46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features

Abstract
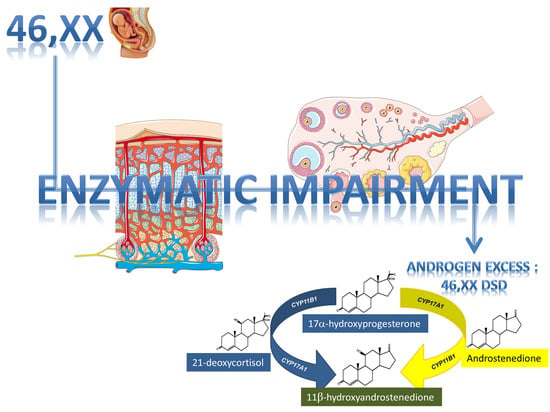
1. Introduction
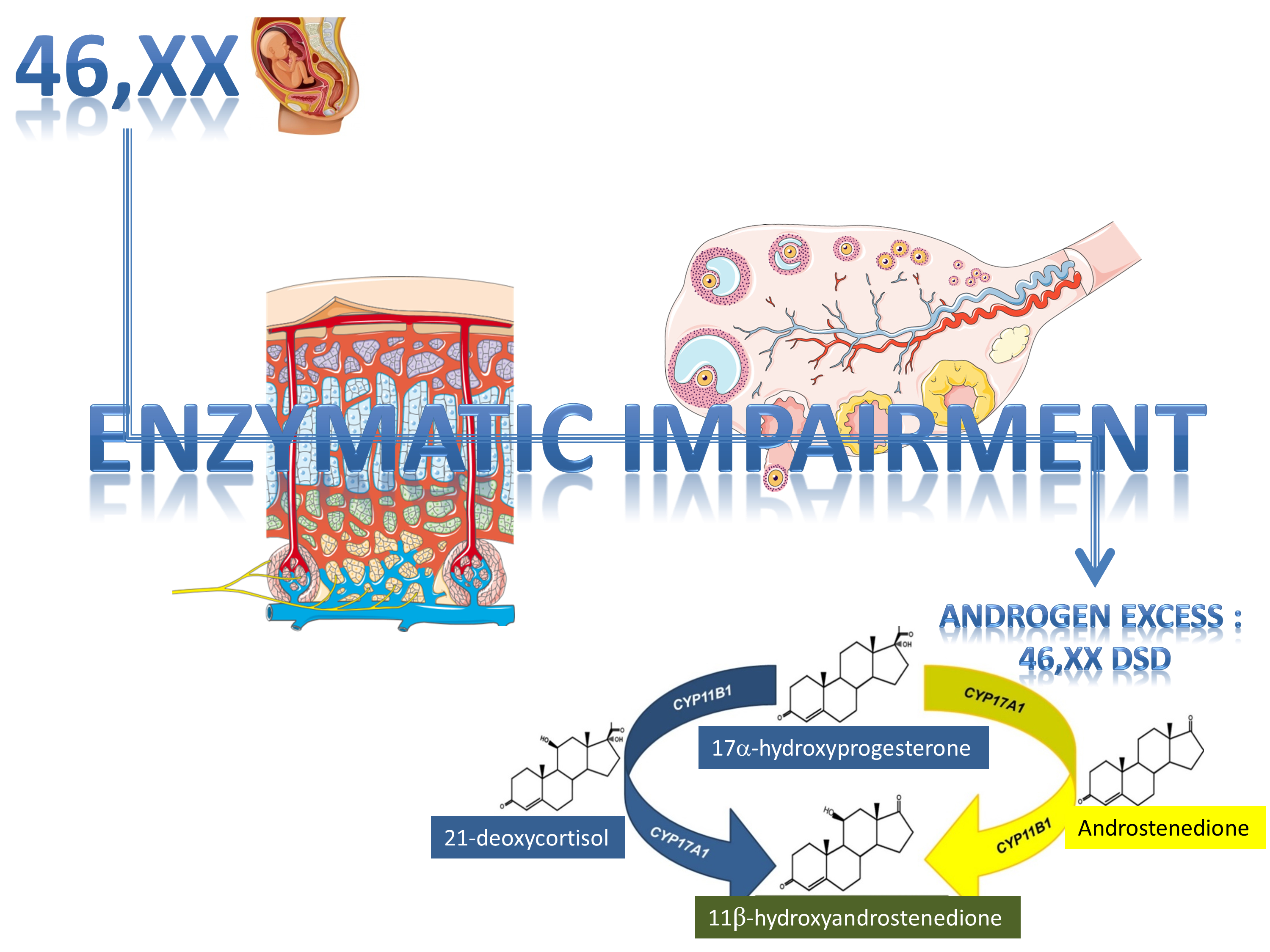
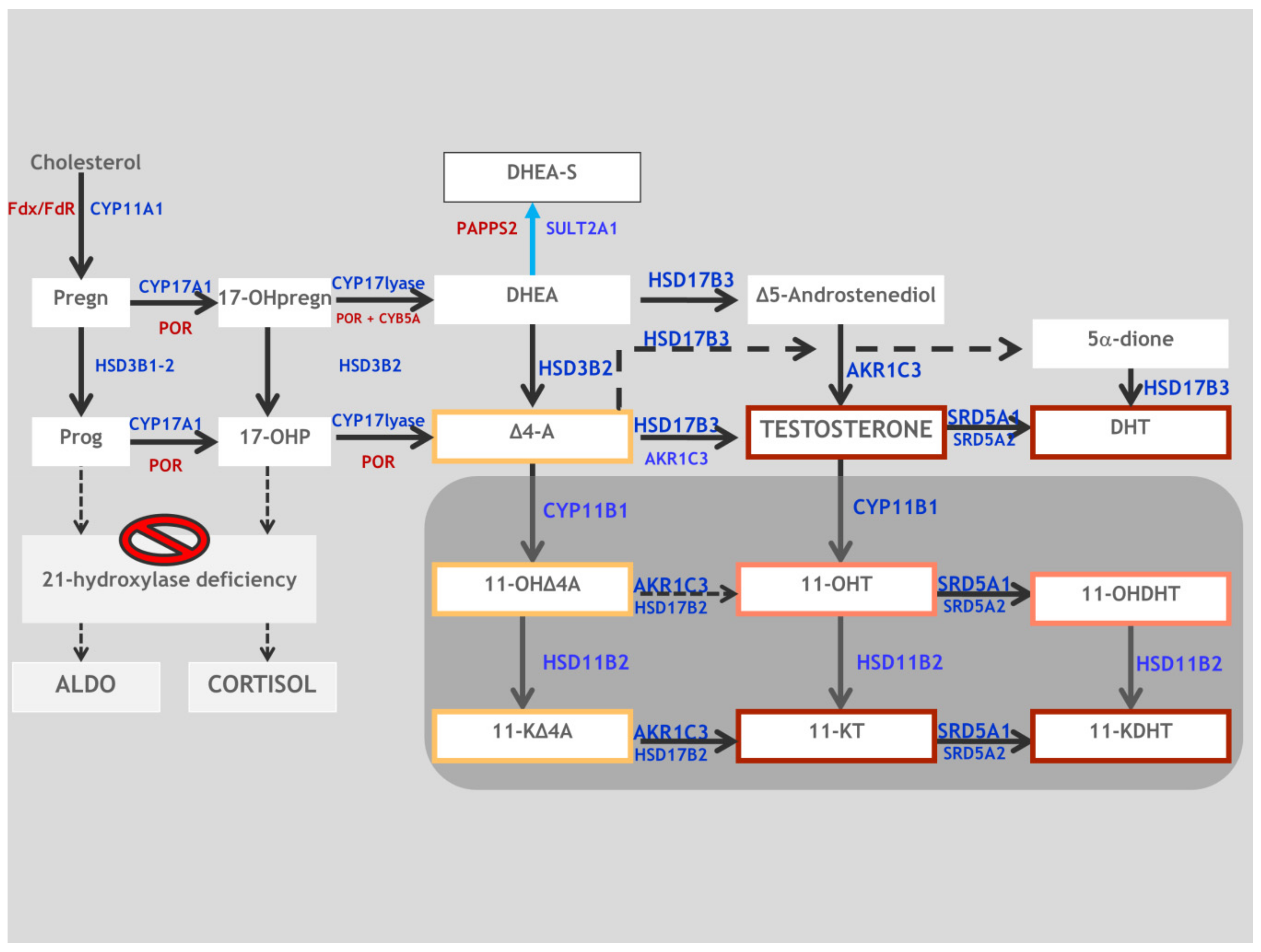
2. Synopsis of Steroidogenesis
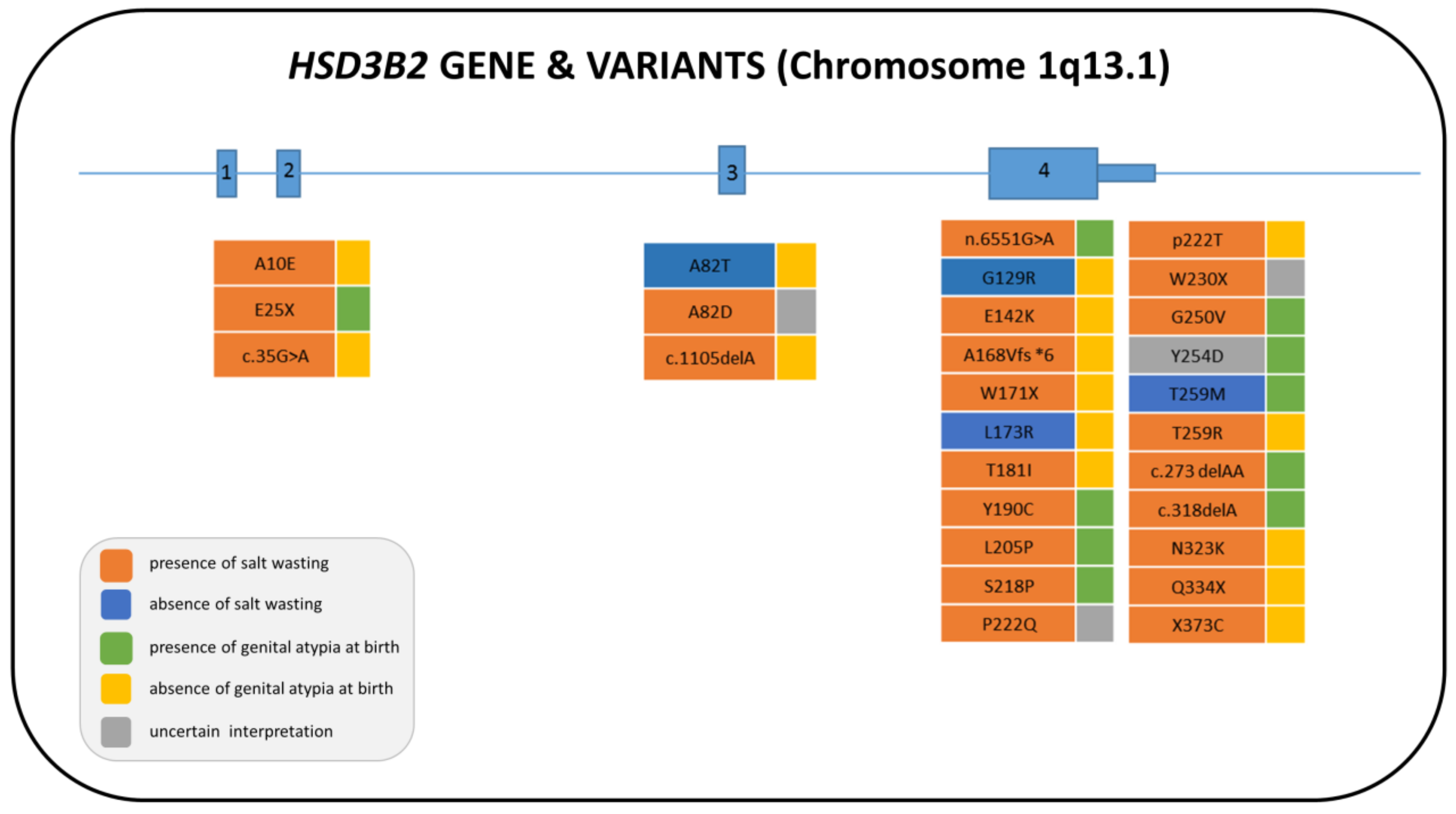
3. 3β-Hydroxysteroid Dehydrogenase Type 2 Deficiency (3β-HSD2D)
3.1. Genetics
3.2. Biochemistry
3.3. Clinical Features and Sex of Rearing
3.4. Therapy and Fertility Prognosis
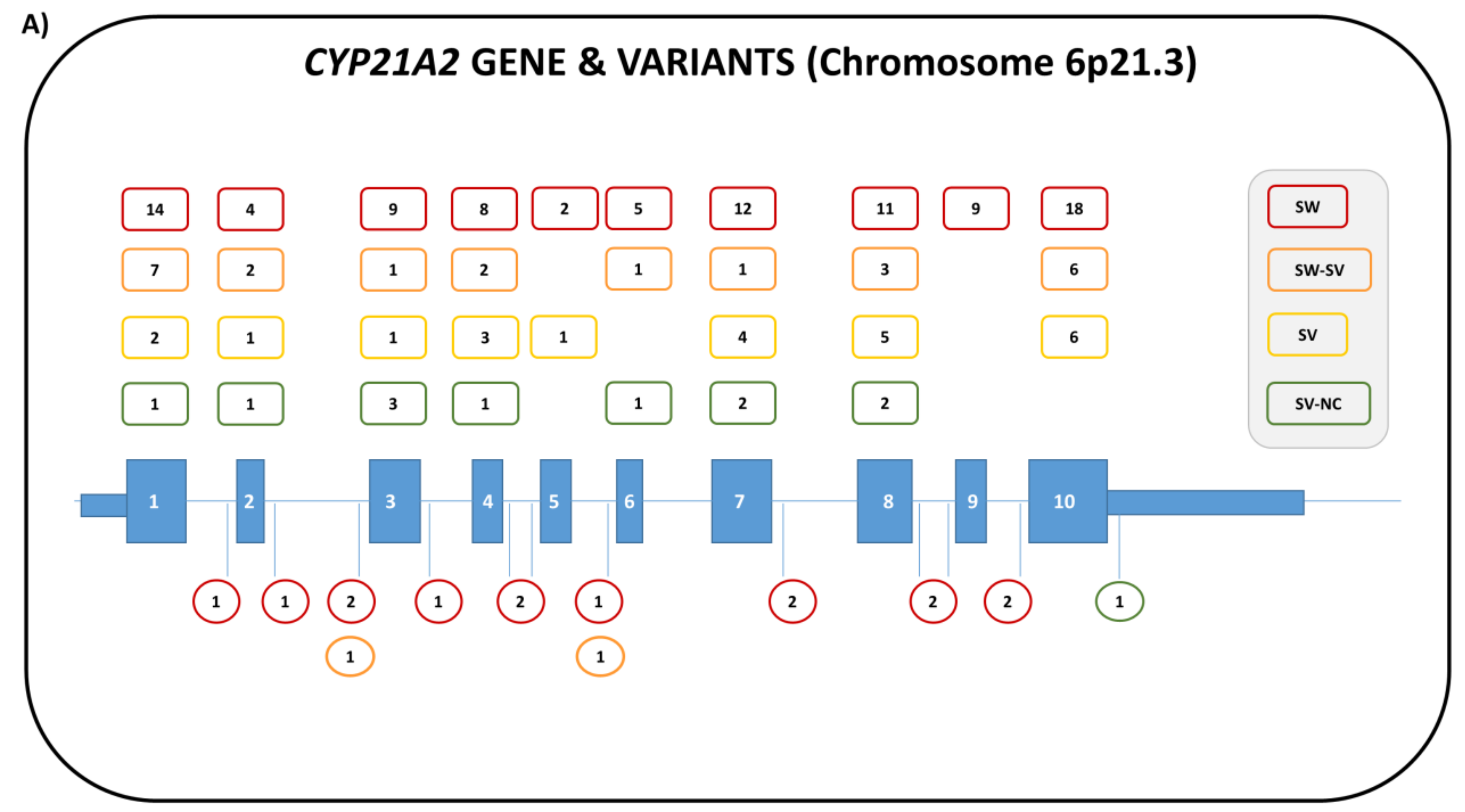
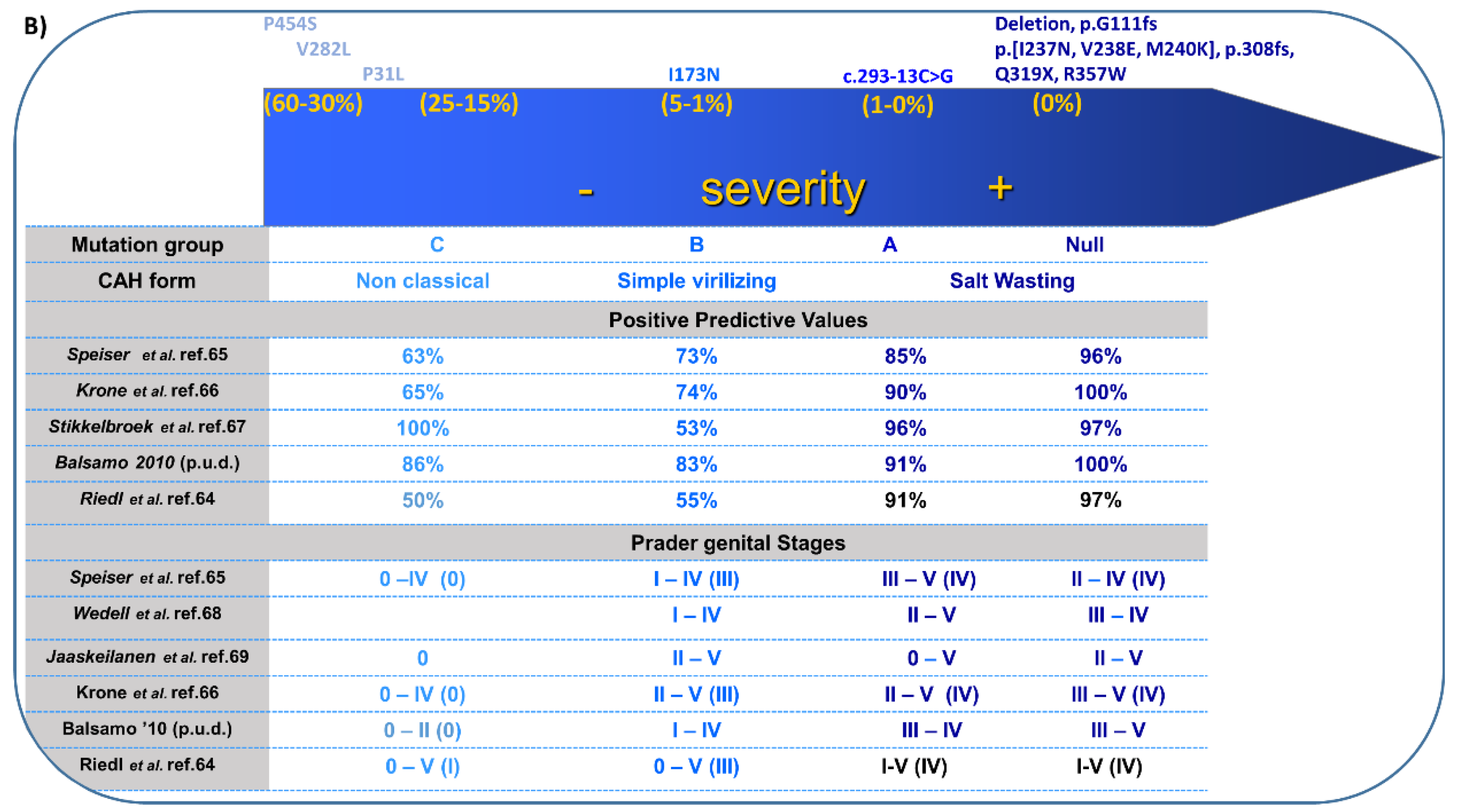
4. 21-Hydroxylase Deficiency (21-OHD)
- (1)
- salt-wasting (<1% enzyme activity);
- (2)
- simple virilizing (1–2%); and
- (3)
- non-classical (20–50%).
4.1. Genetics
4.2. Biochemistry
4.2.1. Neonatal Screening
4.2.2. Prenatal Diagnosis and Treatment of 21-OHD
4.3. Clinical Features and Sex of Rearing
4.4. Therapy and Fertility
Experimental Therapy
5. 11β-Hydroxylase Deficiency (11-OHD)
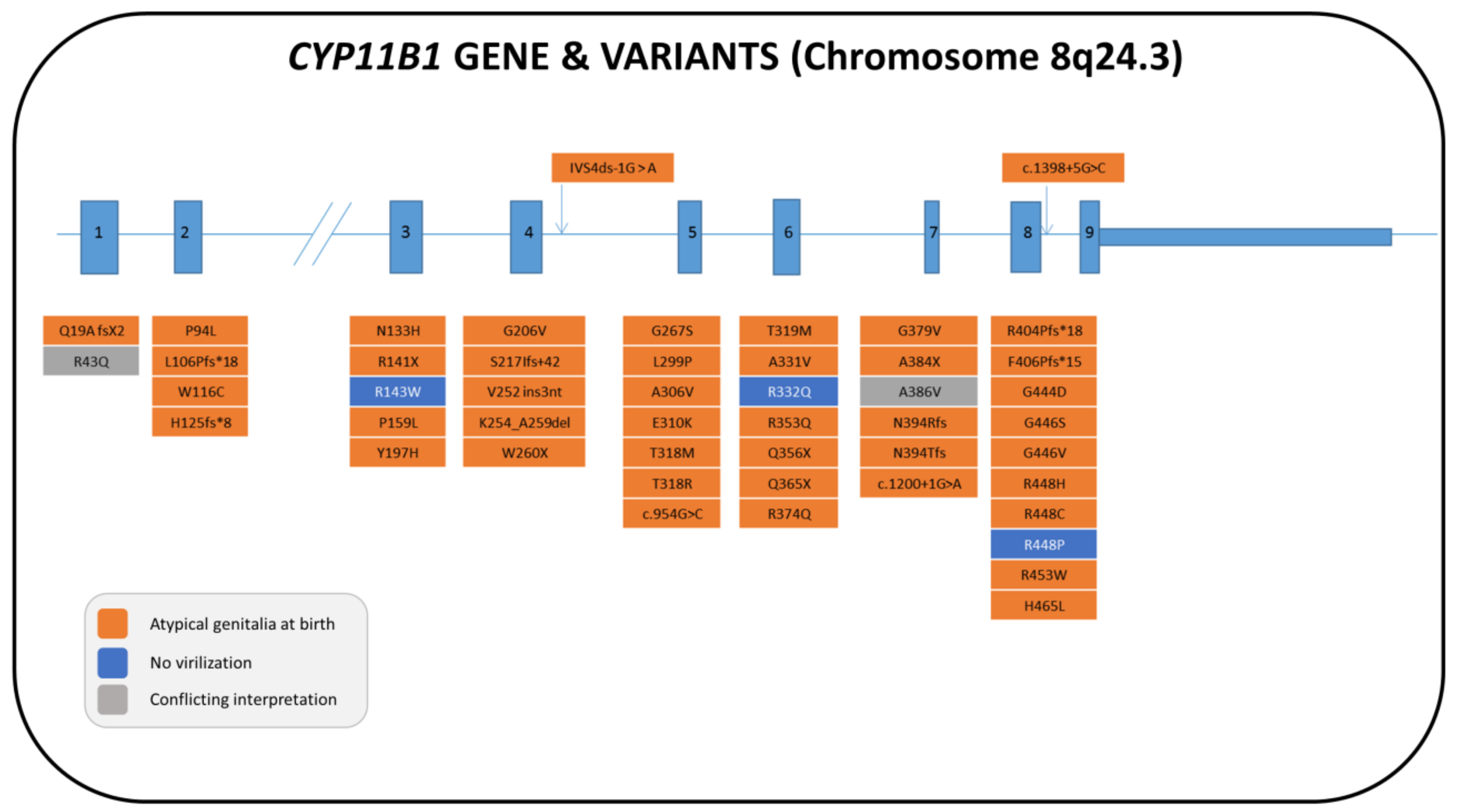
5.1. Genetics
5.2. Biochemistry
5.3. Clinical Features and Sex of Rearing
5.4. Therapy and Fertility Prognosis
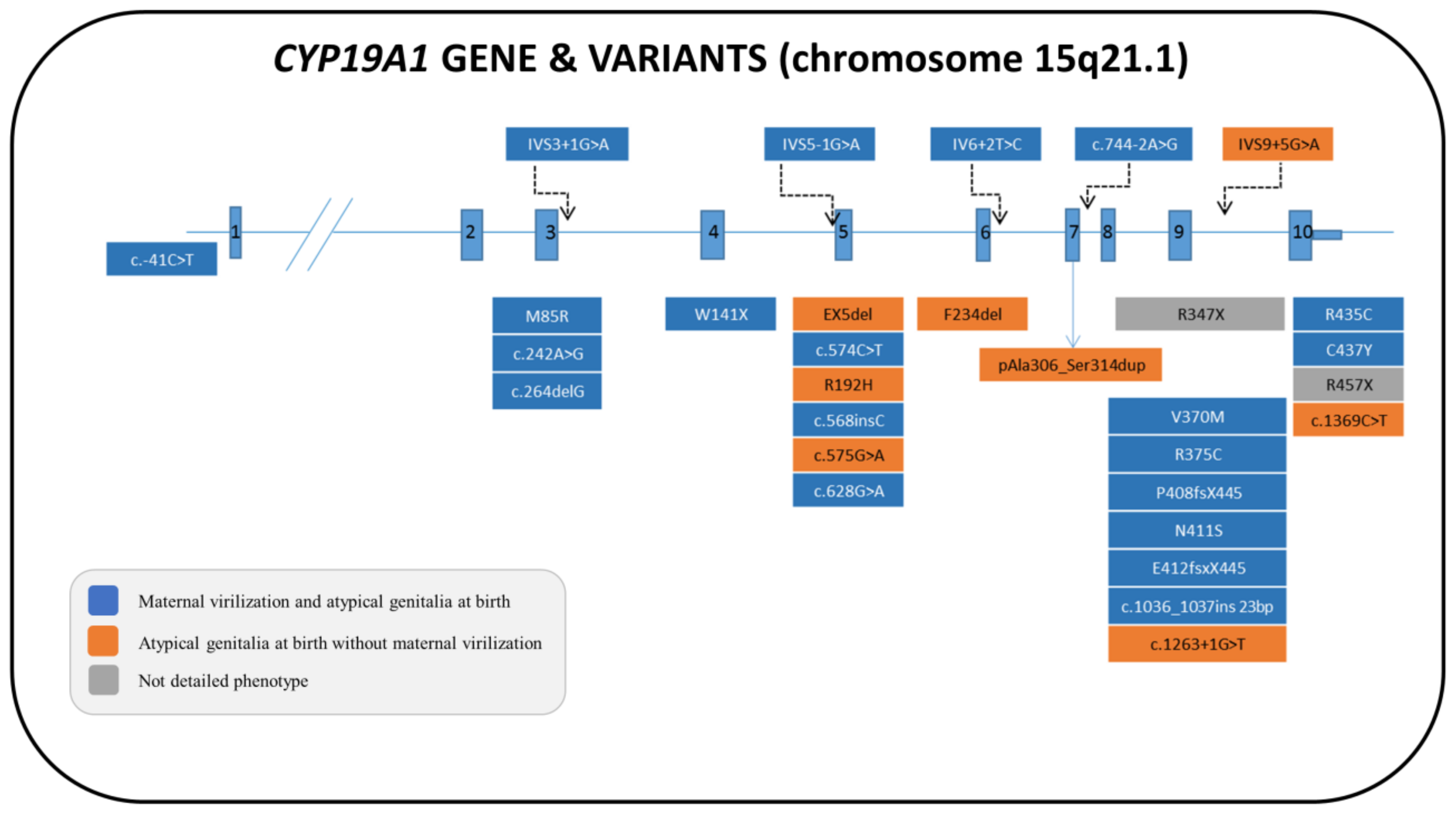
6. Aromatase Deficiency (AroD)
6.1. Genetics
6.2. Biochemistry
6.3. Clinical Features and Sex of Rearing
6.4. Therapy and Fertility Prognosis
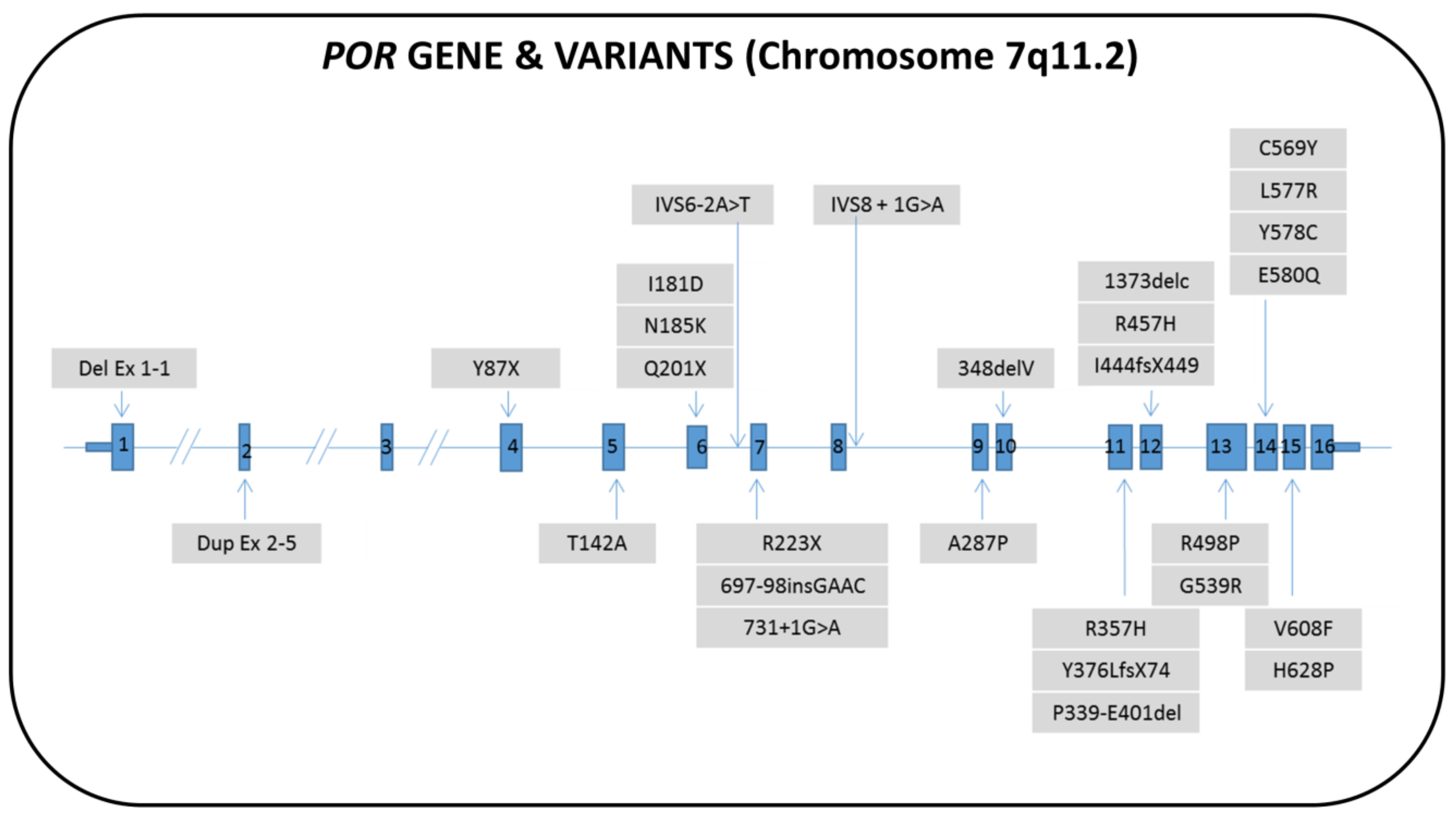
7. P450 Oxidoreductase Deficiency (PORD)
7.1. Genetics
7.2. Biochemistry
7.3. Clinical Features and Sex of Rearing
7.4. Therapy and Fertility Prognosis
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Graphic Abstract Illustration Credits
References
- Hughes, I.A.; Houk, C.; Ahmed, S.F.; Lee, P.A. LWPES Consensus Group; ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch. Dis. Child. 2006, 91, 554–563. [Google Scholar] [CrossRef] [PubMed]
- White, P.C.; Speiser, P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 2000, 21, 245–291. [Google Scholar] [CrossRef] [PubMed]
- Baranowski, E.S.; Arlt, W.; Idkowiak, J. Monogenic Disorders of Adrenal Steroidogenesis. Horm. Res. Paediatr. 2018, 89, 292–310. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; van Karnebeek, C.D.M.; Vockley, J.; Blau, N. A proposed nosology of inborn errors of metabolism. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Miller, W.L. Mechanisms in endocrinology: Rare defects in adrenal steroidogenesis. Eur. J. Endocrinol. 2018, 179, R125–R141. [Google Scholar] [CrossRef] [PubMed]
- Al Alawi, A.M.; Nordenström, A.; Falhammar, H. Clinical perspectives in congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine 2019, 63, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Rhéaume, E.; Simard, J.; Morel, Y.; Mebarki, F.; Zachmann, M.; Forest, M.G.; New, M.I.; Labrie, F. Congenital adrenal hyperplasia due to point mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene. Nat. Genet. 1992, 1, 239–245. [Google Scholar] [CrossRef]
- Schneider, G.; Genel, M.; Bongiovanni, A.M.; Goldman, A.S.; Rosenfield, R.L. Persistent testicular delta5-isomerase-3beta-hydroxysteroid dehydrogenase (delta5-3beta-HSD) deficiency in the delta5-3beta-HSD form of congenital adrenal hyperplasia. J. Clin. Investig. 1975, 55, 681–690. [Google Scholar] [CrossRef]
- Barnes, R.B.; Ehrmann, D.A.; Brigell, D.F.; Rosenfield, R.L. Ovarian steroidogenic responses to gonadotropin-releasing hormone agonist testing with nafarelin in hirsute women with adrenal responses to adrenocorticotropin suggestive of 3 beta-hydroxy-delta 5-steroid dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 1993, 76, 450–455. [Google Scholar]
- Alos, N.; Moisan, A.M.; Ward, L.; Desrochers, M.; Legault, L.; Leboeuf, G.; Van Vliet, G.; Simard, J. A novel A10E homozygous mutation in the HSD3B2 gene causing severe salt-wasting 3beta-hydroxysteroid dehydrogenase deficiency in 46,XX and 46,XY French-Canadians: evaluation of gonadal function after puberty. J. Clin. Endocrinol. Metab. 2000, 85, 1968–1974. [Google Scholar] [PubMed]
- Rhéaume, E.; Sanchez, R.; Simard, J.; Chang, Y.T.; Wang, J.; Pang, S.; Labrie, F. Molecular basis of congenital adrenal hyperplasia in two siblings with classical nonsalt-losing 3 beta-hydroxysteroid dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 1994, 79, 1012–1018. [Google Scholar] [PubMed]
- Moisan, A.M.; Ricketts, M.L.; Tardy, V.; Desrochers, M.; Mébarki, F.; Chaussain, J.L.; Cabrol, S.; Raux-Demay, M.C.; Forest, M.G.; Sippell, W.G.; et al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J. Clin. Endocrinol. Metab. 1999, 84, 4410–4425. [Google Scholar] [PubMed]
- Huang, Y.; Zheng, J.; Xie, T.; Xiao, Q.; Lu, S.; Li, X.; Cheng, J.; Chen, L.; Liu, L. A novel homozygous mutation p.E25X in the HSD3B2 gene causing salt wasting 3β-hydroxysteroid dehydrogenases deficiency in a Chinese pubertal girl: a delayed diagnosis until recurrent ovary cysts. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2014, 52, 948–951. [Google Scholar]
- Benkert, A.R.; Young, M.; Robinson, D.; Hendrickson, C.; Lee, P.A.; Strauss, K.A. Severe Salt-Losing 3β-Hydroxysteroid Dehydrogenase Deficiency: Treatment and Outcomes of HSD3B2 c.35G>A Homozygotes. J. Clin. Endocrinol. Metab. 2015, 100, E1105–E1115. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, B.B.; Russell, A.J.; Vasconcelos-Leite, M.; Arnhold, I.J.; Bloise, W.; Wajchenberg, B.L.; Nicolau, W.; Sutcliffe, R.G.; Wallace, A.M. Mutation in 3 beta-hydroxysteroid dehydrogenase type II associated with pseudohermaphroditism in males and premature pubarche or cryptic expression in females. J. Mol. Endocrinol. 1994, 12, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Nordenström, A.; Forest, M.G.; Wedell, A. A case of 3beta-hydroxysteroid dehydrogenase type II (HSD3B2) deficiency picked up by neonatal screening for 21-hydroxylase deficiency: difficulties and delay in etiologic diagnosis. Horm. Res. 2007, 68, 204–208. [Google Scholar]
- Pang, S.; Levine, L.S.; Stoner, E.; Opitz, J.M.; Pollack, M.S.; Dupont, B.; New, M.I. Nonsalt-losing congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency with normal glomerulosa function. J. Clin. Endocrinol. Metab. 1983, 56, 808–818. [Google Scholar] [CrossRef]
- Chang, Y.T.; Kulin, H.E.; Garibaldi, L.; Suriano, M.J.; Bracki, K.; Pang, S. Hypothalamic-pituitary-gonadal axis function in pubertal male and female siblings with glucocorticoid-treated nonsalt-wasting 3 beta-hydroxysteroid dehydrogenase deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1993, 77, 1251–1257. [Google Scholar]
- Probst-Scheidegger, U.; Udhane, S.S.; l’Allemand, D.; Flück, C.E.; Camats, N. Non-Virilizing Congenital Adrenal Hyperplasia in a Female Patient with a Novel HSD3B2 Mutation. Sex. Dev. Genet. Mol. Biol. Evol. Endocrinol. Embryol. Pathol. Sex Determ. Differ. 2016, 10, 200–204. [Google Scholar] [CrossRef]
- Pang, S.; Wang, W.; Rich, B.; David, R.; Chang, Y.T.; Carbunaru, G.; Myers, S.E.; Howie, A.F.; Smillie, K.J.; Mason, J.I. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2002, 87, 2556–2563. [Google Scholar] [PubMed]
- Russell, A.J.; Wallace, A.M.; Forest, M.G.; Donaldson, M.D.; Edwards, C.R.; Sutcliffe, R.G. Mutation in the human gene for 3 beta-hydroxysteroid dehydrogenase type II leading to male pseudohermaphroditism without salt loss. J. Mol. Endocrinol. 1994, 12, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, T.H.; Mallet, D.; Dige-Petersen, H.; Müller, J.; Main, K.M.; Morel, Y.; Forest, M.G. Delayed diagnosis of congenital adrenal hyperplasia with salt wasting due to type II 3beta-hydroxysteroid dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 2005, 90, 2076–2080. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, K.; Ono, M.; Hijikata, A.; Matsubara, Y.; Katsumata, N.; Takagi, M.; Morio, T.; Ohara, O.; Kashimada, K.; Mizutani, S. Two novel HSD3B2 missense mutations with diverse residual enzymatic activities for Δ5-steroids. Clin. Endocrinol. 2014, 80, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, N.; Tanae, A.; Yasunaga, T.; Horikawa, R.; Tanaka, T.; Hibi, I. A novel missense mutation in the type II 3 beta-hydroxysteroid dehydrogenase gene in a family with classical salt-wasting congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency. Hum. Mol. Genet. 1995, 4, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Baquedano, M.S.; Ciaccio, M.; Marino, R.; Perez Garrido, N.; Ramirez, P.; Maceiras, M.; Turjanski, A.; Defelipe, L.A.; Rivarola, M.A.; Belgorosky, A. A novel missense mutation in the HSD3B2 gene, underlying nonsalt-wasting congenital adrenal hyperplasia. new insight into the structure-function relationships of 3β-hydroxysteroid dehidrogenase type II. J. Clin. Endocrinol. Metab. 2015, 100, E191–E196. [Google Scholar] [CrossRef] [PubMed]
- Rosenfield, R.L.; Rich, B.H.; Wolfsdorf, J.I.; Cassorla, F.; Parks, J.S.; Bongiovanni, A.M.; Wu, C.H.; Shackleton, C.H. Pubertal presentation of congenital delta 5-3 beta-hydroxysteroid dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 1980, 51, 345–353. [Google Scholar] [CrossRef]
- Sanchez, R.; Rhéaume, E.; Laflamme, N.; Rosenfield, R.L.; Labrie, F.; Simard, J. Detection and functional characterization of the novel missense mutation Y254D in type II 3 beta-hydroxysteroid dehydrogenase (3 beta HSD) gene of a female patient with nonsalt-losing 3 beta HSD deficiency. J. Clin. Endocrinol. Metab. 1994, 78, 561–567. [Google Scholar]
- Barnes, R.B.; Rosenfield, R.L.; Burstein, S.; Ehrmann, D.A. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N. Engl. J. Med. 1989, 320, 559–565. [Google Scholar] [CrossRef]
- Paula, F.J.; Dick-de-Paula, I.; Pontes, A.; Schmitt, F.C.; Mendonça, B.B.; Foss, M.C. Hyperandrogenism due to 3 beta-hydroxysteroid dehydrogenase deficiency with accessory adrenocortical tissue: a hormonal and metabolic evaluation. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Medicas E Biol. 1994, 27, 1149–1158. [Google Scholar]
- Marui, S.; Russell, A.J.; Paula, F.J.; Dick-de-Paula, I.; Marcondes, J.A.; Mendonca, B.B. Genotyping of the type II 3beta-hydroxysteroid dehydrogenase gene (HSD3B2) in women with hirsutism and elevated ACTH-stimulated delta(5)-steroids. Fertil. Steril. 2000, 74, 553–557. [Google Scholar] [CrossRef]
- Marui, S.; Castro, M.; Latronico, A.C.; Elias, L.L.; Arnhold, I.J.; Moreira, A.C.; Mendonca, B.B. Mutations in the type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin. Endocrinol. 2000, 52, 67–75. [Google Scholar] [CrossRef]
- Tajima, T.; Fujieda, K.; Nakae, J.; Shinohara, N.; Yoshimoto, M.; Baba, T.; Kinoshita, E.; Igarashi, Y.; Oomura, T. Molecular analysis of type II 3 beta-hydroxysteroid dehydrogenase gene in Japanese patients with classical 3 beta-hydroxysteroid dehydrogenase deficiency. Hum. Mol. Genet. 1995, 4, 969–971. [Google Scholar] [CrossRef]
- Zhang, L.; Sakkal-Alkaddour, H.; Chang, Y.T.; Yang, X.; Pang, S. A new compound heterozygous frameshift mutation in the type II 3 beta-hydroxysteroid dehydrogenase (3 beta-HSD) gene causes salt-wasting 3 beta-HSD deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1996, 81, 291–295. [Google Scholar]
- Scaramuzzo, R.T.; Menabò, S.; Baldazzi, L.; Moscuzza, F.; Saba, A.; Balsamo, A.; Boldrini, A.; Ghirri, P. Two Moroccan Sisters Presenting with a Severe Salt-Wasting Form of Congenital Adrenal Hyperplasia but Normal Female Genitalia. Sex. Dev. Genet. Mol. Biol. Evol. Endocrinol. Embryol. Pathol. Sex Determ. Differ. 2017, 11, 82–85. [Google Scholar] [CrossRef]
- Jeandron, D.D.; Sahakitrungruang, T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic 3β-hydroxysteroid dehydrogenase deficiency: an unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm. Res. Paediatr. 2012, 77, 334–338. [Google Scholar] [CrossRef]
- Alswailem, M.M.; Alzahrani, O.S.; Alhomaidah, D.S.; Alasmari, R.; Qasem, E.; Murugan, A.K.; Alsagheir, A.; Brema, I.; Abbas, B.B.; Almehthel, M.; et al. Mutational analysis of rare subtypes of congenital adrenal hyperplasia in a highly inbred population. Mol. Cell. Endocrinol. 2018, 461, 105–111. [Google Scholar] [CrossRef]
- Flück, C.E.; Pandey, A.V. Steroidogenesis of the testis—New genes and pathways. Ann. Endocrinol. 2014, 75, 40–47. [Google Scholar] [CrossRef]
- Cherradi, N.; Rossier, M.F.; Vallotton, M.B.; Timberg, R.; Friedberg, I.; Orly, J.; Wang, X.J.; Stocco, D.M.; Capponi, A.M. Submitochondrial distribution of three key steroidogenic proteins (steroidogenic acute regulatory protein and cytochrome p450scc and 3beta-hydroxysteroid dehydrogenase isomerase enzymes) upon stimulation by intracellular calcium in adrenal glomerulosa cells. J. Biol. Chem. 1997, 272, 7899–7907. [Google Scholar]
- Rajapaksha, M.; Kaur, J.; Prasad, M.; Pawlak, K.J.; Marshall, B.; Perry, E.W.; Whittal, R.M.; Bose, H.S. An Outer Mitochondrial Translocase, Tom22, Is Crucial for Inner Mitochondrial Steroidogenic Regulation in Adrenal and Gonadal Tissues. Mol. Cell. Biol. 2016, 36, 1032–1047. [Google Scholar] [CrossRef]
- Mermejo, L.M.; Elias, L.L.K.; Marui, S.; Moreira, A.C.; Mendonca, B.B.; de Castro, M. Refining hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. J. Clin. Endocrinol. Metab. 2005, 90, 1287–1293. [Google Scholar] [CrossRef]
- Simard, J.; Ricketts, M.-L.; Gingras, S.; Soucy, P.; Feltus, F.A.; Melner, M.H. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr. Rev. 2005, 26, 525–582. [Google Scholar] [CrossRef]
- Auchus, R.J.; Lee, T.C.; Miller, W.L. Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 1998, 273, 3158–3165. [Google Scholar] [CrossRef]
- Krone, N.; Hughes, B.A.; Lavery, G.G.; Stewart, P.M.; Arlt, W.; Shackleton, C.H.L. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J. Steroid Biochem. Mol. Biol. 2010, 121, 496–504. [Google Scholar] [CrossRef]
- Pang, S.; Carbunaru, G.; Haider, A.; Copeland, K.C.; Chang, Y.T.; Lutfallah, C.; Mason, J.I. Carriers for type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) deficiency can only be identified by HSD3B2 genotype study and not by hormone test. Clin. Endocrinol. 2003, 58, 323–331. [Google Scholar] [CrossRef]
- Lutfallah, C.; Wang, W.; Mason, J.I.; Chang, Y.T.; Haider, A.; Rich, B.; Castro-Magana, M.; Copeland, K.C.; David, R.; Pang, S. Newly proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 2611–2622. [Google Scholar]
- Bongiovanni, A.M. The adrenogenital syndrome with deficiency of 3 beta-hydroxysteroid dehydrogenase. J. Clin. Investig. 1962, 41, 2086–2092. [Google Scholar] [CrossRef]
- Levy-Shraga, Y.; Pinhas-Hamiel, O. High 17-hydroxyprogesterone level in newborn screening test for congenital adrenal hyperplasia. BMJ Case Rep. 2016, 2016, bcr2015213939. [Google Scholar] [CrossRef]
- de Araújo, V.G.B.; de Oliveira, R.S.; Gameleira, K.P.D.; Cruz, C.B.; Lofrano-Porto, A. 3β-hydroxysteroid dehydrogenase type II deficiency on newborn screening test. Arq. Bras. Endocrinol. Metabol. 2014, 58, 650–655. [Google Scholar] [CrossRef]
- Miller, W.L. Steroidogenesis: Unanswered Questions. Trends Endocrinol. Metab. TEM 2017, 28, 771–793. [Google Scholar] [CrossRef]
- Rushworth, R.L.; Torpy, D.J.; Stratakis, C.A.; Falhammar, H. Adrenal Crises in Children: Perspectives and Research Directions. Horm. Res. Paediatr. 2018, 89, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef] [PubMed]
- Krone, N.; Arlt, W. Genetics of congenital adrenal hyperplasia. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Nimkarn, S.; Gangishetti, P.K.; Yau, M.; New, M.I. 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1171/ (accessed on 4 February 2016).
- Merke, D.P.; Bornstein, S.R. Congenital adrenal hyperplasia. Lancet Lond. Engl. 2005, 365, 2125–2136. [Google Scholar] [CrossRef]
- Speiser, P.W.; Dupont, B.; Rubinstein, P.; Piazza, A.; Kastelan, A.; New, M.I. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am. J. Hum. Genet. 1985, 37, 650–667. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Matteson, K.J.; Miller, W.L. Structure of a bovine gene for P-450c21 (steroid 21-hydroxylase) defines a novel cytochrome P-450 gene family. Proc. Natl. Acad. Sci. USA 1986, 83, 4243–4247. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Yoshioka, H.; Yamane, M.; Gotoh, O.; Fujii-Kuriyama, Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc. Natl. Acad. Sci. USA 1986, 83, 2841–2845. [Google Scholar] [CrossRef]
- Simonetti, L.; Bruque, C.D.; Fernández, C.S.; Benavides-Mori, B.; Delea, M.; Kolomenski, J.E.; Espeche, L.D.; Buzzalino, N.D.; Nadra, A.D.; Dain, L. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum. Mutat. 2018, 39, 5–22. [Google Scholar] [CrossRef]
- Balsamo, A.; Baldazzi, L.; Menabò, S.; Cicognani, A. Impact of molecular genetics on congenital adrenal hyperplasia management. Sex. Dev. Genet. Mol. Biol. Evol. Endocrinol. Embryol. Pathol. Sex Determ. Differ. 2010, 4, 233–248. [Google Scholar] [CrossRef]
- New, M.I.; Abraham, M.; Gonzalez, B.; Dumic, M.; Razzaghy-Azar, M.; Chitayat, D.; Sun, L.; Zaidi, M.; Wilson, R.C.; Yuen, T. Genotype-phenotype correlation in 1507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 2013, 110, 2611–2616. [Google Scholar] [CrossRef]
- Tusie-Luna, M.T.; Traktman, P.; White, P.C. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J. Biol. Chem. 1990, 265, 20916–20922. [Google Scholar]
- Balsamo, A.; Cacciari, E.; Baldazzi, L.; Tartaglia, L.; Cassio, A.; Mantovani, V.; Piazzi, S.; Cicognani, A.; Pirazzoli, P.; Mainetti, B.; et al. CYP21 analysis and phenotype/genotype relationship in the screened population of the Italian Emilia-Romagna region. Clin. Endocrinol. 2000, 53, 117–125. [Google Scholar] [CrossRef]
- Riedl, S.; Röhl, F.-W.; Bonfig, W.; Brämswig, J.; Richter-Unruh, A.; Fricke-Otto, S.; Bettendorf, M.; Riepe, F.; Kriegshäuser, G.; Schönau, E.; et al. Genotype/phenotype correlations in 538 congenital adrenal hyperplasia patients from Germany and Austria: discordances in milder genotypes and in screened versus prescreening patients. Endocr. Connect. 2019, 8, 86–94. [Google Scholar] [CrossRef]
- Speiser, P.W.; Dupont, J.; Zhu, D.; Serrat, J.; Buegeleisen, M.; Tusie-Luna, M.T.; Lesser, M.; New, M.I.; White, P.C. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Investig. 1992, 90, 584–595. [Google Scholar] [CrossRef]
- Krone, N.; Braun, A.; Roscher, A.A.; Knorr, D.; Schwarz, H.P. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J. Clin. Endocrinol. Metab. 2000, 85, 1059–1065. [Google Scholar] [CrossRef]
- Stikkelbroeck, N.M.M.L.; Hoefsloot, L.H.; de Wijs, I.J.; Otten, B.J.; Hermus, A.R.M.M.; Sistermans, E.A. CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J. Clin. Endocrinol. Metab. 2003, 88, 3852–3859. [Google Scholar] [CrossRef]
- Wedell, A.; Thilén, A.; Ritzén, E.M.; Stengler, B.; Luthman, H. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J. Clin. Endocrinol. Metab. 1994, 78, 1145–1152. [Google Scholar] [CrossRef]
- Jääskeläinen, J.; Levo, A.; Voutilainen, R.; Partanen, J. Population-wide evaluation of disease manifestation in relation to molecular genotype in steroid 21-hydroxylase (CYP21) deficiency: good correlation in a well defined population. J. Clin. Endocrinol. Metab. 1997, 82, 3293–3297. [Google Scholar] [CrossRef]
- Miller, W.L.; Merke, D.P. Tenascin-X, Congenital Adrenal Hyperplasia, and the CAH-X Syndrome. Horm. Res. Paediatr. 2018, 89, 352–361. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB Is Associated with Hypermobility Type of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef]
- Merke, D.P.; Chen, W.; Morissette, R.; Xu, Z.; Van Ryzin, C.; Sachdev, V.; Hannoush, H.; Shanbhag, S.M.; Acevedo, A.T.; Nishitani, M.; et al. Tenascin-X Haploinsufficiency Associated with Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, E379–E387. [Google Scholar] [CrossRef]
- Morissette, R.; Chen, W.; Perritt, A.F.; Dreiling, J.L.; Arai, A.E.; Sachdev, V.; Hannoush, H.; Mallappa, A.; Xu, Z.; McDonnell, N.B.; et al. Broadening the Spectrum of Ehlers Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2015, 100, E1143–E1152. [Google Scholar] [CrossRef]
- Lao, Q.; Brookner, B.; Merke, D.P. High-Throughput Screening for CYP21A1P-TNXA/TNXB Chimeric Genes Responsible for Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J. Mol. Diagn. JMD 2019, 21, 924–931. [Google Scholar] [CrossRef]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat. Genet. 1997, 17, 104–108. [Google Scholar] [CrossRef]
- Chen, W.; Perritt, A.F.; Morissette, R.; Dreiling, J.L.; Bohn, M.-F.; Mallappa, A.; Xu, Z.; Quezado, M.; Merke, D.P. Ehlers-Danlos Syndrome Caused by Biallelic TNXB Variants in Patients with Congenital Adrenal Hyperplasia. Hum. Mutat. 2016, 37, 893–897. [Google Scholar] [CrossRef]
- Swart, A.C.; Schloms, L.; Storbeck, K.-H.; Bloem, L.M.; du Toit, T.; Quanson, J.L.; Rainey, W.E.; Swart, P. 11β-hydroxyandrostenedione, the product of androstenedione metabolism in the adrenal, is metabolized in LNCaP cells by 5α-reductase yielding 11β-hydroxy-5α-androstanedione. J. Steroid Biochem. Mol. Biol. 2013, 138, 132–142. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Antignac, J.P.; Le Bizec, B.; Morvan, M.-L.; Svechnikov, K.; Söder, O.; Savchuk, I.; Monteiro, A.; Soffientini, U.; Johnston, Z.C.; et al. Alternative (backdoor) androgen production and masculinization in the human fetus. PLoS Biol. 2019, 17. [Google Scholar] [CrossRef]
- Flück, C.E.; Meyer-Böni, M.; Pandey, A.V.; Kempná, P.; Miller, W.L.; Schoenle, E.J.; Biason-Lauber, A. Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am. J. Hum. Genet. 2011, 89, 201–218. [Google Scholar] [CrossRef]
- Kamrath, C.; Hochberg, Z.; Hartmann, M.F.; Remer, T.; Wudy, S.A. Increased activation of the alternative “backdoor” pathway in patients with 21-hydroxylase deficiency: evidence from urinary steroid hormone analysis. J. Clin. Endocrinol. Metab. 2012, 97, E367–E375. [Google Scholar] [CrossRef]
- Lin-Su, K.; Nimkarn, S.; New, M.I. Congenital adrenal hyperplasia in adolescents: diagnosis and management. Ann. N. Y. Acad. Sci. 2008, 1135, 95–98. [Google Scholar] [CrossRef]
- Goto, M.; Piper Hanley, K.; Marcos, J.; Wood, P.J.; Wright, S.; Postle, A.D.; Cameron, I.T.; Mason, J.I.; Wilson, D.I.; Hanley, N.A. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J. Clin. Investig. 2006, 116, 953–960. [Google Scholar] [CrossRef]
- Rege, J.; Nakamura, Y.; Satoh, F.; Morimoto, R.; Kennedy, M.R.; Layman, L.C.; Honma, S.; Sasano, H.; Rainey, W.E. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. J. Clin. Endocrinol. Metab. 2013, 98, 1182–1188. [Google Scholar] [CrossRef]
- Imamichi, Y.; Yuhki, K.-I.; Orisaka, M.; Kitano, T.; Mukai, K.; Ushikubi, F.; Taniguchi, T.; Umezawa, A.; Miyamoto, K.; Yazawa, T. 11-Ketotestosterone Is a Major Androgen Produced in Human Gonads. J. Clin. Endocrinol. Metab. 2016, 101, 3582–3591. [Google Scholar] [CrossRef]
- Rege, J.; Turcu, A.F.; Kasa-Vubu, J.Z.; Lerario, A.M.; Auchus, G.C.; Auchus, R.J.; Smith, J.M.; White, P.C.; Rainey, W.E. 11-Ketotestosterone Is the Dominant Circulating Bioactive Androgen During Normal and Premature Adrenarche. J. Clin. Endocrinol. Metab. 2018, 103, 4589–4598. [Google Scholar] [CrossRef]
- Turcu, A.F.; Nanba, A.T.; Chomic, R.; Upadhyay, S.K.; Giordano, T.J.; Shields, J.J.; Merke, D.P.; Rainey, W.E.; Auchus, R.J. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur. J. Endocrinol. 2016, 174, 601–609. [Google Scholar] [CrossRef]
- Kamrath, C.; Wettstaedt, L.; Boettcher, C.; Hartmann, M.F.; Wudy, S.A. Androgen excess is due to elevated 11-oxygenated androgens in treated children with congenital adrenal hyperplasia. J. Steroid Biochem. Mol. Biol. 2018, 178, 221–228. [Google Scholar] [CrossRef]
- Bacila, I.; Adaway, J.; Hawley, J.; Mahdi, S.; Krone, R.; Patel, L.; Alvi, S.; Randell, T.; Gevers, E.; Dattani, M.; et al. Measurement of salivary adrenal-specific androgens as biomarkers of therapy control in 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Merke, D.P.; Chrousos, G.P.; Eisenhofer, G.; Weise, M.; Keil, M.F.; Rogol, A.D.; Van Wyk, J.J.; Bornstein, S.R. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N. Engl. J. Med. 2000, 343, 1362–1368. [Google Scholar] [CrossRef]
- Varness, T.S.; Allen, D.B.; Hoffman, G.L. Newborn screening for congenital adrenal hyperplasia has reduced sensitivity in girls. J. Pediatr. 2005, 147, 493–498. [Google Scholar] [CrossRef]
- Steigert, M.; Schoenle, E.J.; Biason-Lauber, A.; Torresani, T. High reliability of neonatal screening for congenital adrenal hyperplasia in Switzerland. J. Clin. Endocrinol. Metab. 2002, 87, 4106–4110. [Google Scholar] [CrossRef][Green Version]
- Balsamo, A.; Cacciari, E.; Piazzi, S.; Cassio, A.; Bozza, D.; Pirazzoli, P.; Zappulla, F. Congenital adrenal hyperplasia: neonatal mass screening compared with clinical diagnosis only in the Emilia-Romagna region of Italy, 1980–1995. Pediatrics 1996, 98, 362–367. [Google Scholar]
- Minutti, C.Z.; Lacey, J.M.; Magera, M.J.; Hahn, S.H.; McCann, M.; Schulze, A.; Cheillan, D.; Dorche, C.; Chace, D.H.; Lymp, J.F.; et al. Steroid profiling by tandem mass spectrometry improves the positive predictive value of newborn screening for congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2004, 89, 3687–3693. [Google Scholar] [CrossRef]
- Janzen, N.; Peter, M.; Sander, S.; Steuerwald, U.; Terhardt, M.; Holtkamp, U.; Sander, J. Newborn screening for congenital adrenal hyperplasia: additional steroid profile using liquid chromatography-tandem mass spectrometry. J. Clin. Endocrinol. Metab. 2007, 92, 2581–2589. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet Lond. Engl. 1997, 350, 485–487. [Google Scholar] [CrossRef]
- New, M.I.; Tong, Y.K.; Yuen, T.; Jiang, P.; Pina, C.; Chan, K.C.A.; Khattab, A.; Liao, G.J.W.; Yau, M.; Kim, S.-M.; et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J. Clin. Endocrinol. Metab. 2014, 99, E1022–E1030. [Google Scholar] [CrossRef]
- Eunice, M.; Ammini, A.C. Prenatal treatment of mothers with fetuses at risk for congenital adrenal hyperplasia: How relevant is it to Indian context? Indian J. Endocrinol. Metab. 2013, 17, 373–375. [Google Scholar]
- Simpson, J.L.; Rechitsky, S. Prenatal genetic testing and treatment for congenital adrenal hyperplasia. Fertil. Steril. 2019, 111, 21–23. [Google Scholar] [CrossRef]
- Witchel, S.F.; Miller, W.L. Prenatal treatment of congenital adrenal hyperplasia-not standard of care. J. Genet. Couns. 2012, 21, 615–624. [Google Scholar] [CrossRef]
- New, M.I.; Carlson, A.; Obeid, J.; Marshall, I.; Cabrera, M.S.; Goseco, A.; Lin-Su, K.; Putnam, A.S.; Wei, J.Q.; Wilson, R.C. Prenatal diagnosis for congenital adrenal hyperplasia in 532 pregnancies. J. Clin. Endocrinol. Metab. 2001, 86, 5651–5657. [Google Scholar] [CrossRef]
- Lajic, S.; Nordenström, A.; Ritzén, E.M.; Wedell, A. Prenatal treatment of congenital adrenal hyperplasia. Eur. J. Endocrinol. 2004, 151 (Suppl. 3), U63–U69. [Google Scholar] [CrossRef]
- Hirvikoski, T.; Nordenström, A.; Wedell, A.; Ritzén, M.; Lajic, S. Prenatal Dexamethasone Treatment of Children at Risk for Congenital Adrenal Hyperplasia: The Swedish Experience and Standpoint. J. Clin. Endocrinol. Metab. 2012, 97, 1881–1883. [Google Scholar] [CrossRef]
- Wallensteen, L.; Zimmermann, M.; Thomsen Sandberg, M.; Gezelius, A.; Nordenström, A.; Hirvikoski, T.; Lajic, S. Sex-Dimorphic Effects of Prenatal Treatment With Dexamethasone. J. Clin. Endocrinol. Metab. 2016, 101, 3838–3846. [Google Scholar] [CrossRef]
- Hirvikoski, T.; Nordenström, A.; Lindholm, T.; Lindblad, F.; Ritzén, E.M.; Wedell, A.; Lajic, S. Cognitive functions in children at risk for congenital adrenal hyperplasia treated prenatally with dexamethasone. J. Clin. Endocrinol. Metab. 2007, 92, 542–548. [Google Scholar] [CrossRef]
- Karlsson, L.; Gezelius, A.; Nordenström, A.; Hirvikoski, T.; Lajic, S. Cognitive impairment in adolescents and adults with congenital adrenal hyperplasia. Clin. Endocrinol. 2017, 87, 651–659. [Google Scholar] [CrossRef]
- Karlsson, L.; Barbaro, M.; Ewing, E.; Gomez-Cabrero, D.; Lajic, S. Epigenetic Alterations Associated With Early Prenatal Dexamethasone Treatment. J. Endocrine Soc. 2019, 3, 250–263. [Google Scholar] [CrossRef]
- Zhang, L.H.; Rodriguez, H.; Ohno, S.; Miller, W.L. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: implications for adrenarche and the polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA 1995, 92, 10619–10623. [Google Scholar] [CrossRef]
- El-Maouche, D.; Arlt, W.; Merke, D.P. Congenital adrenal hyperplasia. Lancet Lond. Engl. 2017, 390, 2194–2210. [Google Scholar] [CrossRef]
- González, R.; Ludwikowski, B.M. Should CAH in Females Be Classified as DSD? Front. Pediatr. 2016, 4, 48. [Google Scholar] [CrossRef]
- Salle, J.L.P.; Lorenzo, A.J.; Jesus, L.E.; Leslie, B.; AlSaid, A.; Macedo, F.N.; Jayanthi, V.R.; de Castro, R. Surgical treatment of high urogenital sinuses using the anterior sagittal transrectal approach: a useful strategy to optimize exposure and outcomes. J. Urol. 2012, 187, 1024–1031. [Google Scholar] [CrossRef]
- Ludwikowski, B.M.; González, R. The Surgical Correction of Urogenital Sinus in Patients with DSD: 15 Years after Description of Total Urogenital Mobilization in Children. Front. Pediatr. 2013, 1, 41. [Google Scholar] [CrossRef]
- Merke, D.P.; Poppas, D.P. Management of adolescents with congenital adrenal hyperplasia. Lancet Diabetes Endocrinol. 2013, 1, 341–352. [Google Scholar] [CrossRef]
- Yankovic, F.; Cherian, A.; Steven, L.; Mathur, A.; Cuckow, P. Current practice in feminizing surgery for congenital adrenal hyperplasia; a specialist survey. J. Pediatr. Urol. 2013, 9, 1103–1107. [Google Scholar] [CrossRef]
- Suorsa, K.I.; Mullins, A.J.; Tackett, A.P.; Reyes, K.J.S.; Austin, P.; Baskin, L.; Bernabé, K.; Cheng, E.; Fried, A.; Frimberger, D.; et al. Characterizing Early Psychosocial Functioning of Parents of Children with Moderate to Severe Genital Ambiguity due to Disorders of Sex Development. J. Urol. 2015, 194, 1737–1742. [Google Scholar] [CrossRef]
- Almasri, J.; Zaiem, F.; Rodriguez-Gutierrez, R.; Tamhane, S.U.; Iqbal, A.M.; Prokop, L.J.; Speiser, P.W.; Baskin, L.S.; Bancos, I.; Murad, M.H. Genital Reconstructive Surgery in Females With Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2018, 103, 4089–4096. [Google Scholar] [CrossRef]
- Krege, S.; Eckoldt, F.; Richter-Unruh, A.; Köhler, B.; Leuschner, I.; Mentzel, H.-J.; Moss, A.; Schweizer, K.; Stein, R.; Werner-Rosen, K.; et al. Variations of sex development: The first German interdisciplinary consensus paper. J. Pediatr. Urol. 2019, 15, 114–123. [Google Scholar] [CrossRef]
- Bachelot, A.; Grouthier, V.; Courtillot, C.; Dulon, J.; Touraine, P. Management of endocrine disease: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update on the management of adult patients and prenatal treatment. Eur. J. Endocrinol. 2017, 176, R167–R181. [Google Scholar] [CrossRef]
- Arlt, W.; Willis, D.S.; Wild, S.H.; Krone, N.; Doherty, E.J.; Hahner, S.; Han, T.S.; Carroll, P.V.; Conway, G.S.; Rees, D.A.; et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J. Clin. Endocrinol. Metab. 2010, 95, 5110–5121. [Google Scholar] [CrossRef]
- Wang, L.C.; Poppas, D.P. Surgical outcomes and complications of reconstructive surgery in the female congenital adrenal hyperplasia patient: What every endocrinologist should know. J. Steroid Biochem. Mol. Biol. 2017, 165, 137–144. [Google Scholar] [CrossRef]
- Strandqvist, A.; Falhammar, H.; Lichtenstein, P.; Hirschberg, A.L.; Wedell, A.; Norrby, C.; Nordenskjöld, A.; Frisén, L.; Nordenström, A. Suboptimal psychosocial outcomes in patients with congenital adrenal hyperplasia: epidemiological studies in a nonbiased national cohort in Sweden. J. Clin. Endocrinol. Metab. 2014, 99, 1425–1432. [Google Scholar] [CrossRef]
- Laue, L.; Merke, D.P.; Jones, J.V.; Barnes, K.M.; Hill, S.; Cutler, G.B. A preliminary study of flutamide, testolactone, and reduced hydrocortisone dose in the treatment of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1996, 81, 3535–3539. [Google Scholar]
- Merke, D.P.; Keil, M.F.; Jones, J.V.; Fields, J.; Hill, S.; Cutler, G.B. Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2000, 85, 1114–1120. [Google Scholar] [CrossRef]
- Verma, S.; Vanryzin, C.; Sinaii, N.; Kim, M.S.; Nieman, L.K.; Ravindran, S.; Calis, K.A.; Arlt, W.; Ross, R.J.; Merke, D.P. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin. Endocrinol. 2010, 72, 441–447. [Google Scholar] [CrossRef]
- Porter, J.; Withe, M.; Ross, R.J. Immediate-release granule formulation of hydrocortisone, Alkindi®, for treatment of paediatric adrenal insufficiency (Infacort development programme). Expert Rev. Endocrinol. Metab. 2018, 13, 119–124. [Google Scholar] [CrossRef]
- Chabraoui, L.; Abid, F.; Menassa, R.; Gaouzi, A.; El Hessni, A.; Morel, Y. Three novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11Beta-hydroxylase deficiency in a moroccan population. Horm. Res. Paediatr. 2010, 74, 182–189. [Google Scholar] [CrossRef]
- Zachmann, M.; Tassinari, D.; Prader, A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. A study of 25 patients. J. Clin. Endocrinol. Metab. 1983, 56, 222–229. [Google Scholar] [CrossRef]
- Curnow, K.M.; Slutsker, L.; Vitek, J.; Cole, T.; Speiser, P.W.; New, M.I.; White, P.C.; Pascoe, L. Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc. Natl. Acad. Sci. USA 1993, 90, 4552–4556. [Google Scholar] [CrossRef]
- Geley, S.; Kapelari, K.; Jöhrer, K.; Peter, M.; Glatzl, J.; Vierhapper, H.; Schwarz, S.; Helmberg, A.; Sippell, W.G.; White, P.C.; et al. CYP11B1 mutations causing congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 1996, 81, 2896–2901. [Google Scholar]
- Merke, D.P.; Tajima, T.; Chhabra, A.; Barnes, K.; Mancilla, E.; Baron, J.; Cutler, G.B. Novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11 beta-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 1998, 83, 270–273. [Google Scholar]
- Solyom, J.; Racz, K.; Peter, F.; Homoki, J.; Sippell, W.G.; Peter, M. Clinical, Hormonal and Molecular Genetic Characterization of Hungarian Patients with 11β-Hydroxylase Deficiency. Int. J. Disabil. Hum. Dev. 2011, 2, 37–44. [Google Scholar] [CrossRef]
- Motaghedi, R.; Betensky, B.P.; Slowinska, B.; Cerame, B.; Cabrer, M.; New, M.I.; Wilson, R.C. Update on the prenatal diagnosis and treatment of congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. J. Pediatr. Endocrinol. Metab. JPEM 2005, 18, 133–142. [Google Scholar] [CrossRef]
- Krone, N.; Riepe, F.G.; Götze, D.; Korsch, E.; Rister, M.; Commentz, J.; Partsch, C.-J.; Grötzinger, J.; Peter, M.; Sippell, W.G. Congenital adrenal hyperplasia due to 11-hydroxylase deficiency: functional characterization of two novel point mutations and a three-base pair deletion in the CYP11B1 gene. J. Clin. Endocrinol. Metab. 2005, 90, 3724–3730. [Google Scholar] [CrossRef]
- Nimkarn, S.; New, M.I. Steroid 11beta- hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol. Metab. TEM 2008, 19, 96–99. [Google Scholar] [CrossRef]
- Soardi, F.C.; Penachioni, J.Y.; Justo, G.Z.; Bachega, T.A.S.S.; Inácio, M.; Mendonça, B.B.; de Castro, M.; de Mello, M.P. Novel mutations in CYP11B1 gene leading to 11 beta-hydroxylase deficiency in Brazilian patients. J. Clin. Endocrinol. Metab. 2009, 94, 3481–3485. [Google Scholar] [CrossRef]
- Parajes, S.; Loidi, L.; Reisch, N.; Dhir, V.; Rose, I.T.; Hampel, R.; Quinkler, M.; Conway, G.S.; Castro-Feijóo, L.; Araujo-Vilar, D.; et al. Functional consequences of seven novel mutations in the CYP11B1 gene: Four mutations associated with nonclassic and three mutations causing classic 11{beta}-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 779–788. [Google Scholar] [CrossRef]
- Dumic, K.; Wilson, R.; Thanasawat, P.; Grubic, Z.; Kusec, V.; Stingl, K.; New, M.I. Steroid 11-beta hydroxylase deficiency caused by compound heterozygosity for a novel mutation in intron 7 (IVS 7 DS+4A to G) in one CYP11B1 allele and R448H in exon 8 in the other. Eur. J. Pediatr. 2010, 169, 891–894. [Google Scholar] [CrossRef]
- Ben Charfeddine, I.; Riepe, F.G.; Kahloul, N.; Kulle, A.E.; Adala, L.; Mamaï, O.; Amara, A.; Mili, A.; Amri, F.; Saad, A.; et al. Two novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11β hydroxylase deficiency in a Tunisian family. Gen. Comp. Endocrinol. 2012, 175, 514–518. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Y.; Sun, S.; Zhang, H.; Wang, W.; Ning, G.; Li, X. A prevalent and three novel mutations in CYP11B1 gene identified in Chinese patients with 11-beta hydroxylase deficiency. J. Steroid Biochem. Mol. Biol. 2013, 133, 25–29. [Google Scholar] [CrossRef]
- Bin-Abbas, B.; Al-Humaida, D.; Al-Sagheir, A.; Qasem, E.; Almohanna, M.; Alzahrani, A.S. Divergent gender identity in three siblings with 46XX karyotype and severely virilizing congenital adrenal hyperplasia caused by a novel CYP11B1 mutation. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2014, 20, e191–e197. [Google Scholar]
- Dumic, K.; Yuen, T.; Grubic, Z.; Kusec, V.; Barisic, I.; New, M.I. Two Novel CYP11B1 Gene Mutations in Patients from Two Croatian Families with 11β-Hydroxylase Deficiency. Int. J. Endocrinol. 2014, 2014. [Google Scholar] [CrossRef]
- Menabò, S.; Polat, S.; Baldazzi, L.; Kulle, A.E.; Holterhus, P.-M.; Grötzinger, J.; Fanelli, F.; Balsamo, A.; Riepe, F.G. Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: functional consequences of four CYP11B1 mutations. Eur. J. Hum. Genet. EJHG 2014, 22, 610–616. [Google Scholar] [CrossRef]
- Charnwichai, P.; Yeetong, P.; Suphapeetiporn, K.; Supornsilchai, V.; Sahakitrungruang, T.; Shotelersuk, V. Splicing analysis of CYP11B1 mutation in a family affected with 11β-hydroxylase deficiency: case report. BMC Endocr. Disord. 2016, 16, 37. [Google Scholar] [CrossRef]
- Matallana-Rhoades, A.M.; Corredor-Castro, J.D.; Bonilla-Escobar, F.J.; Mecias-Cruz, B.V.; Mejia de Beldjena, L. Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: description of a new mutation, R384X. Colomb. Méd. 2016, 47, 172–175. [Google Scholar]
- Kandemir, N.; Yilmaz, D.Y.; Gonc, E.N.; Ozon, A.; Alikasifoglu, A.; Dursun, A.; Ozgul, R.K. Novel and prevalent CYP11B1 gene mutations in Turkish patients with 11-β hydroxylase deficiency. J. Steroid Biochem. Mol. Biol. 2017, 165, 57–63. [Google Scholar] [CrossRef]
- Khattab, A.; Haider, S.; Kumar, A.; Dhawan, S.; Alam, D.; Romero, R.; Burns, J.; Li, D.; Estatico, J.; Rahi, S.; et al. Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 2017, 114, E1933–E1940. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Tong, T.; Yang, Q. Non-classical 11β-hydroxylase deficiency caused by compound heterozygous mutations: a case study and literature review. J. Ovarian Res. 2018, 11, 82. [Google Scholar] [CrossRef]
- Baş, F.; Toksoy, G.; Ergun-Longmire, B.; Uyguner, Z.O.; Abalı, Z.Y.; Poyrazoğlu, Ş.; Karaman, V.; Avcı, Ş.; Altunoğlu, U.; Bundak, R.; et al. Prevalence, clinical characteristics and long-term outcomes of classical 11 β-hydroxylase deficiency (11BOHD) in Turkish population and novel mutations in CYP11B1 gene. J. Steroid Biochem. Mol. Biol. 2018, 181, 88–97. [Google Scholar] [CrossRef]
- Breil, T.; Yakovenko, V.; Inta, I.; Choukair, D.; Klose, D.; Mittnacht, J.; Schulze, E.; Alrajab, A.; Grulich-Henn, J.; Bettendorf, M. Typical characteristics of children with congenital adrenal hyperplasia due to 11β-hydroxylase deficiency: a single-centre experience and review of the literature. J. Pediatr. Endocrinol. Metab. JPEM 2019, 32, 259–267. [Google Scholar] [CrossRef]
- Valadares, L.P.; Pfeilsticker, A.C.V.; de Brito Sousa, S.M.; Cardoso, S.C.; de Moraes, O.L.; Gonçalves de Castro, L.C.; de Oliveira, R.S.; Lofrano-Porto, A. Insights on the phenotypic heterogenity of 11β-hydroxylase deficiency: clinical and genetic studies in two novel families. Endocrine 2018, 62, 326–332. [Google Scholar] [CrossRef]
- Hampf, M.; Dao, N.T.; Hoan, N.T.; Bernhardt, R. Unequal crossing-over between aldosterone synthase and 11beta-hydroxylase genes causes congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2001, 86, 4445–4452. [Google Scholar]
- Menabò, S.; Boccassini, S.; Gambineri, A.; Balsamo, A.; Pasquali, R.; Prontera, O.; Mazzanti, L.; Baldazzi, L. Improving the diagnosis of 11β-hydroxylase deficiency using home-made MLPA probes: identification of a novel chimeric CYP11B2/CYP11B1 gene in a Sicilian patient. J. Endocrinol. Investig. 2016, 39, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Holcombe, J.H.; Keenan, B.S.; Nichols, B.L.; Kirkland, R.T.; Clayton, G.W. Neonatal salt loss in the hypertensive form of congenital adrenal hyperplasia. Pediatrics 1980, 65, 777–781. [Google Scholar] [PubMed]
- Joehrer, K.; Geley, S.; Strasser-Wozak, E.M.; Azziz, R.; Wollmann, H.A.; Schmitt, K.; Kofler, R.; White, P.C. CYP11B1 mutations causing non-classic adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Hum. Mol. Genet. 1997, 6, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.J.; Nugent, T.; Perry, L.A.; Davies, K.; Morel, Y.; Drake, W.M.; Savage, M.O.; Johnston, L.B. Cosegregation of a novel homozygous CYP11B1 mutation with the phenotype of non-classical congenital adrenal hyperplasia in a consanguineous family. Horm. Res. 2007, 67, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Razzaghy-Azar, M.; Karimi, S.; Shirazi, E. Gender Identity in Patients with Congenital Adrenal Hyperplasia. Int. J. Endocrinol. Metab. 2017, 15, e12537. [Google Scholar] [CrossRef] [PubMed]
- Simm, P.J.; Zacharin, M.R. Successful pregnancy in a patient with severe 11-beta-hydroxylase deficiency and novel mutations in CYP11B1 gene. Horm. Res. 2007, 68, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Belgorosky, A.; Guercio, G.; Pepe, C.; Saraco, N.; Rivarola, M.A. Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm. Res. 2009, 72, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Shozu, M.; Akasofu, K.; Harada, T.; Kubota, Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J. Clin. Endocrinol. Metab. 1991, 72, 560–566. [Google Scholar] [CrossRef]
- Harada, N.; Ogawa, H.; Shozu, M.; Yamada, K. Genetic studies to characterize the origin of the mutation in placental aromatase deficiency. Am. J. Hum. Genet. 1992, 51, 666–672. [Google Scholar] [PubMed]
- Conte, F.A.; Grumbach, M.M.; Ito, Y.; Fisher, C.R.; Simpson, E.R. A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J. Clin. Endocrinol. Metab. 1994, 78, 1287–1292. [Google Scholar] [PubMed]
- Ito, Y.; Fisher, C.R.; Conte, F.A.; Grumbach, M.M.; Simpson, E.R. Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc. Natl. Acad. Sci. USA 1993, 90, 11673–11677. [Google Scholar] [CrossRef] [PubMed]
- Morishima, A.; Grumbach, M.M.; Simpson, E.R.; Fisher, C.; Qin, K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J. Clin. Endocrinol. Metab. 1995, 80, 3689–3698. [Google Scholar] [PubMed]
- Portrat-Doyen, S.; Forest, M.G.; Nicolino, M.; Morel, Y.; Chatelain, P.C. Female pseudohermaphroditism (FPH) resulting from aromatase (P450arom) deficiency associated with a novel mutation (R457X) in the CYP19 gene. Hormone Res. 1996, 46/S2, 4, Oral Presentation n. 14. [Google Scholar]
- Mullis, P.E.; Yoshimura, N.; Kuhlmann, B.; Lippuner, K.; Jaeger, P.; Harada, H. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J. Clin. Endocrinol. Metab. 1997, 82, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Beck, A.; Wickert, L.; Bolkenius, U.; Tittel, B.; Hinkel, K.; Bidlingmaier, F. Female pseudohermaphroditism associated with a novel homozygous G-to-A (V370-to-M) substitution in the P-450 aromatase gene. J. Pediatr. Endocrinol. Metab. JPEM 1998, 11, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Belgorosky, A.; Pepe, C.; Marino, R.; Guercio, G.; Saraco, N.; Vaiani, E.; Rivarola, M.A. Hypothalamic-pituitary-ovarian axis during infancy, early and late prepuberty in an aromatase-deficient girl who is a compound heterocygote for two new point mutations of the CYP19 gene. J. Clin. Endocrinol. Metab. 2003, 88, 5127–5131. [Google Scholar] [CrossRef] [PubMed]
- Pepe, C.M.; Saraco, N.I.; Baquedano, M.S.; Guercio, G.; Vaiani, E.; Marino, R.; Pandey, A.V.; Flück, C.E.; Rivarola, M.A.; Belgorosky, A. The cytochrome P450 aromatase lacking exon 5 is associated with a phenotype of nonclassic aromatase deficiency and is also present in normal human steroidogenic tissues. Clin. Endocrinol. 2007, 67, 698–705. [Google Scholar] [CrossRef]
- Lin, L.; Ercan, O.; Raza, J.; Burren, C.P.; Creighton, S.M.; Auchus, R.J.; Dattani, M.T.; Achermann, J.C. Variable phenotypes associated with aromatase (CYP19) insufficiency in humans. J. Clin. Endocrinol. Metab. 2007, 92, 982–990. [Google Scholar] [CrossRef]
- Richter-Unruh, A.; Schmidt, M.; Verhoef-Post, M.; Themmen, A. Aromatase deficiency: delayed puberty in a German girl is due to 2 new mutations of the CYP19 gene. In Proceedings of the ENDO 2008: The Endocrine Society 90th Annual Meeting, San Francisco, CA, USA, 15–18 June 2008; p. Abstr P2-590. [Google Scholar]
- Hauri-Hohl, A.; Meyer-Böni, M.; Lang-Muritano, M.; Hauri-Hohl, M.; Schoenle, E.J.; Biason-Lauber, A. Aromatase deficiency owing to a functional variant in the placenta promoter and a novel missense mutation in the CYP19A1 gene. Clin. Endocrinol. 2011, 75, 39–43. [Google Scholar] [CrossRef]
- Bouchoucha, N.; Samara-Boustani, D.; Pandey, A.V.; Bony-Trifunovic, H.; Hofer, G.; Aigrain, Y.; Polak, M.; Flück, C.E. Characterization of a novel CYP19A1 (aromatase) R192H mutation causing virilization of a 46,XX newborn, undervirilization of the 46,XY brother, but no virilization of the mother during pregnancies. Mol. Cell. Endocrinol. 2014, 390, 8–17. [Google Scholar] [CrossRef]
- Gagliardi, L.; Scott, H.S.; Feng, J.; Torpy, D.J. A case of Aromatase deficiency due to a novel CYP19A1 mutation. BMC Endocr. Disord. 2014, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Marino, R.; Perez Garrido, N.; Costanzo, M.; Guercio, G.; Juanes, M.; Rocco, C.; Ramirez, P.; Warman, D.M.; Ciaccio, M.; Pena, G.; et al. Five new cases of 46,XX aromatase deficiency: clinical follow-up from birth to puberty, a novel mutation, and a founder effect. J. Clin. Endocrinol. Metab. 2015, 100, E301–E307. [Google Scholar] [CrossRef] [PubMed]
- Saraco, N.; Nesi-Franca, S.; Sainz, R.; Marino, R.; Marques-Pereira, R.; La Pastina, J.; Perez Garrido, N.; Sandrini, R.; Rivarola, M.A.; de Lacerda, L.; et al. An Intron 9 CYP19 Gene Variant (IVS9+5G>A), Present in an Aromatase-Deficient Girl, Affects Normal Splicing and Is Also Present in Normal Human Steroidogenic Tissues. Horm. Res. Paediatr. 2015, 84, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.-J.; Cheng, T.; Zhu, H.; Han, B.; Fan, M.-X.; Gu, T.; Zhao, S.-X.; Liu, Y.; Cheng, K.-X.; Song, H.-D.; et al. Aromatase deficiency: a novel compound heterozygous mutation identified in a Chinese girl with severe phenotype and obvious maternal virilization. Mol. Cell. Endocrinol. 2016, 433, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Akçurin, S.; Türkkahraman, D.; Kim, W.-Y.; Durmaz, E.; Shin, J.-G.; Lee, S.-J. A Novel Null Mutation in P450 Aromatase Gene (CYP19A1) Associated with Development of Hypoplastic Ovaries in Humans. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 205–210. [Google Scholar] [CrossRef]
- Mazen, I.; McElreavey, K.; Elaidy, A.; Kamel, A.K.; Abdel-Hamid, M.S. Aromatase Deficiency due to a Homozygous CYP19A1 Mutation in a 46,XX Egyptian Patient with Ambiguous Genitalia. Sex. Dev. Genet. Mol. Biol. Evol. Endocrinol. Embryol. Pathol. Sex Determ. Differ. 2017, 11, 275–279. [Google Scholar] [CrossRef]
- Unal, E.; Yıldırım, R.; Taş, F.F.; Demir, V.; Onay, H.; Haspolat, Y.K. Aromatase Deficiency due to a Novel Mutation in CYP19A1 Gene. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 377–381. [Google Scholar]
- Ludwikowski, B.; Heger, S.; Datz, N.; Richter-Unruh, A.; González, R. Aromatase deficiency: Rare cause of virilization. Eur. J. Pediatr. Surg. 2013, 23, 418–422. [Google Scholar] [CrossRef]
- Verma, N.; Jain, V.; Birla, S.; Jain, R.; Sharma, A. Growth and hormonal profile from birth to adolescence of a girl with aromatase deficiency. J. Pediatr. Endocrinol. Metab. JPEM 2012, 25, 1185–1190. [Google Scholar] [CrossRef]
- Janner, M.; Flück, C.E.; Mullis, P.E. Impact of estrogen replacement throughout childhood on growth, pituitary-gonadal axis and bone in a 46,XX patient with CYP19A1 deficiency. Horm. Res. Paediatr. 2012, 78, 261–268. [Google Scholar] [CrossRef]
- Flück, C.E.; Tajima, T.; Pandey, A.V.; Arlt, W.; Okuhara, K.; Verge, C.F.; Jabs, E.W.; Mendonça, B.B.; Fujieda, K.; Miller, W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004, 36, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Pandey, A.V.; Agrawal, V.; Reardon, W.; Lapunzina, P.D.; Mowat, D.; Jabs, E.W.; Van Vliet, G.; Sack, J.; Flück, C.E.; et al. Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am. J. Hum. Genet. 2005, 76, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Arlt, W.; Walker, E.A.; Draper, N.; Ivison, H.E.; Ride, J.P.; Hammer, F.; Chalder, S.M.; Borucka-Mankiewicz, M.; Hauffa, B.P.; Malunowicz, E.M.; et al. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet Lond. Engl. 2004, 363, 2128–2135. [Google Scholar] [CrossRef]
- Peterson, R.E.; Imperato-McGinley, J.; Gautier, T.; Shackleton, C. Male pseudohermaphroditism due to multiple defects in steroid-biosynthetic microsomal mixed-function oxidases. A new variant of congenital adrenal hyperplasia. N. Engl. J. Med. 1985, 313, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Idkowiak, J.; O’Riordan, S.; Reisch, N.; Malunowicz, E.M.; Collins, F.; Kerstens, M.N.; Köhler, B.; Graul-Neumann, L.M.; Szarras-Czapnik, M.; Dattani, M.; et al. Pubertal presentation in seven patients with congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J. Clin. Endocrinol. Metab. 2011, 96, E453–E462. [Google Scholar] [CrossRef]
- Krone, N.; Reisch, N.; Idkowiak, J.; Dhir, V.; Ivison, H.E.; Hughes, B.A.; Rose, I.T.; O’Neil, D.M.; Vijzelaar, R.; Smith, M.J.; et al. Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J. Clin. Endocrinol. Metab. 2012, 97, E257–E267. [Google Scholar] [CrossRef] [PubMed]
- Fukami, M.; Horikawa, R.; Nagai, T.; Tanaka, T.; Naiki, Y.; Sato, N.; Okuyama, T.; Nakai, H.; Soneda, S.; Tachibana, K.; et al. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J. Clin. Endocrinol. Metab. 2005, 90, 414–426. [Google Scholar] [CrossRef]
- Homma, K.; Hasegawa, T.; Nagai, T.; Adachi, M.; Horikawa, R.; Fujiwara, I.; Tajima, T.; Takeda, R.; Fukami, M.; Ogata, T. Urine steroid hormone profile analysis in cytochrome P450 oxidoreductase deficiency: implication for the backdoor pathway to dihydrotestosterone. J. Clin. Endocrinol. Metab. 2006, 91, 2643–2649. [Google Scholar] [CrossRef]
- Scott, R.R.; Gomes, L.G.; Huang, N.; Van Vliet, G.; Miller, W.L. Apparent manifesting heterozygosity in P450 oxidoreductase deficiency and its effect on coexisting 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2007, 92, 2318–2322. [Google Scholar] [CrossRef]
- Fukami, M.; Nishimura, G.; Homma, K.; Nagai, T.; Hanaki, K.; Uematsu, A.; Ishii, T.; Numakura, C.; Sawada, H.; Nakacho, M.; et al. Cytochrome P450 oxidoreductase deficiency: identification and characterization of biallelic mutations and genotype-phenotype correlations in 35 Japanese patients. J. Clin. Endocrinol. Metab. 2009, 94, 1723–1731. [Google Scholar] [CrossRef]
- Sahakitrungruang, T.; Huang, N.; Tee, M.K.; Agrawal, V.; Russell, W.E.; Crock, P.; Murphy, N.; Migeon, C.J.; Miller, W.L. Clinical, genetic, and enzymatic characterization of P450 oxidoreductase deficiency in four patients. J. Clin. Endocrinol. Metab. 2009, 94, 4992–5000. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Mallet, D.; Hofer, G.; Samara-Boustani, D.; Leger, J.; Polak, M.; Morel, Y.; Pandey, A.V. Deletion of P399_E401 in NADPH cytochrome P450 oxidoreductase results in partial mixed oxidase deficiency. Biochem. Biophys. Res. Commun. 2011, 412, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Tachibana, K.; Asakura, Y.; Yamamoto, T.; Hanaki, K.; Oka, A. Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am. J. Med. Genet. A. 2004, 128, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Hinney, B.; Peter, M.; Steinberger, D.; Lakomek, M. Features of Antley-Bixler syndrome in an infant born to a mother with pregnancy luteoma. Eur. J. Pediatr. 2000, 159, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Cragun, D.L.; Trumpy, S.K.; Shackleton, C.H.L.; Kelley, R.I.; Leslie, N.D.; Mulrooney, N.P.; Hopkin, R.J. Undetectable maternal serum uE3 and postnatal abnormal sterol and steroid metabolism in Antley-Bixler syndrome. Am. J. Med. Genet. A. 2004, 129, 1–7. [Google Scholar] [CrossRef]
- Shackleton, C.; Marcos, J.; Malunowicz, E.M.; Szarras-Czapnik, M.; Jira, P.; Taylor, N.F.; Murphy, N.; Crushell, E.; Gottschalk, M.; Hauffa, B.; et al. Biochemical diagnosis of Antley-Bixler syndrome by steroid analysis. Am. J. Med. Genet. A. 2004, 128, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.; Arlt, W.; Shackleton, C.; Kelley, R.I.; Braddock, S.R. Linking Antley-Bixler syndrome and congenital adrenal hyperplasia: a novel case of P450 oxidoreductase deficiency. Am. J. Med. Genet. A. 2006, 140A, 1797–1803. [Google Scholar] [CrossRef]
- Ko, J.M.; Cheon, C.-K.; Kim, G.-H.; Yoo, H.-W. A case of Antley-Bixler syndrome caused by compound heterozygous mutations of the cytochrome P450 oxidoreductase gene. Eur. J. Pediatr. 2009, 168, 877–880. [Google Scholar] [CrossRef]
- But, W.M.; Lo, I.F.M.; Shek, C.C.; Tse, W.Y.; Lam, S.T.S. Ambiguous genitalia, impaired steroidogenesis, and Antley-Bixler syndrome in a patient with P450 oxidoreductase deficiency. Hong Kong Med. J. Xianggang Yi Xue Za Zhi 2010, 16, 59–62. [Google Scholar]
- Guaragna-Filho, G.; Castro, C.C.T.D.S.; De Carvalho, R.R.; Coeli, F.B.; Ferraz, L.F.C.; Petroli, R.J.; De Mello, M.P.; Sewaybricker, L.E.; Lemos-Marini, S.H.V.; D’Souza-Li, L.F.R.; et al. 46,XX DSD and Antley-Bixler syndrome due to novel mutations in the cytochrome P450 oxidoreductase gene. Arq. Bras. Endocrinol. Metabol. 2012, 56, 578–585. [Google Scholar] [CrossRef]
- Oldani, E.; Garel, C.; Bucourt, M.; Carbillon, L. Prenatal Diagnosis of Antley-Bixler Syndrome and POR Deficiency. Am. J. Case Rep. 2015, 16, 882–885. [Google Scholar] [PubMed]
- Burkhard, F.Z.; Parween, S.; Udhane, S.S.; Flück, C.E.; Pandey, A.V. P450 Oxidoreductase deficiency: Analysis of mutations and polymorphisms. J. Steroid Biochem. Mol. Biol. 2017, 165, 38–50. [Google Scholar] [CrossRef]
- Oh, J.; Song, J.S.; Park, J.E.; Jang, S.Y.; Ki, C.S.; Kim, D.K. A Case of Antley-Bixler Syndrome With a Novel Likely Pathogenic Variant (c.529G>C) in the POR Gene. Ann. Lab. Med. 2017, 37, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Puiu, M.; Pienar, A.; Emandi, A.C.; Arghirescu, S.; Popa, C.; Micle, I. A case of antley bixler syndrome: diagnosis and outcome. Acta Endocrinol. 2012, 8, 479–484. [Google Scholar] [CrossRef]
- Fukami, M.; Hasegawa, T.; Horikawa, R.; Ohashi, T.; Nishimura, G.; Homma, K.; Ogata, T. Cytochrome P450 oxidoreductase deficiency in three patients initially regarded as having 21-hydroxylase deficiency and/or aromatase deficiency: diagnostic value of urine steroid hormone analysis. Pediatr. Res. 2006, 59, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Agrawal, V.; Sandee, D.; Tee, M.K.; Huang, N.; Choi, J.H.; Morrissey, K.; Giacomini, K.M. Consequences of POR mutations and polymorphisms. Mol. Cell. Endocrinol. 2011, 336, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Kempná, P.; Hofer, G.; Mullis, P.E.; Flück, C.E. Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Mol. Endocrinol. Baltim. Md 2007, 21, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Idkowiak, J.; Cragun, D.; Hopkin, R.J.; Arlt, W. Cytochrome P450 Oxidoreductase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1419/ (accessed on 3 August 2017).
- Reisch, N.; Idkowiak, J.; Hughes, B.A.; Ivison, H.E.; Abdul-Rahman, O.A.; Hendon, L.G.; Olney, A.H.; Nielsen, S.; Harrison, R.; Blair, E.M.; et al. Prenatal diagnosis of congenital adrenal hyperplasia caused by P450 oxidoreductase deficiency. J. Clin. Endocrinol. Metab. 2013, 98, E528–E536. [Google Scholar] [CrossRef]
- Miller, W.L. Minireview: regulation of steroidogenesis by electron transfer. Endocrinology 2005, 146, 2544–2550. [Google Scholar] [CrossRef]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Hu, L.; Zhuo, W.; He, Y.-J.; Zhou, H.-H.; Fan, L. Pharmacogenetics of P450 oxidoreductase: Implications in drug metabolism and therapy. Pharmacogenet. Genomics 2012, 22, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Tomalik-Scharte, D.; Maiter, D.; Kirchheiner, J.; Ivison, H.E.; Fuhr, U.; Arlt, W. Impaired hepatic drug and steroid metabolism in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. Eur. J. Endocrinol. 2010, 163, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.R.; Miller, W.L. Genetic and clinical features of p450 oxidoreductase deficiency. Horm. Res. 2008, 69, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Laue, K.; Pogoda, H.-M.; Daniel, P.B.; van Haeringen, A.; Alanay, Y.; von Ameln, S.; Rachwalski, M.; Morgan, T.; Gray, M.J.; Breuning, M.H.; et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am. J. Hum. Genet. 2011, 89, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet Lond. Engl. 2002, 360, 1155–1162. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group/Name of IEM | Enzyme | Gene/Inheritance | Cellular/Tissue Localization | OMIM Gene Number |
|---|---|---|---|---|
| 3β-hydroxysteroid dehydrogenase 2 deficiency (3β-HSD2D) | 3βHSD2 or HSD3B2 | HSD3B2/AR | Mitochondria/each adrenal zone and gonads | 613,890 |
| 21-hydroxylase deficiency (21-OHD) | P450c21 or CYP21A2 | CYP21A2/AR | Endoplasmic reticulum/each adrenal zone | 613,815 |
| 11β-hydroxylase deficiency (11-OHD) | P450c11β or CYP11B1 | CYP11B1/AR | Mitochondria/adrenal fasciculata | 610,613 |
| Aromatase deficiency (AroD) | P450aro or CYP19A1 | CYP19A1/AR | Endoplasmic reticulum/gonads (granulosa cells) | 107,910 |
| Cytochrome P450 oxidoreductase deficiency (PORD) | P450-OR or POR | POR/AR | Endoplasmic reticulum/each adrenal zone and gonads | 124,015 |
| Enzymatic Deficiency | Clinical Deatures at Birth | Other Vlinical Features | Biochemical Markers for Diagnosis | Clinical Course | Therapy | Sex of Rearing | Fertility |
|---|---|---|---|---|---|---|---|
| 3β-HSD2D | Salt wasting (70%); mild genital atipicity (25%) | Precocious adrenarche; | Serum: ↑ stimulated ratio of Δ4 over Δ5 steroids; Urine: ↑ ratios DHEA/GC metabolites and 5PT/GC metabolites | Hirsutism, polycystic ovaries, primary amenorrhea, or irregular menses; osteopenia/osteoporosis | Hydrocortisone Fludrocortisone +/− Rapid rehydration, correction of hypoglycemia +/− Sex hormone | female | Unknown |
| 21-OHD | Classical: salt wasting + atypical genitalia (75%); simple virilization (25%). Non-classical: asymptomatic | Enlarged adrenals; pseudo-precocious puberty; adrenal rest in the ovaries | Serum: ↑17-OHP and 21-DOF; 11-KΔ4A, 11-KT Urine: ↑ metabolites of 17-OHP (17HP,PT) and 21-DOF (P’TONE) Saliva: ↑11-KΔ4A and 11-KT | Precocious puberty; hirsutism; irregular menses; polycystic ovaries | Hydrocortisone Fludrocortisone +/− Rapid rehydration, correction of hypoglycemia | Prader 1–3: female. Prader 4–5: early diagnosis tentatively female; late diagnosis: possible male | Generally reduced according to the severity of enzymatic deficiency; improvement with therapeutic optimization and compliance |
| 11-OHD | Classical: atypical genitalia (100%), high blood pressure (~10%) Non-classical: asymptomatic | Pseudo-precocious puberty; high blood pressure in infancy (20%) | Serum: ↑ S and DOC Urine: ↑ metabolites of S (THS) and DOC (THDOC) | Precocious puberty; hirsutism; irregular menses; polycystic ovaries; high blood pressure (50%) | Hydrocortisone +/− Mineralocorticoid receptor antagonists | As above | As above; caution with spironolattone due to teratogenic effects |
| AroD | Various degrees of external genitalia atipicity (100%) | Gestational maternal virilization; abdominal signs of ovarian cysts | Serum: ↑ T; +/− ↑ LH and FSH | Delayed puberty; primary amenorrhea; osteopenia/osteoporosis; dislypidemic pattern | Increasing doses of estrogens; progestins added to induce menache | female | Unknown |
| PORD | Various degrees of external genitalia atipicity (~75%) | Gestational maternal virilization; skeletal malformations of the Antley–Bixler phenotype | Serum: unspecific mild ↑ of 17-OHP; ↑ Pregn, Prog, 17-OHP Urine: combined impairment of diagnostic ratios for CYP17A1D and CYP21A2D; ↑ of Pregn metabolites (PD) | Declining androgenization after birth; hypergonadotropic hypogonadism with delayed puberty; large ovarian cysts | +/− Hydrocortisone +/− Sex hormone | female (declining virilization after birth); concerns about prenatal androgenic imprinting | Unknown |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baronio, F.; Ortolano, R.; Menabò, S.; Cassio, A.; Baldazzi, L.; Di Natale, V.; Tonti, G.; Vestrucci, B.; Balsamo, A. 46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features. Int. J. Mol. Sci. 2019, 20, 4605. https://doi.org/10.3390/ijms20184605
Baronio F, Ortolano R, Menabò S, Cassio A, Baldazzi L, Di Natale V, Tonti G, Vestrucci B, Balsamo A. 46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features. International Journal of Molecular Sciences. 2019; 20(18):4605. https://doi.org/10.3390/ijms20184605
Chicago/Turabian StyleBaronio, Federico, Rita Ortolano, Soara Menabò, Alessandra Cassio, Lilia Baldazzi, Valeria Di Natale, Giacomo Tonti, Benedetta Vestrucci, and Antonio Balsamo. 2019. "46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features" International Journal of Molecular Sciences 20, no. 18: 4605. https://doi.org/10.3390/ijms20184605
APA StyleBaronio, F., Ortolano, R., Menabò, S., Cassio, A., Baldazzi, L., Di Natale, V., Tonti, G., Vestrucci, B., & Balsamo, A. (2019). 46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features. International Journal of Molecular Sciences, 20(18), 4605. https://doi.org/10.3390/ijms20184605