Heterogeneity in Malignant Pleural Mesothelioma


Abstract
1. Introduction
2. Inter-Tumor Heterogeneity
3. Spatial Intra-Tumor Heterogeneity
3.1. Spatial Genetic Heterogeneity
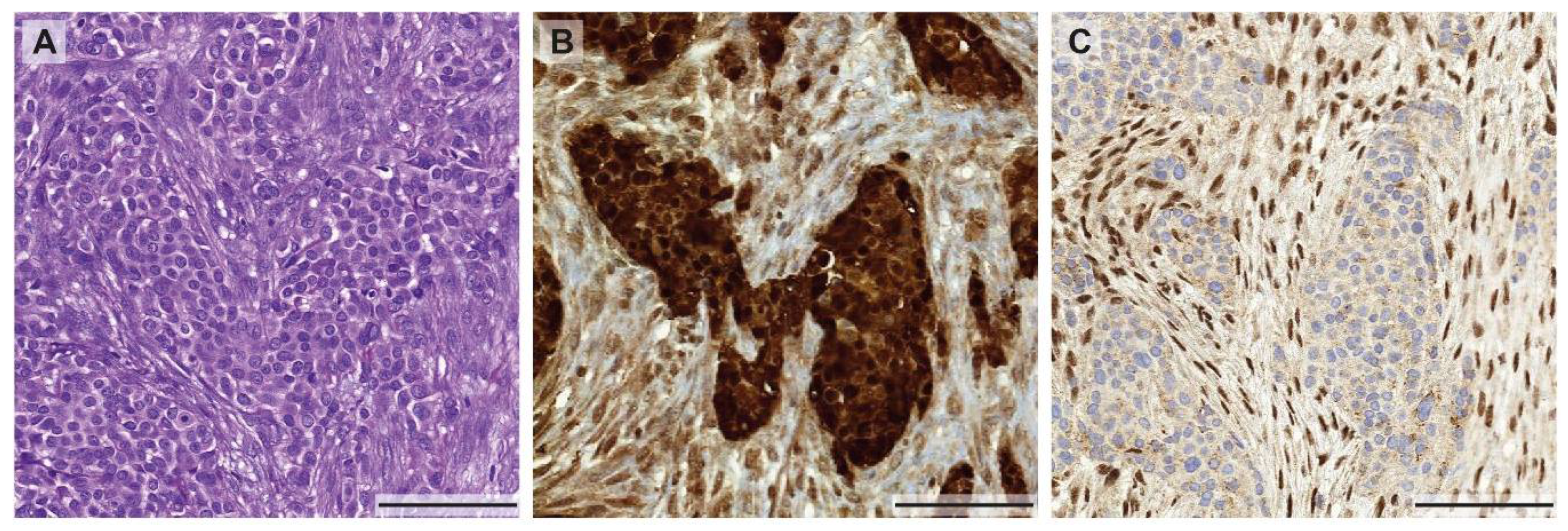
3.2. Spatial Phenotypic and Tumor Microenvironment Heterogeneity
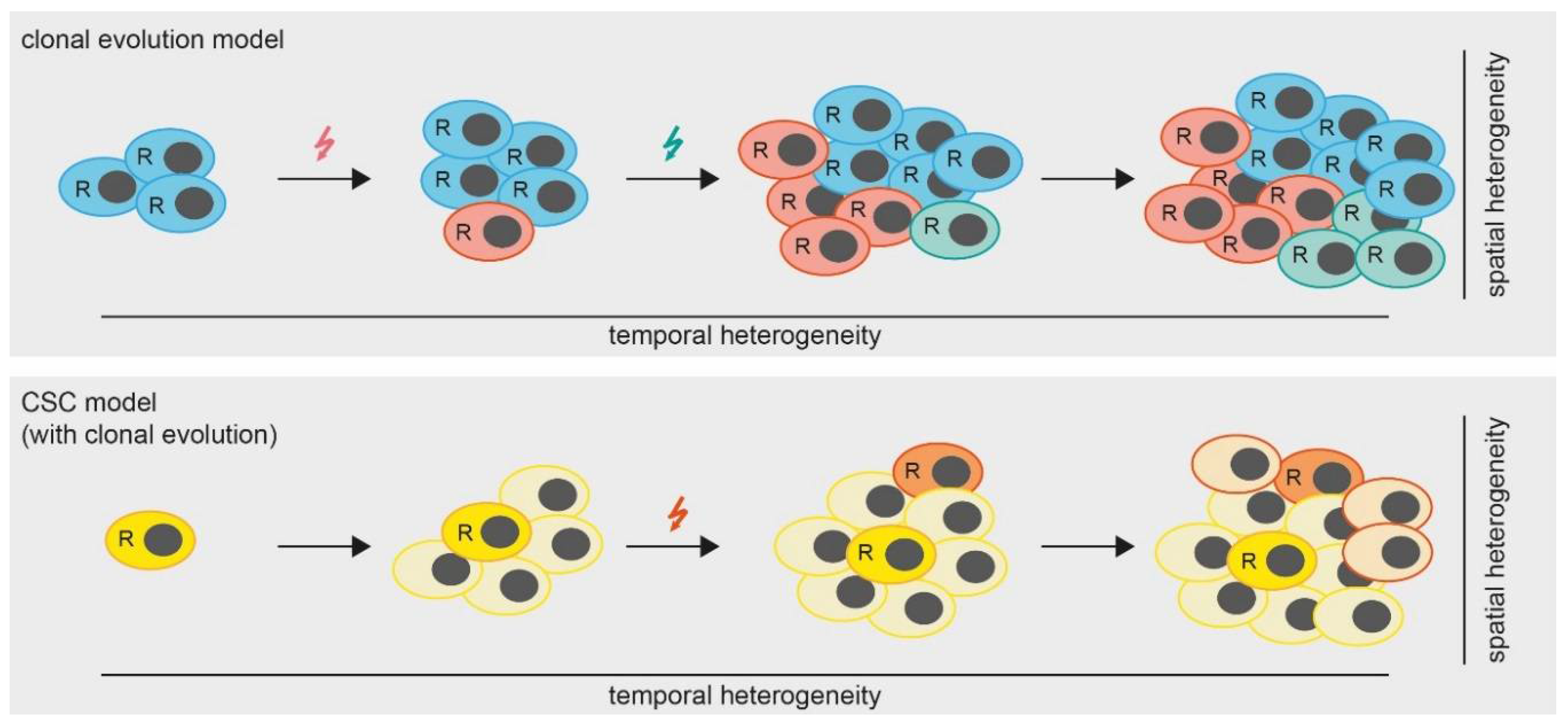
4. Temporal Intra-Tumor Heterogeneity
4.1. Chemotherapy and Tumor Heterogeneity
4.2. MPM Cancer Stem Cell
5. Implications for Therapy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jaurand, M.C.; Fleury-Feith, J. Pathogenesis of malignant pleural mesothelioma. Respirology 2005, 10, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Opitz, I. Management of malignant pleural mesothelioma-the european experience. J. Thorac. Dis. 2014, 6 (Suppl. 2), S238–S252. [Google Scholar] [PubMed]
- Opitz, I.; Friess, M.; Kestenholz, P.; Schneiter, D.; Frauenfelder, T.; Nguyen-Kim, T.D.; Seifert, B.; Hoda, M.A.; Klepetko, W.; Stahel, R.A.; et al. A new prognostic score supporting treatment allocation for multimodality therapy for malignant pleural mesothelioma: A review of 12 years’ experience. J. Thorac. Oncol. 2015, 10, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Buikhuisen, W.A.; Hiddinga, B.I.; Baas, P.; van Meerbeeck, J.P. Second line therapy in malignant pleural mesothelioma: A systematic review. Lung Cancer 2015, 89, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the mesothelioma avastin cisplatin pemetrexed study (maps): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Hiddinga, B.I.; Rolfo, C.; van Meerbeeck, J.P. Mesothelioma treatment: Are we on target? A review. J. Adv. Res. 2015, 6, 319–330. [Google Scholar] [CrossRef] [PubMed]
- COSMIC database, w.s.i. Available online: Https://cancer.Sanger.Ac.Uk/cosmic (accessed on 30 April 2018).
- Dahabreh, I.J.; Linardou, H.; Siannis, F.; Kosmidis, P.; Bafaloukos, D.; Murray, S. Somatic egfr mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.E.; Karrison, T.; Ananthanarayanan, V.; Gallan, A.J.; Adusumilli, P.S.; Alchami, F.S.; Attanoos, R.; Brcic, L.; Butnor, K.J.; Galateau-Salle, F.; et al. Nuclear grade and necrosis predict prognosis in malignant epithelioid pleural mesothelioma: A multi-institutional study. Mod. Pathol. 2018, 31, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Vrugt, B.; Felley-Bosco, E.; Simmler, S.; Storz, M.; Friess, M.; Meerang, M.; Soltermann, A.; Moch, H.; Stahel, R.; Weder, W.; et al. Sarcomatoid differentiation during progression of malignant pleural mesothelioma. Zent. Chir. Z. Allg. Vis. Thorac. Gefäßch. 2015, 140, FV21. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-exome sequencing reveals frequent genetic alterations in bap1, nf2, cdkn2a, and cul1 in malignant pleural mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase bap1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Borczuk, A.C.; Pei, J.; Taub, R.N.; Levy, B.; Nahum, O.; Chen, J.; Chen, K.; Testa, J.R. Genome-wide analysis of abdominal and pleural malignant mesothelioma with DNA arrays reveals both common and distinct regions of copy number alteration. Cancer Biol. Ther. 2016, 17, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Miller, J.; Lucito, R.; Tang, C.; Ivanova, A.V.; Pei, J.; Carbone, M.; Cruz, C.; Beck, A.; Webb, C.; et al. Genomic events associated with progression of pleural malignant mesothelioma. Int. J. Cancer 2009, 124, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Illei, P.B.; Rusch, V.W.; Zakowski, M.F.; Ladanyi, M. Homozygous deletion of cdkn2a and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin. Cancer Res. 2003, 9, 2108–2113. [Google Scholar] [PubMed]
- Lo Iacono, M.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Maki-Nevala, S.; Sarhadi, V.K.; Knuuttila, A.; Scheinin, I.; Ellonen, P.; Lagstrom, S.; Ronty, M.; Kettunen, E.; Husgafvel-Pursiainen, K.; Wolff, H.; et al. Driver gene and novel mutations in asbestos-exposed lung adenocarcinoma and malignant mesothelioma detected by exome sequencing. Lung 2016, 194, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ugurluer, G.; Chang, K.; Gamez, M.E.; Arnett, A.L.; Jayakrishnan, R.; Miller, R.C.; Sio, T.T. Genome-based mutational analysis by next generation sequencing in patients with malignant pleural and peritoneal mesothelioma. Anticancer Res. 2016, 36, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tomson, B.N.; Buys, T.P.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. Genomic landscape of malignant mesotheliomas. Mol. Cancer Ther. 2016, 15, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Shukuya, T.; Serizawa, M.; Watanabe, M.; Akamatsu, H.; Abe, M.; Imai, H.; Tokito, T.; Ono, A.; Taira, T.; Kenmotsu, H.; et al. Identification of actionable mutations in malignant pleural mesothelioma. Lung Cancer 2014, 86, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Destro, A.; Ceresoli, G.L.; Falleni, M.; Zucali, P.A.; Morenghi, E.; Bianchi, P.; Pellegrini, C.; Cordani, N.; Vaira, V.; Alloisio, M.; et al. Egfr overexpression in malignant pleural mesothelioma. An immunohistochemical and molecular study with clinico-pathological correlations. Lung Cancer 2006, 51, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Kasai, T.; Takeda, M.; Takano, M.; Morita, K.; Kadota, E.; Iizuka, N.; Maruyama, H.; Haratake, J.; Kojima, Y.; et al. A comparison of epidermal growth factor receptor expression in malignant peritoneal and pleural mesothelioma. Pathol. Int. 2012, 62, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.E. Predictive factors for epidermal growth factor receptor inhibitors—The bull’s-eye hits the arrow. Cancer Cell 2004, 5, 411–415. [Google Scholar] [CrossRef]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase ii study of erlotinib in patients with malignant pleural mesothelioma: A southwest oncology group study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E., 2nd; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Gefitinib in patients with malignant mesothelioma: A phase II study by the cancer and leukemia group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef] [PubMed]
- Paepe, A.D.; Vermaelen, K.Y.; Cornelissen, R.; Germonpre, P.R.; Janssens, A.; Lambrechts, M.; Bootsma, G.; Meerbeeck, J.P.V.; Surmont, V. Cetuximab plus platinum-based chemotherapy in patients with malignant pleural mesothelioma: A single arm phase ii trial. J. Clin. Oncol. 2017, 35, e20030. [Google Scholar]
- Sheffield, B.S.; Lorette, J.; Shen, Y.; Marra, M.A.; Churg, A. Immunohistochemistry for nf2, lats1/2, and yap/taz fails to separate benign from malignant mesothelial proliferations. Arch. Pathol. Lab. Med. 2016, 140, 391. [Google Scholar] [CrossRef] [PubMed]
- Hylebos, M.; Van Camp, G.; van Meerbeeck, J.P.; Op de Beeck, K. The genetic landscape of malignant pleural mesothelioma: Results from massively parallel sequencing. J. Thorac. Oncol. 2016, 11, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Varesano, S.; Leo, C.; Boccardo, S.; Salvi, S.; Truini, M.; Ferro, P.; Fedeli, F.; Canessa, P.A.; Dessanti, P.; Pistillo, M.P.; et al. Status of anaplastic lymphoma kinase (alk) in malignant mesothelioma. Anticancer Res. 2014, 34, 2589–2592. [Google Scholar] [PubMed]
- Mönch, D.; Bode-Erdmann, S.; Kalla, J.; Sträter, J.; Schwänen, C.; Falkenstern-Ge, R.; Klumpp, S.; Friedel, G.; Ott, G.; Kalla, C. A subgroup of pleural mesothelioma expresses alk protein and may be targetable by combined rapamycin and crizotinib therapy. Oncotarget 2018, 9, 20781. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, R.; Ma, P.C.; Seiwert, T.Y.; Jagadeeswaran, S.; Zumba, O.; Nallasura, V.; Ahmed, S.; Filiberti, R.; Paganuzzi, M.; Puntoni, R.; et al. Functional analysis of c-met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 2006, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Bois, M.C.; Mansfield, A.S.; Sukov, W.R.; Jenkins, S.M.; Moser, J.C.; Sattler, C.A.; Smith, C.Y.; Molina, J.R.; Peikert, T.; Roden, A.C. C-met expression and met amplification in malignant pleural mesothelioma. Ann. Diagn. Pathol. 2016, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Politi, L.; Borzellino, G. Second surgery for recurrence of malignant pleural mesothelioma after extrapleural pneumonectomy. Ann. Thorac. Surg. 2010, 89, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Baldini, E.H.; Richards, W.G.; Gill, R.R.; Goodman, B.M.; Winfrey, O.K.; Eisen, H.M.; Mak, R.H.; Chen, A.B.; Kozono, D.E.; Bueno, R.; et al. Updated patterns of failure after multimodality therapy for malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2015, 149, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Comertpay, S.; Pastorino, S.; Tanji, M.; Mezzapelle, R.; Strianese, O.; Napolitano, A.; Baumann, F.; Weigel, T.; Friedberg, J.; Sugarbaker, P.; et al. Evaluation of clonal origin of malignant mesothelioma. J. Transl. Med. 2014, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hiroshima, K.; Matsumoto, S.; Nabeshima, K.; Yusa, T.; Ozaki, D.; Fujino, M.; Yamakawa, H.; Nakatani, Y.; Tada, Y.; et al. Diagnostic usefulness of p16/cdkn2a fish in distinguishing between sarcomatoid mesothelioma and fibrous pleuritis. Am. J. Clin. Pathol. 2013, 139, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Park, J.H.; Inoue, H.; Husain, A.; Olugbile, S.; Zewde, M.; Nakamura, Y.; Vigneswaran, W.T. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology 2017, 6, e1278330. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57 (Suppl. 1), i90–i98. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.J.; Segard, T.; Morandeau, L.; Lee, Y.C.; Millward, M.J.; Segal, A.; Nowak, A.K. Characterization of hypoxia in malignant pleural mesothelioma with fmiso pet-ct. Lung Cancer 2015, 90, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Terra, S.B.S.P.; Mansfield, A.S.; Dong, H.; Peikert, T.; Roden, A.C. Temporal and spatial heterogeneity of programmed cell death 1-ligand 1 expression in malignant mesothelioma. Oncoimmunology 2017, 6, e1356146. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Cheng, Y.K.; Randles, A.; Itzkovitz, S.; Marusyk, A.; Ametller, E.; Gonzalez-Farre, X.; Munoz, M.; Russnes, H.G.; Helland, A.; et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014, 6, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Oehl, K.; Kresoja-Rakic, J.; Opitz, I.; Vrugt, B.; Weder, W.; Stahel, R.; Wild, P.; Felley-Bosco, E. Live-cell mesothelioma biobank to explore mechanisms of tumor progression. Front. Oncol. 2018, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.; Meerang, M.; Friess, M.; Soltermann, A.; Frischknecht, L.; Thies, S.; Felley-Bosco, E.; Tsao, M.S.; Allo, G.; de Perrot, M.; et al. Pi3k/mtor signaling in mesothelioma patients treated with induction chemotherapy followed by extrapleural pneumonectomy. J. Thorac. Oncol. 2014, 9, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Berard, K.; Friess, M.; Bitanihirwe, B.K.; Soltermann, A.; Vrugt, B.; Felley-Bosco, E.; Bueno, R.; Richards, W.G.; Seifert, B.; et al. Low merlin expression and high survivin labeling index are indicators for poor prognosis in patients with malignant pleural mesothelioma. Mol. Oncol. 2016, 10, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Sidi, R.; Pasello, G.; Opitz, I.; Soltermann, A.; Tutic, M.; Rehrauer, H.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Induction of senescence markers after neo-adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: An exploratory analysis. Eur. J. Cancer 2011, 47, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; D’Costa, S.; Yoon, B.I.; Brody, A.R.; Sills, R.C.; Kim, Y. Characterization of side population cells in human malignant mesothelioma cell lines. Lung Cancer 2010, 70, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Frei, C.; Opitz, I.; Soltermann, A.; Fischer, B.; Moura, U.; Rehrauer, H.; Weder, W.; Stahel, R.; Felley-Bosco, E. Pleural mesothelioma side populations have a precursor phenotype. Carcinogenesis 2011, 32, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Kim, N.Y.; Seo, Y.R.; Kim, Y. An integrated analysis of the genome-wide profiles of DNA methylation and mrna expression defining the side population of a human malignant mesothelioma cell line. J. Cancer 2016, 7, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts fak inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237ra268. [Google Scholar] [CrossRef] [PubMed]
- Lachaud, C.C.; Lopez-Beas, J.; Soria, B.; Hmadcha, A. Egf-induced adipose tissue mesothelial cells undergo functional vascular smooth muscle differentiation. Cell Death Dis. 2014, 5, e1304. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Asthana, S.; Chan, E.; Bandyopadhyay, S.; Martins, M.M.; Olivas, V.; Yan, J.J.; Pham, L.; Wang, M.M.; Bollag, G.; et al. Mapping the molecular determinants of braf oncogene dependence in human lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E748–E757. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Chen, C.; Hulbert, A.; Brock, M.V.; Yu, F. Non-blood circulating tumor DNA detection in cancer. Oncotarget 2017, 8, 69162–69173. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage ii colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Wu, X.; Tong, X.; Wang, X.; Chang, Z.; Mao, Y.; Chen, X.; Sun, J.; Wang, Z.; Hong, Z.; et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci. Rep. 2017, 7, 583. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Schmitt, M.; Loeb, L. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Drug | Study | Year of Completion | Status | # Patients | Results | Marker | Mutational Rate in MPM | Expression in MPM |
|---|---|---|---|---|---|---|---|---|---|
| mTor | Everolimus | NCT00770120 | 2014 | completed | 61 | primary endpoint not reached | NF2 (Merlin) | 17% (105/629) | 4% [31]–8% [32] negative |
| NCT01024946 | 2012 | completed | 11 | none published | |||||
| FAK | Defactinib | NCT01870609 | 2016 | terminated | 344 | lack of efficiency | |||
| ALK1 | PF-03446962 | NCT01486368 | 2015 | completed | 17 | primary endpoint not reached | ALK | 0% (1/343) | 0% [33]–20% [34] positive |
| EGFR | Erlotinib | NCT00039182 | 2007 | completed | 55 | primary endpoints not reached | EGFR | 1% (8/652) | 15% [25], 50% [26], 75% [28] high |
| Cetuximab | NCT00996567 | 2015 | completed | 22 | primary endpoint not reached | ||||
| c-Met | Tivantinib | NCT01861301 | 2015 | terminated | 18 | lack of efficiency | MET | 1% (3/448) | 17% [35]–40% [36] high |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oehl, K.; Vrugt, B.; Opitz, I.; Meerang, M. Heterogeneity in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 1603. https://doi.org/10.3390/ijms19061603
Oehl K, Vrugt B, Opitz I, Meerang M. Heterogeneity in Malignant Pleural Mesothelioma. International Journal of Molecular Sciences. 2018; 19(6):1603. https://doi.org/10.3390/ijms19061603
Chicago/Turabian StyleOehl, Kathrin, Bart Vrugt, Isabelle Opitz, and Mayura Meerang. 2018. "Heterogeneity in Malignant Pleural Mesothelioma" International Journal of Molecular Sciences 19, no. 6: 1603. https://doi.org/10.3390/ijms19061603
APA StyleOehl, K., Vrugt, B., Opitz, I., & Meerang, M. (2018). Heterogeneity in Malignant Pleural Mesothelioma. International Journal of Molecular Sciences, 19(6), 1603. https://doi.org/10.3390/ijms19061603