ruegg, M.A.; Hall, M.N.; Ma, L.; Long, F. WNT7B promotes bone formation in part through mTORC1. PLoS Genet. 2014, 10, e1004145. [Google Scholar] [CrossRef]
- Moverare-Skrtic, S.; Henning, P.; Liu, X.; Nagano, K.; Saito, H.; Borjesson, A.E.; Sjogren, K.; Windahl, S.H.; Farman, H.; Kindlund, B.; et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 2014, 20, 1279–1288. [Google Scholar] [CrossRef]
- Glass, D.A., II; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sanudo, C.; Bolado, A.; Fernandez, A.F.; Arozamena, J.; Pascual-Carra, M.A.; Rodriguez-Rey, J.C.; Fraga, M.F.; Bonewald, L.; Riancho, J.A. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J. Bone Miner. Res. 2012, 27, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Kramer, I.; Collette, N.M.; Loots, G.G.; Natt, F.; Kneissel, M.; Keller, H. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J. Bone Miner. Res. 2007, 22, 1957–1967. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 2008, 3, e2942. [Google Scholar] [CrossRef]
- Drake, M.T.; Srinivasan, B.; Modder, U.I.; Peterson, J.M.; McCready, L.K.; Riggs, B.L.; Dwyer, D.; Stolina, M.; Kostenuik, P.; Khosla, S. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J. Clin. Endocrinol. Metab. 2010, 95, 5056–5062. [Google Scholar] [CrossRef]
- Baertschi, S.; Baur, N.; Lueders-Lefevre, V.; Voshol, J.; Keller, H. Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation. J. Biol. Chem. 2014, 289, 24995–25009. [Google Scholar] [CrossRef]
- Wein, M.N.; Spatz, J.; Nishimori, S.; Doench, J.; Root, D.; Babij, P.; Nagano, K.; Baron, R.; Brooks, D.; Bouxsein, M.; et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J. Bone Miner. Res. 2015, 30, 400–411. [Google Scholar] [CrossRef]
- Stegen, S.; Stockmans, I.; Moermans, K.; Thienpont, B.; Maxwell, P.H.; Carmeliet, P.; Carmeliet, G. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat. Commun. 2018, 9, 2557. [Google Scholar] [CrossRef]
- Wein, M.N.; Liang, Y.; Goransson, O.; Sundberg, T.B.; Wang, J.; Williams, E.A.; O’Meara, M.J.; Govea, N.; Beqo, B.; Nishimori, S.; et al. SIKs control osteocyte responses to parathyroid hormone. Nat. Commun. 2016, 7, 13176. [Google Scholar] [CrossRef]
- Lombardi, M.S.; Gillieron, C.; Berkelaar, M.; Gabay, C. Salt-inducible kinases (SIK) inhibition reduces RANKL-induced osteoclastogenesis. PLoS ONE 2017, 12, e0185426. [Google Scholar] [CrossRef] [PubMed]
- Taub, M. Salt Inducible Kinase Signaling Networks: Implications for Acute Kidney Injury and Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 3219. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J. Biol. Chem. 2009, 284, 35939–35950. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Conway, S.J.; Ferrari, S.L. Regulation of beta catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc. Natl. Acad. Sci. USA 2012, 109, 15048–15053. [Google Scholar] [CrossRef]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.G.; Nicola, N.A.; et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef]
- Masuki, H.; Li, M.; Hasegawa, T.; Suzuki, R.; Ying, G.; Zhusheng, L.; Oda, K.; Yamamoto, T.; Kawanami, M.; Amizuka, N. Immunolocalization of DMP1 and sclerostin in the epiphyseal trabecule and diaphyseal cortical bone of osteoprotegerin deficient mice. BioMed Res. 2010, 31, 307–318. [Google Scholar] [CrossRef]
- Koide, M.; Kobayashi, Y.; Yamashita, T.; Uehara, S.; Nakamura, M.; Hiraoka, B.Y.; Ozaki, Y.; Iimura, T.; Yasuda, H.; Takahashi, N.; et al. Bone Formation Is Coupled to Resorption Via Suppression of Sclerostin Expression by Osteoclasts. J. Bone Miner. Res. 2017, 32, 2074–2086. [Google Scholar] [CrossRef]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.A.; Novick, S.J.; et al. Irisin Mediates Effects on Bone and Fat via alphaV Integrin Receptors. Cell 2018, 175, 1756–1768.e17. [Google Scholar] [CrossRef]
- Rivadeneira, F.; Styrkarsdottir, U.; Estrada, K.; Halldorsson, B.V.; Hsu, Y.H.; Richards, J.B.; Zillikens, M.C.; Kavvoura, F.K.; Amin, N.; Aulchenko, Y.S.; et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 2009, 41, 1199–1206. [Google Scholar]
- Zisimopoulou, P.; Evangelakou, P.; Tzartos, J.; Lazaridis, K.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; Deymeer, F.; et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J. Autoimmun. 2014, 52, 139–145. [Google Scholar] [CrossRef]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.; Amin, N.; Kemp, J.P.; et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef]
- Tu, X.; Delgado-Calle, J.; Condon, K.W.; Maycas, M.; Zhang, H.; Carlesso, N.; Taketo, M.M.; Burr, D.B.; Plotkin, L.I.; Bellido, T. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc. Natl. Acad. Sci. USA 2015, 112, E478–E486. [Google Scholar] [CrossRef] [PubMed]
- Styrkarsdottir, U.; Thorleifsson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Center, J.R.; Lee, S.H.; Nguyen, T.V.; Kwok, T.C.Y.; Lee, J.S.W.; Ho, S.C.; et al. Sequence variants in the PTCH1 gene associate with spine bone mineral density and osteoporotic fractures. Nat. Commun. 2016, 7, 10129. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.P.; Morris, J.A.; Medina-Gomez, C.; Forgetta, V.; Warrington, N.M.; Youlten, S.E.; Zheng, J.; Gregson, C.L.; Grundberg, E.; Trajanoska, K.; et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat. Genet. 2017, 49, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Trajanoska, K.; Morris, J.A.; Oei, L.; Zheng, H.F.; Evans, D.M.; Kiel, D.P.; Ohlsson, C.; Richards, J.B.; Rivadeneira, F.; consortium, G.G.; et al. Assessment of the genetic and clinical determinants of fracture risk: Genome wide association and mendelian randomisation study. BMJ 2018, 362, k3225. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.X.; Mao, W.W.; Zheng, X.F.; Jiang, L.S. The role of R-spondins and their receptors in bone metabolism. Prog. Biophys. Mol. Biol. 2016, 122, 93–100. [Google Scholar] [CrossRef]
- Sharma, A.R.; Choi, B.S.; Park, J.M.; Lee, D.H.; Lee, J.E.; Kim, H.S.; Yoon, J.K.; Song, D.K.; Nam, J.S.; Lee, S.S. Rspo 1 promotes osteoblast differentiation via Wnt signaling pathway. Indian J. Biochem. Biophys. 2013, 50, 19–25. [Google Scholar]
- Wang, H.; Brennan, T.A.; Russell, E.; Kim, J.H.; Egan, K.P.; Chen, Q.; Israelite, C.; Schultz, D.C.; Johnson, F.B.; Pignolo, R.J. R-Spondin 1 promotes vibration-induced bone formation in mouse models of osteoporosis. J. Mol. Med. 2013, 91, 1421–1429. [Google Scholar] [CrossRef]
- Shi, G.X.; Zheng, X.F.; Zhu, C.; Li, B.; Wang, Y.R.; Jiang, S.D.; Jiang, L.S. Evidence of the Role of R-Spondin 1 and Its Receptor Lgr4 in the Transmission of Mechanical Stimuli to Biological Signals for Bone Formation. Int. J. Mol. Sci. 2017, 18, 564. [Google Scholar] [CrossRef]
- Mahasarakham, C.P.A.; Ezura, Y.; Kawasaki, M.; Smriti, A.; Moriya, S.; Yamada, T.; Izu, Y.; Nifuji, A.; Nishimori, K.; Izumi, Y.; et al. BMP-2 Enhances Lgr4 Gene Expression in Osteoblastic Cells. J. Cell Physiol. 2016, 231, 887–895. [Google Scholar] [CrossRef]
- Friedman, M.S.; Oyserman, S.M.; Hankenson, K.D. Wnt11 promotes osteoblast maturation and mineralization through R-spondin 2. J. Biol. Chem. 2009, 284, 14117–14125. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, N.S.; Divito, J.K.; Kupper, T.S.; Huang, J.T.; Divito, S.J. Cross-Sectional Study Evaluating Skin, Hair, Nail, and Bone Disease in Patients with Focal Dermal Hypoplasia. Pediatr. Dermatol. 2017, 34, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Barrott, J.J.; Cash, G.M.; Smith, A.P.; Barrow, J.R.; Murtaugh, L.C. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 12752–12757. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shaver, T.M.; Balasa, A.; Ljungberg, M.C.; Wang, X.; Wen, S.; Nguyen, H.; Van den Veyver, I.B. Deletion of Porcn in mice leads to multiple developmental defects and models human focal dermal hypoplasia (Goltz syndrome). PLoS ONE 2012, 7, e32331. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Ruan, M.; Pederson, L.; Hachfeld, C.; Davey, R.A.; Zajac, J.D.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Osteoclast TGF-beta Receptor Signaling Induces Wnt1 Secretion and Couples Bone Resorption to Bone Formation. J. Bone Miner. Res. 2016, 31, 76–85. [Google Scholar] [CrossRef]
- Tasca, A.; Astleford, K.; Blixt, N.C.; Jensen, E.D.; Gopalakrishnan, R.; Mansky, K.C. SMAD1/5 signaling in osteoclasts regulates bone formation via coupling factors. PLoS ONE 2018, 13, e0203404. [Google Scholar] [CrossRef]
- Weske, S.; Vaidya, M.; Reese, A.; von Wnuck Lipinski, K.; Keul, P.; Bayer, J.K.; Fischer, J.W.; Flogel, U.; Nelsen, J.; Epple, M.; et al. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat. Med. 2018, 24, 667–678. [Google Scholar] [CrossRef]
- Zhang, L.; Choi, H.J.; Estrada, K.; Leo, P.J.; Li, J.; Pei, Y.F.; Zhang, Y.; Lin, Y.; Shen, H.; Liu, Y.Z.; et al. Multistage genome-wide association meta-analyses identified two new loci for bone mineral density. Hum. Mol. Genet. 2014, 23, 1923–1933. [Google Scholar] [CrossRef]
- Ozeki, N.; Mogi, M.; Hase, N.; Hiyama, T.; Yamaguchi, H.; Kawai, R.; Kondo, A.; Nakata, K. Wnt16 Signaling Is Required for IL-1beta-Induced Matrix Metalloproteinase-13-Regulated Proliferation of Human Stem Cell-Derived Osteoblastic Cells. Int. J. Mol. Sci. 2016, 17, 221. [Google Scholar] [CrossRef]
- Chang, J.; Sonoyama, W.; Wang, Z.; Jin, Q.; Zhang, C.; Krebsbach, P.H.; Giannobile, W.; Shi, S.; Wang, C.Y. Noncanonical Wnt-4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 MAPK. J. Biol. Chem. 2007, 282, 30938–30948. [Google Scholar] [CrossRef]
- Santiago, F.; Oguma, J.; Brown, A.M.; Laurence, J. Noncanonical Wnt signaling promotes osteoclast differentiation and is facilitated by the human immunodeficiency virus protease inhibitor ritonavir. Biochem. Biophys. Res. Commun. 2012, 417, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Ko, N.L.; Lee, M.J.; Yao, Y.; Waldmann, T.A.; Trepel, J.B.; Nicot, C. Adult T-cell leukemia cells overexpress Wnt5a and promote osteoclast differentiation. Blood 2013, 121, 5045–5054. [Google Scholar] [CrossRef] [PubMed]
- Lories, R.J.; Corr, M.; Lane, N.E. To Wnt or not to Wnt: The bone and joint health dilemma. Nat. Rev. Rheumatol. 2013, 9, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Udagawa, N.; Kobayashi, Y. Non-canonical Wnt signals regulate cytoskeletal remodeling in osteoclasts. Cell. Mol. Life Sci. 2018, 75, 3683–3692. [Google Scholar] [CrossRef]
- Solling, A.S.K.; Harslof, T.; Langdahl, B. The clinical potential of romosozumab for the prevention of fractures in postmenopausal women with osteoporosis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 105–115. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef]
- Lewiecki, E.M.; Blicharski, T.; Goemaere, S.; Lippuner, K.; Meisner, P.D.; Miller, P.D.; Miyauchi, A.; Maddox, J.; Chen, L.; Horlait, S. A Phase III Randomized Placebo-Controlled Trial to Evaluate Efficacy and Safety of Romosozumab in Men With Osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 3183–3193. [Google Scholar] [CrossRef]
- Gao, Y.; Huang, E.; Zhang, H.; Wang, J.; Wu, N.; Chen, X.; Wang, N.; Wen, S.; Nan, G.; Deng, F.; et al. Crosstalk between Wnt/beta-catenin and estrogen receptor signaling synergistically promotes osteogenic differentiation of mesenchymal progenitor cells. PLoS ONE 2013, 8, e82436. [Google Scholar] [CrossRef]
- Kondoh, S.; Inoue, K.; Igarashi, K.; Sugizaki, H.; Shirode-Fukuda, Y.; Inoue, E.; Yu, T.; Takeuchi, J.K.; Kanno, J.; Bonewald, L.F.; et al. Estrogen receptor alpha in osteocytes regulates trabecular bone formation in female mice. Bone 2014, 60, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Roforth, M.M.; Fujita, K.; McGregor, U.I.; Kirmani, S.; McCready, L.K.; Peterson, J.M.; Drake, M.T.; Monroe, D.G.; Khosla, S. Effects of age on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in humans. Bone 2014, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M. Sclerostin and Bone Aging: A Mini-Review. Gerontology 2016, 62, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Youssef, S.J.; Oursler, M.J. Sclerostin expression and functions beyond the osteocyte. Bone 2017, 96, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.S.; Padhi, I.D.; Raisz, L.G.; Lorenzo, J.A. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J. Clin. Endocrinol. Metab. 2010, 95, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.E.; Lee, S.H.; Lee, S.Y.; Kim, S.Y.; Kim, H.H.; Mirza, F.S.; Lee, S.K.; Lorenzo, J.A.; Kim, G.S.; Koh, J.M. Long-term treatment with raloxifene, but not bisphosphonates, reduces circulating sclerostin levels in postmenopausal women. Osteoporos. Int. 2012, 23, 1235–1243. [Google Scholar] [CrossRef]
- Reid, I.R. Short-term and long-term effects of osteoporosis therapies. Nat. Rev. Endocrinol. 2015, 11, 418–428. [Google Scholar] [CrossRef]
- MacNabb, C.; Patton, D.; Hayes, J.S. Sclerostin Antibody Therapy for the Treatment of Osteoporosis: Clinical Prospects and Challenges. J. Osteoporos. 2016, 2016, 6217286. [Google Scholar] [CrossRef]
- Fukumoto, S.; Matsumoto, T. Recent advances in the management of osteoporosis. F1000Res. 2017, 6, 625. [Google Scholar] [CrossRef]
- Appelman-Dijkstra, N.M.; Papapoulos, S.E. Clinical advantages and disadvantages of anabolic bone therapies targeting the WNT pathway. Nat. Rev. Endocrinol. 2018, 14, 605–623. [Google Scholar] [CrossRef]
- van Geel, T.A.; van Helden, S.; Geusens, P.P.; Winkens, B.; Dinant, G.J. Clinical subsequent fractures cluster in time after first fractures. Ann. Rheum. Dis. 2009, 68, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Warmington, K.S.; Morony, S.; Gong, J.; Cao, J.; Gao, Y.; Shalhoub, V.; Tipton, B.; Haldankar, R.; et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J. Bone Miner. Res. 2009, 24, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Warmington, K.S.; Niu, Q.T.; Asuncion, F.J.; Barrero, M.; Grisanti, M.; Dwyer, D.; Stouch, B.; Thway, T.M.; Stolina, M.; et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J. Bone Miner. Res. 2010, 25, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Ominsky, M.S.; Boyce, R.W.; Li, X.; Ke, H.Z. Effects of sclerostin antibodies in animal models of osteoporosis. Bone 2017, 96, 63–75. [Google Scholar] [CrossRef]
- Seefried, L.; Baumann, J.; Hemsley, S.; Hofmann, C.; Kunstmann, E.; Kiese, B.; Huang, Y.; Chivers, S.; Valentin, M.A.; Borah, B.; et al. Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia. J. Clin. Investig. 2017, 127, 2148–2158. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Devogelaer, J.P.; Durigova, M.; Goemaere, S.; Hemsley, S.; Jakob, F.; Junker, U.; Ruckle, J.; Seefried, L.; Winkle, P.J. BPS804 Anti-Sclerostin Antibody in Adults With Moderate Osteogenesis Imperfecta: Results of a Randomized Phase 2a Trial. J. Bone Miner. Res. 2017, 32, 1496–1504. [Google Scholar] [CrossRef]
- New Therapy to Cost One-Third Less Than Other Bone-Building Agents Over Full Course of Therapy. Available online: https://www.amgen.com/media/news-releases/2019/04/evenity-romosozumabaqqg-now-available-in-the-united-states-for-the-treatment-of-osteoporosis-in-postmenopausal-women-at-high-risk-for-fracture/ (accessed on 29 October 2019).
- van Lierop, A.H.; Moester, M.J.; Hamdy, N.A.; Papapoulos, S.E. Serum Dickkopf 1 levels in sclerostin deficiency. J. Clin. Endocrinol. Metab. 2014, 99, E252–E256. [Google Scholar] [CrossRef]
- Stolina, M.; Dwyer, D.; Niu, Q.T.; Villasenor, K.S.; Kurimoto, P.; Grisanti, M.; Han, C.Y.; Liu, M.; Li, X.; Ominsky, M.S.; et al. Temporal changes in systemic and local expression of bone turnover markers during six months of sclerostin antibody administration to ovariectomized rats. Bone 2014, 67, 305–313. [Google Scholar] [CrossRef]
- Witcher, P.C.; Miner, S.E.; Horan, D.J.; Bullock, W.A.; Lim, K.E.; Kang, K.S.; Adaniya, A.L.; Ross, R.D.; Loots, G.G.; Robling, A.G. Sclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition. JCI Insight. 2018, 3, 98673. [Google Scholar] [CrossRef]
- Florio, M.; Gunasekaran, K.; Stolina, M.; Li, X.; Liu, L.; Tipton, B.; Salimi-Moosavi, H.; Asuncion, F.J.; Li, C.; Sun, B.; et al. A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat. Commun. 2016, 7, 11505. [Google Scholar] [CrossRef]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt signaling in cartilage development and diseases: Lessons from animal studies. Lab. Investig. 2016, 96, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Cell Death in Chondrocytes, Osteoblasts, and Osteocytes. Int. J. Mol. Sci. 2016, 17, 2045. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nawata, M.; Wakitani, S. Expression profiles and functional analyses of Wnt-related genes in human joint disorders. Am. J. Pathol. 2005, 167, 97–105. [Google Scholar] [CrossRef]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.Y.; Fuller, E.S.; Russell, A.K.; Smith, S.M.; Smith, M.M.; Jackson, M.T.; Cake, M.A.; Read, R.A.; Bateman, J.F.; Sambrook, P.N.; et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr. Cartil. 2011, 19, 874–885. [Google Scholar] [CrossRef]
- Bouaziz, W.; Funck-Brentano, T.; Lin, H.; Marty, C.; Ea, H.K.; Hay, E.; Cohen-Solal, M. Loss of sclerostin promotes osteoarthritis in mice via beta-catenin-dependent and -independent Wnt pathways. Arthritis Res. Ther. 2015, 17, 24. [Google Scholar] [CrossRef]
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; Kc, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef]
- Lietman, C.; Wu, B.; Lechner, S.; Shinar, A.; Sehgal, M.; Rossomacha, E.; Datta, P.; Sharma, A.; Gandhi, R.; Kapoor, M.; et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight 2018, 3, 96308. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef]
- Swierkot, J.; Gruszecka, K.; Matuszewska, A.; Wiland, P. Assessment of the Effect of Methotrexate Therapy on Bone Metabolism in Patients with Rheumatoid Arthritis. Arch. Immunol. Ther. Exp. 2015, 63, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.G.; Yang, Y.Y.; He, X.; Li, X.F.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.W.; Jin, Y.; Li, J. Wnt signaling pathway in rheumatoid arthritis, with special emphasis on the different roles in synovial inflammation and bone remodeling. Cell Signal. 2013, 25, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Bottcher, M.; Cromme, C.; Konig, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 2016, 8, 330ra335. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Baum, W.; Dwyer, D.; Stock, M.; Schwabe, K.; Ke, H.Z.; Stolina, M.; Schett, G.; Bozec, A. Sclerostin inhibition reverses systemic, periarticular and local bone loss in arthritis. Ann. Rheum. Dis. 2013, 72, 1732–1736. [Google Scholar] [CrossRef] [PubMed]
- Marenzana, M.; Vugler, A.; Moore, A.; Robinson, M. Effect of sclerostin-neutralising antibody on periarticular and systemic bone in a murine model of rheumatoid arthritis: A microCT study. Arthritis Res. Ther. 2013, 15, R125. [Google Scholar] [CrossRef]
- Sen, M.; Lauterbach, K.; El-Gabalawy, H.; Firestein, G.S.; Corr, M.; Carson, D.A. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 2791–2796. [Google Scholar] [CrossRef]
- Sen, M.; Carson, D.A. Wnt signaling in rheumatoid synoviocyte activation. Mod. Rheumatol. 2002, 12, 5–9. [Google Scholar] [CrossRef]
- Sen, M.; Chamorro, M.; Reifert, J.; Corr, M.; Carson, D.A. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001, 44, 772–781. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a induces endothelial inflammation via beta-catenin-independent signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef]
- Rauner, M.; Stein, N.; Winzer, M.; Goettsch, C.; Zwerina, J.; Schett, G.; Distler, J.H.; Albers, J.; Schulze, J.; Schinke, T.; et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 2012, 27, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kayama, H.; Shojima, K.; Matsumoto, S.; Koyama, H.; Minami, Y.; Nojima, S.; Morii, E.; Honda, H.; Takeda, K.; et al. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-gamma in colitis. Sci. Rep. 2015, 5, 10536. [Google Scholar] [CrossRef] [PubMed]
- Miao, P.; Zhou, X.W.; Wang, P.; Zhao, R.; Chen, N.; Hu, C.Y.; Chen, X.H.; Qian, L.; Yu, Q.W.; Zhang, J.Y.; et al. Regulatory effect of anti-gp130 functional mAb on IL-6 mediated RANKL and Wnt5a expression through JAK-STAT3 signaling pathway in FLS. Oncotarget 2018, 9, 20366–20376. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.J.; Lee, S.W.; Park, Y.B.; Lee, S.K.; Park, M.C. Secreted frizzled-related protein 5 suppresses inflammatory response in rheumatoid arthritis fibroblast-like synoviocytes through down-regulation of c-Jun N-terminal kinase. Rheumatology 2014, 53, 1704–1711. [Google Scholar] [CrossRef]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trumper, L.; Binder, C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef]
- Enomoto, M.; Hayakawa, S.; Itsukushima, S.; Ren, D.Y.; Matsuo, M.; Tamada, K.; Oneyama, C.; Okada, M.; Takumi, T.; Nishita, M.; et al. Autonomous regulation of osteosarcoma cell invasiveness by Wnt5a/Ror2 signaling. Oncogene 2009, 28, 3197–3208. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewe, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- MacLauchlan, S.; Zuriaga, M.A.; Fuster, J.J.; Cuda, C.M.; Jonason, J.; Behzadi, F.; Duffen, J.P.; Haines, G.K., III; Aprahamian, T.; Perlman, H.; et al. Genetic deficiency of Wnt5a diminishes disease severity in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 166. [Google Scholar] [CrossRef]
- Cao, W.; Niu, M.; Tong, Y.; Du, Y.; Lou, W.; Mao, Y.; Dou, Y.; Yuan, H.; Zhao, W. Depleting the carboxy-terminus of human Wnt5a attenuates collagen-induced arthritis in DBA/1 mice. Biochem. Biophys. Res. Commun. 2018, 504, 679–685. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tanaka, Y.; Soen, S.; Yamanaka, H.; Yoneda, T.; Tanaka, S.; Nitta, T.; Okubo, N.; Genant, H.K.; van der Heijde, D. Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): A randomised, double-blind, placebo-controlled phase 3 trial. Ann. Rheum. Dis. 2019, 78, 899–907. [Google Scholar]
- Tanaka, S. RANKL is a therapeutic target of bone destruction in rheumatoid arthritis. F1000Res. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y. Clinical immunity in bone and joints. J. Bone Miner. Metab. 2019, 37, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fukuyo, S.; Yamaoka, K.; Sonomoto, K.; Oshita, K.; Okada, Y.; Saito, K.; Yoshida, Y.; Kanazawa, T.; Minami, Y.; Tanaka, Y. IL-6-accelerated calcification by induction of ROR2 in human adipose tissue-derived mesenchymal stem cells is STAT3 dependent. Rheumatology 2014, 53, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Sonomoto, K.; Yamaoka, K.; Oshita, K.; Fukuyo, S.; Zhang, X.; Nakano, K.; Okada, Y.; Tanaka, Y. Interleukin-1beta induces differentiation of human mesenchymal stem cells into osteoblasts via the Wnt-5a/receptor tyrosine kinase-like orphan receptor 2 pathway. Arthritis Rheum. 2012, 64, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, M.G.; Grohar, P.J.; Steensma, M.R.; Williams, B.O. Wnt Signaling in Ewing Sarcoma, Osteosarcoma, and Malignant Peripheral Nerve Sheath Tumors. Curr. Osteoporos. Rep. 2017, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Tsang, M.; Kodjabachian, L.; Sims, N.A.; Trivett, M.K.; Ehrich, M.; Dobrovic, A.; Slavin, J.; Choong, P.F.; Simmons, P.J.; et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J. Clin. Investig. 2009, 119, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; Taylor, S.; Gupte, A.; Chalk, A.M.; Bhattacharya, S.; Green, A.C.; Martin, T.J.; Strbenac, D.; Robinson, M.D.; Purton, L.E.; et al. Wnt inhibitory factor 1 (WIF1) is a marker of osteoblastic differentiation stage and is not silenced by DNA methylation in osteosarcoma. Bone 2015, 73, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.H.; Kubo, T.; Healey, J.H.; Yang, R.; Nathan, S.S.; Kolb, E.A.; Mazza, B.; Meyers, P.A.; Gorlick, R. Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the Wnt-beta-catenin pathway. Cancer Res. 2004, 64, 2734–2739. [Google Scholar] [CrossRef]
- Lin, C.H.; Guo, Y.; Ghaffar, S.; McQueen, P.; Pourmorady, J.; Christ, A.; Rooney, K.; Ji, T.; Eskander, R.; Zi, X.; et al. Dkk-3, a secreted wnt antagonist, suppresses tumorigenic potential and pulmonary metastasis in osteosarcoma. Sarcoma 2013, 2013, 147541. [Google Scholar] [CrossRef]
- Techavichit, P.; Gao, Y.; Kurenbekova, L.; Shuck, R.; Donehower, L.A.; Yustein, J.T. Secreted Frizzled-Related Protein 2 (sFRP2) promotes osteosarcoma invasion and metastatic potential. BMC Cancer 2016, 16, 869. [Google Scholar] [CrossRef]
- Danieau, G.; Morice, S.; Redini, F.; Verrecchia, F.; Royer, B.B. New Insights about the Wnt/beta-Catenin Signaling Pathway in Primary Bone Tumors and Their Microenvironment: A Promising Target to Develop Therapeutic Strategies? Int. J. Mol. Sci. 2019, 20, 3751. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Melot, T.; Garau, X.S.; Zucker, J.M.; Lenoir, G.M.; Ambros, P.F.; Sheer, D.; Turc-Carel, C.; Triche, T.J.; et al. The Ewing family of tumors—A subgroup of small-round-cell tumors defined by specific chimeric transcripts. N. Engl. J. Med. 1994, 331, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Kauer, M.; Ban, J.; Kofler, R.; Walker, B.; Davis, S.; Meltzer, P.; Kovar, H. A molecular function map of Ewing’s sarcoma. PLoS ONE 2009, 4, e5415. [Google Scholar] [CrossRef]
- Tanaka, M.; Yamazaki, Y.; Kanno, Y.; Igarashi, K.; Aisaki, K.; Kanno, J.; Nakamura, T. Ewing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J. Clin. Investig. 2014, 124, 3061–3074. [Google Scholar] [CrossRef]
- Crompton, J.G.; Ogura, K.; Bernthal, N.M.; Kawai, A.; Eilber, F.C. Local Control of Soft Tissue and Bone Sarcomas. J. Clin. Oncol. 2018, 36, 111–117. [Google Scholar] [CrossRef]
- Shimozaki, S.; Yamamoto, N.; Domoto, T.; Nishida, H.; Hayashi, K.; Kimura, H.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Kato, T.; et al. Efficacy of glycogen synthase kinase-3beta targeting against osteosarcoma via activation of beta-catenin. Oncotarget 2016, 7, 77038–77051. [Google Scholar] [CrossRef][Green Version]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.; Cleary, M.L. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef]
- Xiao, H.; Jensen, P.E.; Chen, X. Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies. Int. J. Mol. Sci. 2019, 20, 4360. [Google Scholar] [CrossRef]
- Abe, M.; Harada, T.; Matsumoto, T. Concise review: Defining and targeting myeloma stem cell-like cells. Stem. Cells 2014, 32, 1067–1073. [Google Scholar] [CrossRef]
- Marino, S.; Roodman, G.D. Multiple Myeloma and Bone: The Fatal Interaction. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Eda, H.; Santo, L.; Wein, M.N.; Hu, D.Z.; Cirstea, D.D.; Nemani, N.; Tai, Y.T.; Raines, S.E.; Kuhstoss, S.A.; Munshi, N.C.; et al. Regulation of Sclerostin Expression in Multiple Myeloma by Dkk-1: A Potential Therapeutic Strategy for Myeloma Bone Disease. J. Bone Miner. Res. 2016, 31, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat. Cell Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2019, 15. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Longerich, T.; Endris, V.; Neumann, O.; Rempel, E.; Kirchner, M.; Abadi, Z.; Uhrig, S.; Kriegsmann, M.; Weiss, K.H.; Breuhahn, K.; et al. RSPO2 gene rearrangement: A powerful driver of beta-catenin activation in liver tumours. Gut 2019, 68, 1287–1296. [Google Scholar] [CrossRef]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef]
- Giraudet, A.L.; Cassier, P.A.; Iwao-Fukukawa, C.; Garin, G.; Badel, J.N.; Kryza, D.; Chabaud, S.; Gilles-Afchain, L.; Clapisson, G.; Desuzinges, C.; et al. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer 2018, 18, 646. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef]
- Lenz, H.J.; Kahn, M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014, 105, 1087–1092. [Google Scholar] [CrossRef]
- Manegold, P.; Lai, K.K.Y.; Wu, Y.; Teo, J.L.; Lenz, H.J.; Genyk, Y.S.; Pandol, S.J.; Wu, K.; Lin, D.P.; Chen, Y.; et al. Differentiation Therapy Targeting the beta-Catenin/CBP Interaction in Pancreatic Cancer. Cancers 2018, 10, 95. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.; Cui, B.; Widhopf, G.F., II; Shen, Z.; Wu, R.; Zhang, L.; Zhang, S.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J. Clin. Investig. 2016, 126, 585–598. [Google Scholar] [CrossRef]
- Choi, M.Y.; Widhopf, G.F., II; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959.e3. [Google Scholar] [CrossRef] [PubMed]
- Funck-Brentano, T.; Nilsson, K.H.; Brommage, R.; Henning, P.; Lerner, U.H.; Koskela, A.; Tuukkanen, J.; Cohen-Solal, M.; Moverare-Skrtic, S.; Ohlsson, C. Porcupine inhibitors impair trabecular and cortical bone mass and strength in mice. J. Endocrinol. 2018, 238, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; McDonald, M.J.; Foxa, G.E.; Diegel, C.R.; Williams, B.O.; Virshup, D.M. Bone loss from Wnt inhibition mitigated by concurrent alendronate therapy. Bone Res. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Van Bezooijen, R.L.; Deruiter, M.C.; Vilain, N.; Monteiro, R.M.; Visser, A.; van der Wee-Pals, L.; van Munsteren, C.J.; Hogendoorn, P.C.; Aguet, M.; Mummery, C.L.; et al. SOST expression is restricted to the great arteries during embryonic and neonatal cardiovascular development. Dev. Dyn. 2007, 236, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Yuan, L.; Zhang, J.J.; Hao, L.; Wang, D.G. Serum sclerostin values are associated with abdominal aortic calcification and predict cardiovascular events in patients with chronic kidney disease stages 3-5D. Nephrology 2017, 22, 286–292. [Google Scholar] [CrossRef]
- Sato, M.; Hanafusa, N.; Kawaguchi, H.; Tsuchiya, K.; Nitta, K. A Prospective Cohort Study Showing No Association Between Serum Sclerostin Level and Mortality in Maintenance Hemodialysis Patients. Kidney Blood Press. Res. 2018, 43, 1023–1033. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol (Location) | Type of mutation (Genetic Inheritance) | Phenotype | OMIM | Clinical Features | Refs |
|---|---|---|---|---|---|
| LRP5 (11q13.2) | Loss-of-function (AR) | OPPG synd. | 259770 | osteoporosis, visual impairment | [4,14] |
| Gain-of-function (AD) | endosteal hyperostosis, AD osteosclerosis, AD | 144750 | high BMD, cranial nerve palsies, torus palatinus | [14,28] | |
| SOST (17q21.31) | Loss-of-function (AR) | SOST1 | 269500 | high BMD, thick cortical bone, cranial nerve palsies, syndactyly | [18,19,20] |
| Loss-of-function (AR) | VBCH | 239100 | high BMD, thick cortical bone, cranial nerve palsies | [20,29,30] | |
| LRP4 (11p11.2) | Loss-of-function (AR) | CLSS | 212780 | syndactyly, mild facial dysmorphism, agenesis of kidneys | [31,32] |
| Loss-of-function (AD, AR) | SOST2 | 614305 | See SOST1 | [25] | |
| SFRP4 (7p14.1) | Loss-of-function (AR) | Pyle disease metaphyseal dysplasia | 265900 | wide trabecular metaphyses, thin cortical bone, bone fragility | [33] |
| RSPO2 (8q23.1) | Loss-of-function (AR) | Tetraamelia synd.2 | 618021 | symmetric absence of the limbs, agenesis of lungs | [34] |
| PORCN (Xp11.23) | Loss-of-function (XLD) | FDH Goltz-Gorlin synd. | 305600 | linear skin lesions, asymmetric bone defects, striation of bones | [35] |
| WNT1 (12q13.12) | Loss-of-function (AR) | OI, type15 | 615220 | recurrent fractures, bone deformity, low BMD, short stature learning delays and brain anomalies in some patient | [36,37,38,39,40,41] |
| Loss-of-function (AD) | osteoporosis, early-onset, susceptibility to, AD | 615221 | |||
| WNT5A (3p14.3) | Loss-of-function (AD) | Robinow synd., AD1 | 180700 | resembling a fetal face, mesomelic limb shortening, micro penis in males, renal and vertebral anomalies | [42,43] |
| ROR2 (9q22.31) | Loss-of-function (AR) | Robinow synd., AR | 268310 | [42] |
| Gene Symbol | Type of Genetic Modification | Bone Volume | Bone Formation | Bone Resorption | Refs |
|---|---|---|---|---|---|
| Lrp4 | Obl cKO (OCN-Cre) | ↑ | ↑ | ↓ | [54,55,56] |
| Lrp5 | KO | ↓ | ↓ | - | [57] |
| Gut cKO (Vil1-Cre) | ↓ | ↓ | → | [58] | |
| Ocy cKO (Dmp1-Cre) | ↓ | - | - | [59] | |
| Ocy HBM KI | ↑ | ↑ | → | ||
| Gut HBM KI | → | - | - | ||
| Lrp6 | Obl cKO (OCN-Cre) | ↓ | ↓ | → | [60] |
| Lrp5/6 | Ocp cKO (RANK-Cre) | ↓ | ↓ | ↓ | [61] |
| Fzd8 | KO | ↓ | → | ↑ | [62] |
| Fzd9 | KO | ↓ | ↓ | → | [63] |
| Ror2 | het KO | ↑ | → | ↓ | [64] |
| Ocp cKO (RANK-Cre) | ↑ | → | ↓ | ||
| Ocl cKO (Ctsk-Cre) | ↑ | → | ↓(function) | [65] | |
| Sost | KO | ↑ | ↑ | → | [66] |
| Sfrp4 | KO | ↓* | ↓ | ↑ | [33] |
| LacZ KI | ↑,↓* | ↑ | ↓ | [67] | |
| Dkk1 | het KO | ↑ | ↑ | → | [68] |
| Obl cKO (OSX-Cre) | ↑ | ↑ | → | [69] | |
| Ocy cKO (Dmp1-Cre) | ↑ | ↑ | → | ||
| Rspo2 | Obl cKO (OCN-Cre) | ↓ | ↓ | → | [70] |
| Lgr4 | KO | ↓ | ↓ | ↑, - | [71,72] |
| Ocp cKO (Lyz2-Cre) | ↓ | → | ↑ | [73] | |
| Wls | Obl cKO (OCN-Cre) | ↓ | ↓ | ↑ | [74] |
| Wnt1 | Ocy cKO (Dmp1-Cre) | ↓ | ↓(function) | → | [75] |
| Obl cKO (Runx2-Cre) | ↓ | - | - | [76] | |
| Obl TG (Col1a1-tTA) | ↑ | ↑ | → | ||
| Ocp cKO (Lyz2-Cre) | → | - | - | ||
| Wnt4 | Obl TG (Col2.3) | ↑ | ↑ | ↓ | [77] |
| Wnt5a | het KO | ↓ | ↓ | ↓ | [64,78] |
| Obl cKO (OSX-Cre) | ↓ | ↓ | ↓ | [64] | |
| Wnt7b | Obl TG (Col1-Cre;R26-Wnt7b) | ↑ | ↑ | ↑ | [79] |
| Obl TG (OSX-Cre;R26-Wnt7b) | ↑** | ↑ | ↑ | ||
| Wnt10b | KO | ↓ | ↓ | → | [50,51] |
| Obl TG (OC) | ↑ | ↑ | ↑(function) | ||
| Wnt16 | KO | ↓* | → | ↑ | [80] |
| Ocy cKO (Dmp1-Cre) | →* | - | - | ||
| Obl cKO (Runx2-Cre) | ↓* | - | - |
| Study | Patient | Group | Participants | Age | Protocol | Length | BMD (%) | New Fracture (%) | Bone Turnover Marker (Maximum %) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| [Ref No.] | (N) | (Year) | Lumber Spine | Total Hip | P1NP | CTX | |||||
| FRAME [125,126] | PMO | All | 7180 | 70.8 | 12months | after 24 months | on day 14 | ||||
| Treatment | 3589 | 70.9 | 210mg romo./1M ⇒60mg deno./6M | ⇩ | +17.6 | +8.8 | 0.6 | +150 | −50 | ||
| Control | 3591 | 70.8 | placebo/1M ⇒60mg deno./6M | 12months | +5.0 | +2.9 | 2.5 | no change | no change | ||
| STRUCTURE [125,127] | PMO | All | 436 | 71.5 | 12months | no data | on day 14 | ||||
| Treatment | 218 | 71.8 | 210mg romo./1M | +9.8 | +2.9 | +180 | +30 | ||||
| Control | 218 | 71.2 | 20μg TPTD/1D | +5.4 | −0.5 | +30 | no change | ||||
| ARCH [125,128] | PMO | All | 4093 | 74.3 | 12months | after 24 months | on 1 month | ||||
| Treatment | 2046 | 74.4 | 210mg romo./1M ⇒70mg ALN./1W | ⇩ | +14.9 | +8.5 | 6.2 | +80 | −40 | ||
| Control | 2047 | 74.2 | 70mg ALN./1W ⇒70mg ALN./1W | 24months | +7.0 | +3.6 | 11.9 | −10 | −60 | ||
| BRIDGE [129] | M with OP | All | 245 | 72.1 | no data | on 1 month | |||||
| Treatment | 163 | 72.4 | 210mg romo./1M | 12months | +12.1 | +2.5 | +85.8 | −30.8 | |||
| Control | 82 | 71.5 | placebo/1M | +1.2 | −0.5 | +1.2 | −1.7 | ||||
| Target | Mechanism of Action | Agent | Stage of Clinical Development (Identifier) | Tumor Hystotype | Refs |
|---|---|---|---|---|---|
| WNT | Porc inhibitor | ETC-159 | Phase1 (NCT 02521844) | solid tumors | [213] |
| LGK974 | Phase1 (NCT 01351103) | pancreatic cancer, colorectal cancer, melanoma (and 5 more...) | [214] | ||
| Soluble FZD8 | OMP-54F28 (ipafricept) | Phase1 (NCT 02069145) | hepatocellular cancer | [13,212,215] | |
| Phase1 (NCT 02092363) | ovarian cancer | ||||
| Phase1 (NCT 02050178) | pancreatic cancer | ||||
| Phase1 (NCT 01608867) | solid tumors | [216] | |||
| FZD10 | Anti-FZD 10 mAb | OTSA101 | Phase1 (NCT 01469975) | synovial sarcoma | [217] |
| FZDs | Anti-FZD 1/2/5/7/8 mAb | OMP-18R5 (vantictumab) | Phase1 (NCT 01345201) | solid tumors | [13,212,218] |
| Phase1 (NCT 02005315) | pancreatic cancer | ||||
| Phase1 (NCT 01957007) | solid tumors | ||||
| Phase1 (NCT 01973309) | metastatic breast cancer | ||||
| RSPO3 | Anti-RSPO3 mAb | OMP-131R10 (rosmantuzumab) | Phase1 (NCT 02482441) | solid tumors | [212] |
| b-catenin | b-catenin/CBP Inhibitor | PRI-724 | Phase1 (NCT 01764477) | metastatic pancreatic cancer | [13,212,219,220] |
| Phase1/2 (NCT 01606579) | advanced myeloid malignancies | ||||
| ROR1 | Anti-ROR1 mAb | UC-961 (cirmtuzumab) | Phase1 (NCT 02860676) | chronic lymphocytic leukemia | [212,221,222] |
| Phase1/2 (NCT 03088878) | B-cell lymphoid malignancies | ||||
| Phase1 (NCT 02776917) | breast neoplasms |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. https://doi.org/10.3390/ijms20225525
Maeda K, Kobayashi Y, Koide M, Uehara S, Okamoto M, Ishihara A, Kayama T, Saito M, Marumo K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. International Journal of Molecular Sciences. 2019; 20(22):5525. https://doi.org/10.3390/ijms20225525
Chicago/Turabian StyleMaeda, Kazuhiro, Yasuhiro Kobayashi, Masanori Koide, Shunsuke Uehara, Masanori Okamoto, Akihiro Ishihara, Tomohiro Kayama, Mitsuru Saito, and Keishi Marumo. 2019. "The Regulation of Bone Metabolism and Disorders by Wnt Signaling" International Journal of Molecular Sciences 20, no. 22: 5525. https://doi.org/10.3390/ijms20225525
APA StyleMaeda, K., Kobayashi, Y., Koide, M., Uehara, S., Okamoto, M., Ishihara, A., Kayama, T., Saito, M., & Marumo, K. (2019). The Regulation of Bone Metabolism and Disorders by Wnt Signaling. International Journal of Molecular Sciences, 20(22), 5525. https://doi.org/10.3390/ijms20225525