Perspectives of RAS and RHEB GTPase Signaling Pathways in Regenerating Brain Neurons

 , , , and
, , , and 
Abstract
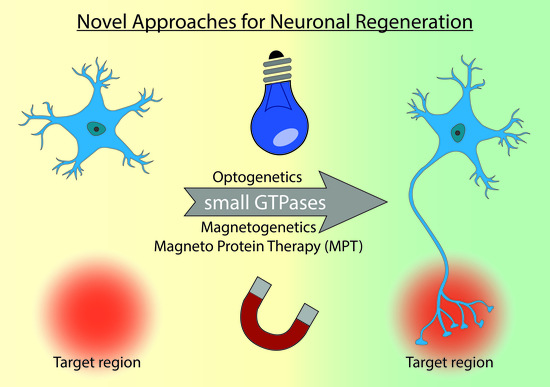
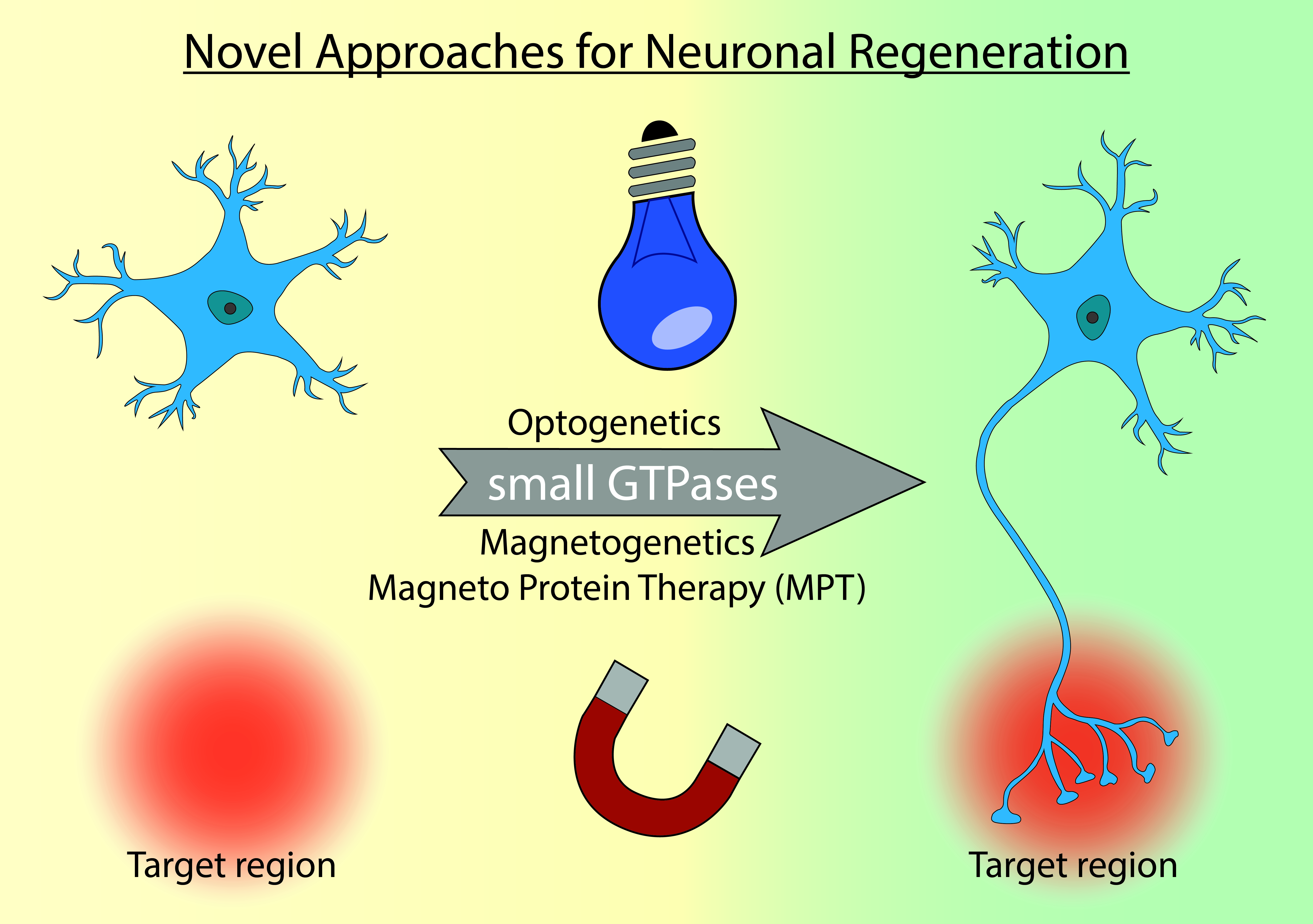{kind=link}
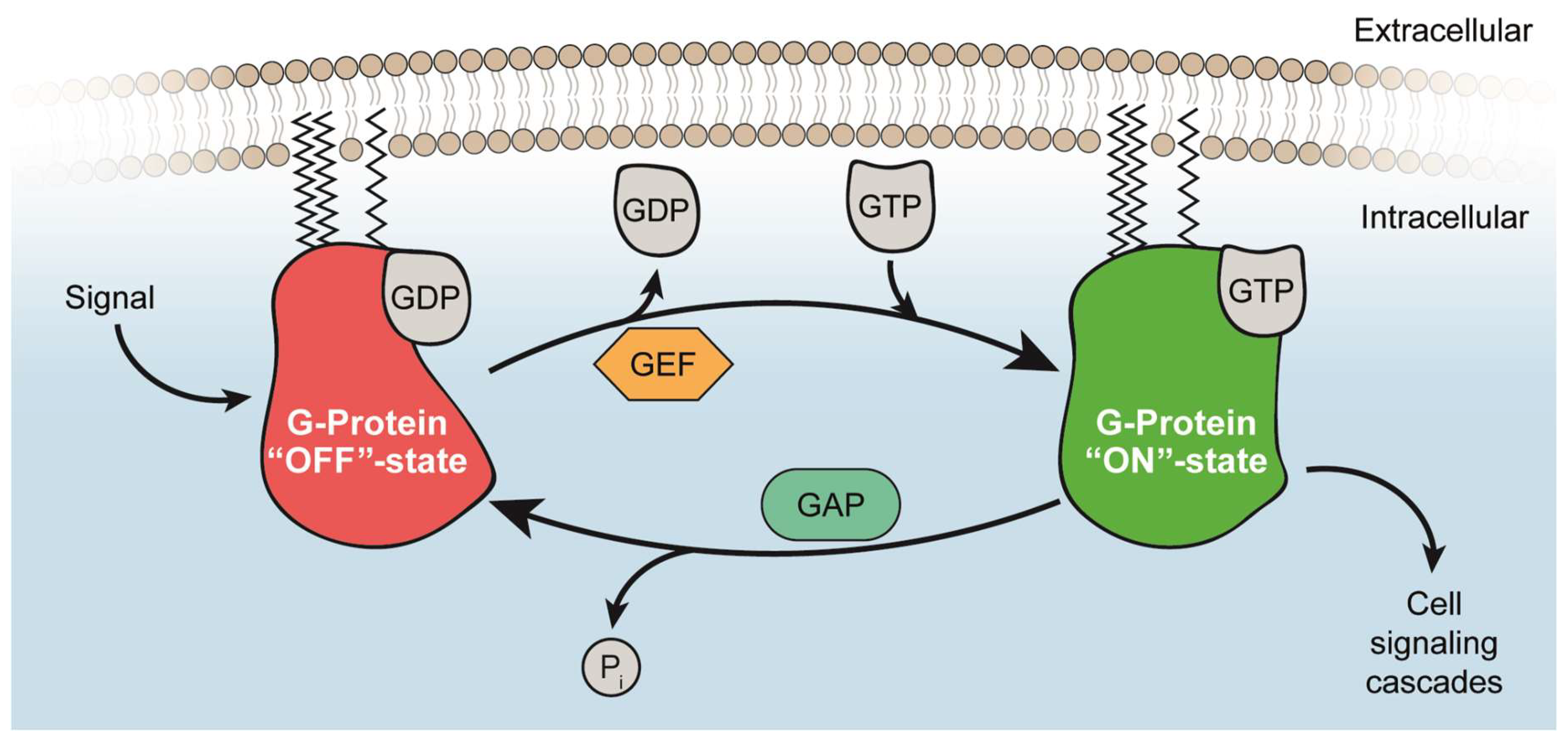{kind=link}
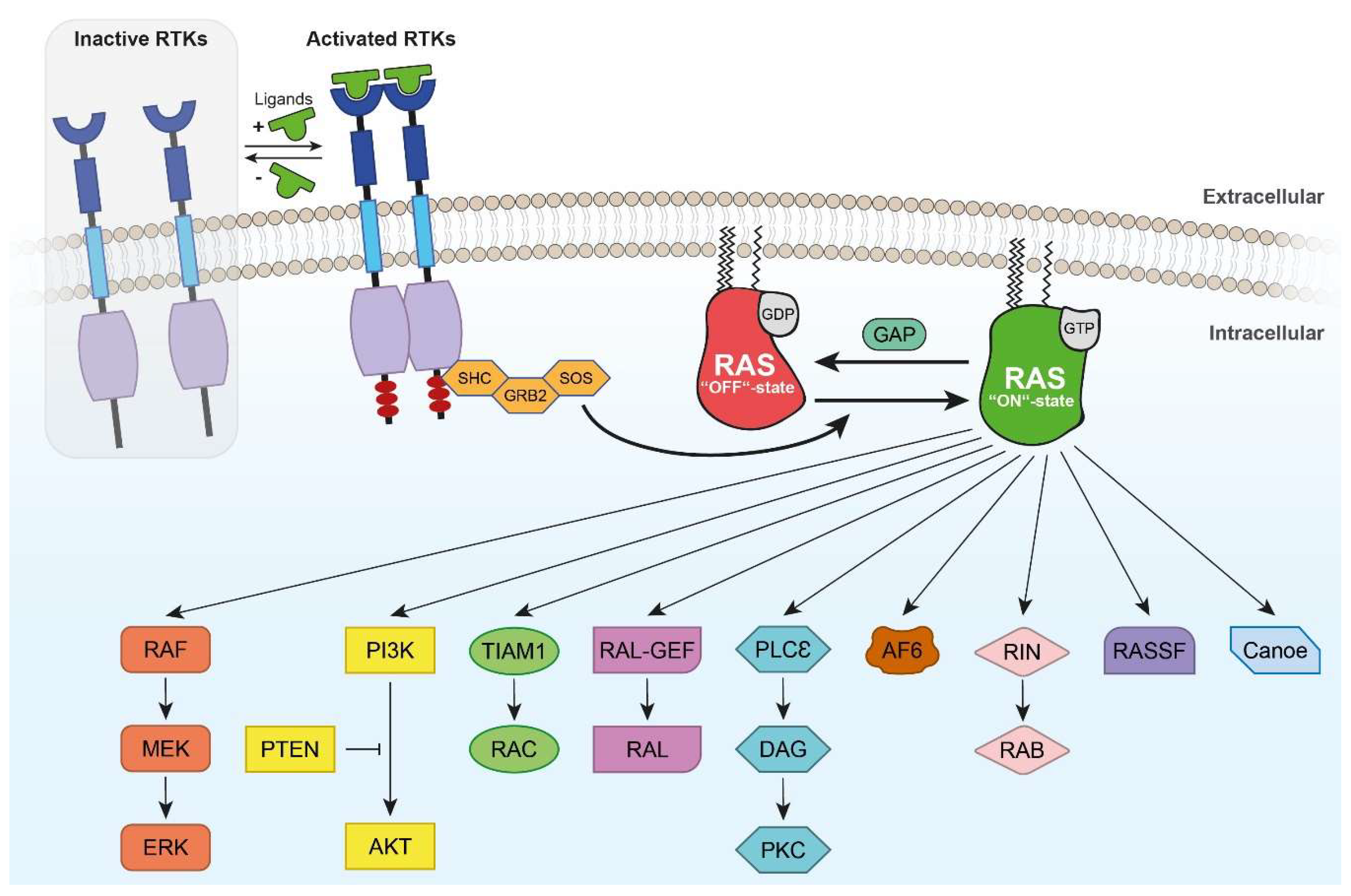{kind=link}
{kind=link}
{kind=link}
1. Introduction
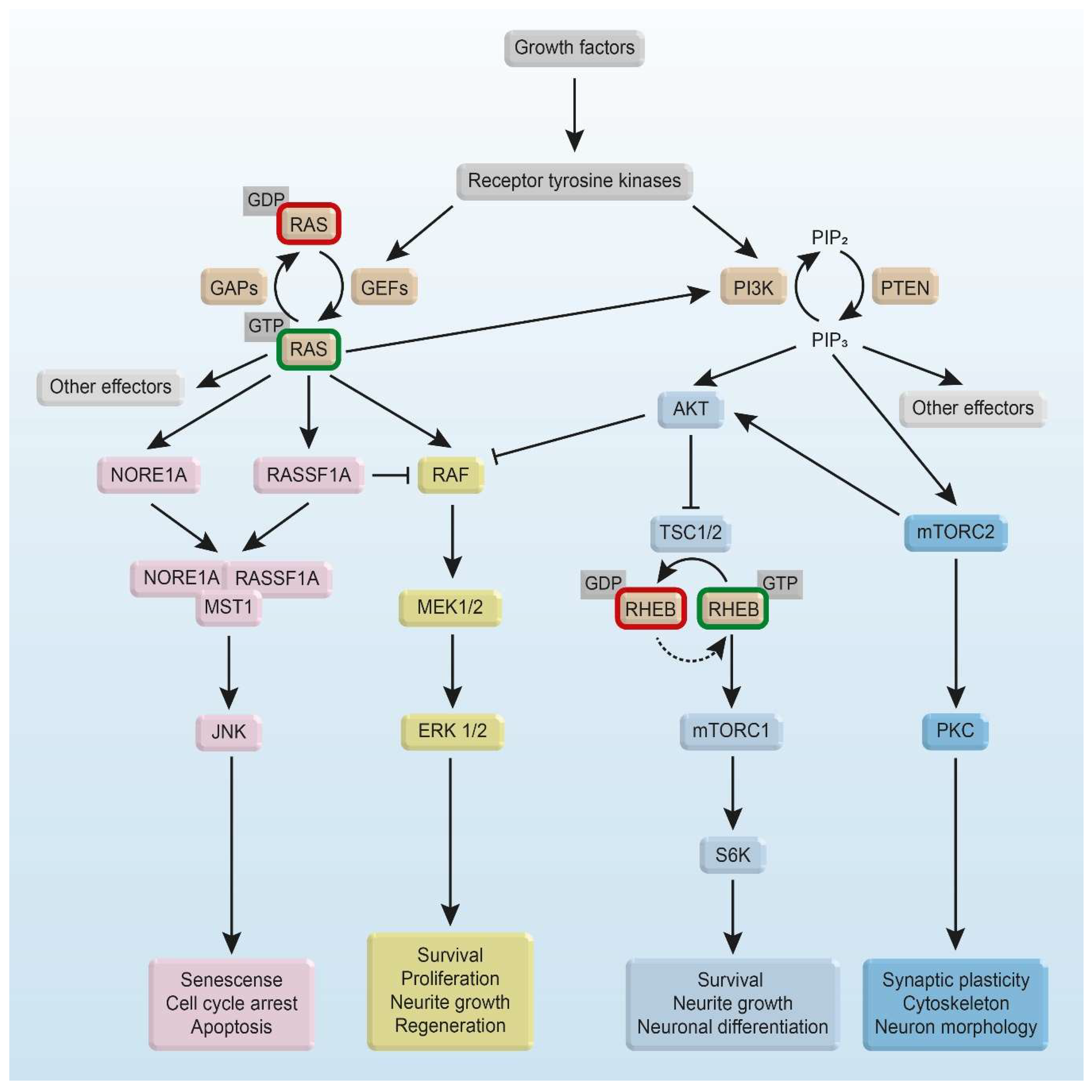
2. Structural Basis of the RAS and RHEB GTPase Switch
2.1. Guanine Nucleotide-Binding
2.2. Activation of RAS and RHEB by GEFs
2.3. Inactivation of RAS and RHEB by GAPs
2.4. Membrane Anchoring of RAS and RHEB
3. Intracellular RAS and RHEB Effector Mechanisms
3.1. RAS/RAF Effector Mechanisms
3.2. RHEB Effector Mechanisms
3.3. Other RAS Effectors
4. RAS and RHEB Signaling in Survival, Apoptosis, and Neurodegenerative Diseases
4.1. RAS Signaling for Survival
4.2. RAS Signaling for Apoptosis
4.3. Subcellular Localization-Dependent Signaling of RHEB
4.4. RHEB-Mediated Enhancement of Apoptosis
4.5. RHEB in Neurodegenerative Disease
4.6. RAS Signaling and Neurite Growth
4.7. TRK Receptor-Mediated RAS/RAF/ERK Pathway
4.8. Other Intracellular Mechanisms Signaling for Regeneration
5. Optogenetics
5.1. Light-Directed Protein-Protein Interaction Devices
5.2. Light-Mediated Activation of RAS/RAF/ERK
5.3. Light-Mediated Activation of PI3-Kinase
5.4. Combined Activation of RAS- and PI3K-Pathways
5.5. Activating the RHO Family of Proteins
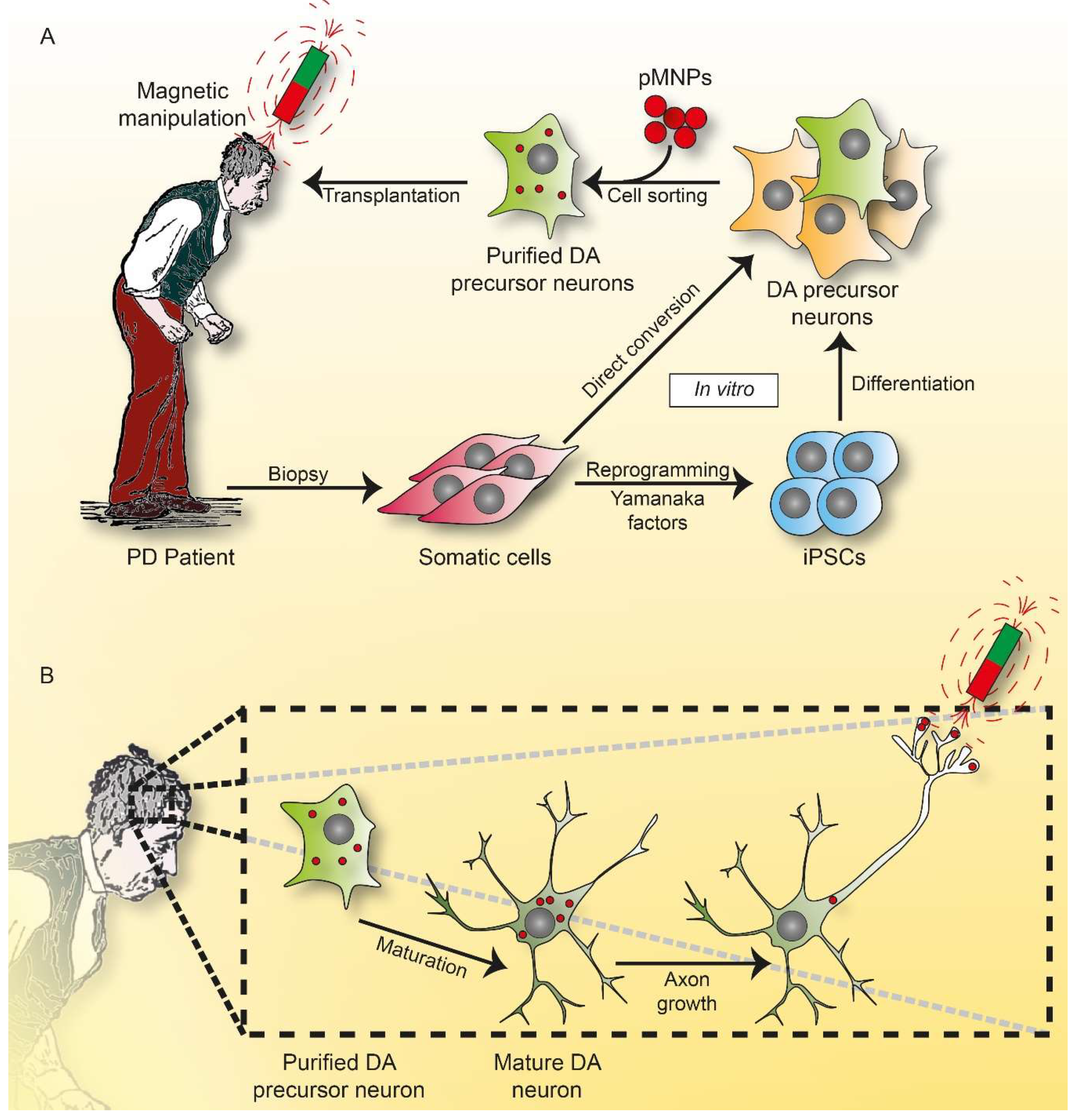
6. Magnetogenetics
6.1. Functionalized Magnetic Nanoparticles in Regenerative Medicine
6.2. Magneto-Thermal Approaches
6.3. Magnetic Control of Receptor Clustering
6.4. Magnetic Activation of Intracellular Signaling
7. Outlook and Perspectives: The Dawn of Magneto Protein Therapy in Brain?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4E-BP1 | Eukaryotic translation initiation factor 4E (EIF4E)-binding protein |
| 6-OHDA | 6-hydroxydopamine |
| AD | Alzheimer disease |
| AF6 | Afadin 6 |
| AKT | Protein kinase B |
| AP-1 | Activator protein 1 |
| APC | Adenomatous-polyposis-coli |
| APP | Amyloid precursor protein |
| ARF | ADP ribosylation factor |
| ASK-1 | Apoptosis signal-regulating kinase-1 |
| ATG13 | Autophagy-related gene 13 |
| Aβ | Amyloid beta |
| BAD | Bcl-2-associated death |
| BAG1 | BCL-2-associated athanogene-1 |
| BAX | Bcl-2-associated X protein |
| BCL-2 | B-cell lymphoma 2 |
| BCL-xL | B-cell lymphoma-extra large |
| BDNF | Brain-derived neurotrophic factor |
| BIM | Bcl-2-like protein 11 |
| BNIP3 | BCL-2/adenovirus E1B 19 kDa interacting protein 3 |
| CA | Conserved area |
| CAAX | C = Cys, A = aliphatic and X = any amino acid |
| CDC | Cell division cycle |
| CDC25 | Cell division cycle 25 |
| CGN | Cerebellar granule neuron |
| CIB1 | Cryptochrome-interacting basic helix-loop-helix 1 |
| CIBN | Cryptochrome-interacting basic-helix-loop-helix, N-terminal domain |
| CK2 | Casein kinase 2 |
| CNK1 | Connector enhancer of kinase suppressor of RAS 1 |
| CNS | Central nervous system |
| CREB | cAMP response element-binding protein |
| CRMP-2 | Collapsin response mediator protein-2 |
| CRY2 | Cryptochrome 2 |
| CSPG | Chondroitin sulfate proteoglycan |
| DCC | Deleted in colorectal cancer |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| DH | Dbl-homology domain |
| DLK | Dual leucine zipper-bearing kinase |
| DR4 | Death receptor 4 |
| DREZ | Dorsal root entry zone |
| DRG | Dorsal root ganglion |
| EGF | Epidermal growth factor |
| EIF4E | Eukaryotic translation initiation factor 4E |
| ER | Endoplasmatic reticulum |
| ERK | Extracellular-signal-regulated-kinase |
| ETS | E26 transformation-specific |
| ETS | E26 transformation-specific |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FKF1 | Flavin-binding, kelch repeat, f box 1 |
| FKHR | Forkhead |
| FMN | Flavin mononucleotide |
| GAP | GTPase-activating protein |
| GDP | Guanosine diphosphate |
| GEF | Guanine nucleotide exchange factor |
| GNBP | Guanine nucleotide binding protein |
| GTP | Guanosine triphosphate |
| GTPases | Guanosine triphosphatases |
| HD | Huntington’s disease |
| hMSC | Human mesenchymal stem cells |
| H-RAS | Harvey-RAS |
| HTT | Huntingtin |
| HVR | Hypervariable region |
| ICMT | Isoprenylcysteine carboxylmethyl transferase |
| iLID | Improved light-inducible dimer |
| ILK | Integrin-linked kinase |
| ITSN | Intersectin |
| KLF | Krüppel-like family of transcription factors |
| KO | Knock out |
| K-RAS | Kirsten-RAS |
| KSR | Kinase suppressor of RAS |
| LOV | Light-oxygen-voltage |
| MAG | Myelin-associated glycoprotein |
| MAPK | Mitogen activated protein kinase |
| MEK | MAPK/ERK kinase |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| MNPs | Magnetic nanoparticle |
| MPT | Magneto Protein Therapy |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridin |
| mSIN1 | Mammalian stress-activated map kinase-interacting protein 1 |
| MST | Mammalian sterile |
| mTOR | Mammalian target of rapamycin |
| MUPP1 | Multi-PDZ domain protein 1 |
| NF-κB | Nuclear factor κB |
| NGF | Nerve growth factor |
| NIR | Near-infrared |
| NMDA | N-Methyl-d-aspartic acid |
| NOGO | Neurite outgrowth inhibitor |
| NORE1 | Novel RAS effector 1 |
| NOTCH | Neurogenic locus notch homolog protein |
| N-RAS | Neuroblastoma-RAS |
| NURR1 | Nuclear receptor-related 1 protein |
| PAK | p21-activated kinase |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| PC12 | Pheochromocytoma cells |
| PCB | Phycocyanobilin |
| PDK1 | Phosphoinositide-dependent kinase1 |
| PDZ | PSD-95 Dlg1 ZO-1 |
| PERK | Protein kinase-like ER kinase |
| PH | Pleckstrin-homology domain |
| PHYB | Phytochrome B |
| PI3K | Phosphoinositide-3 kinase |
| PIF | Phytochromes interacting factor |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PNS | Peripheral nervous system |
| PRAS40 | Proline-rich AKT substrate 40 kDa |
| PTEN | Phosphatase and tensin homolog |
| RA | RAS association |
| RAB | RAS-like proteins in brain |
| RAC | RAS-related C3 botulinum toxin substrate |
| RAF | Rapidly accelerated fibrosarcoma |
| RAG | Recombination-activating gene |
| RAL | RAS-like |
| RALGDS | RAL guanine nucleotide dissociation stimulator |
| RAN | RAS-like nuclear |
| RAP1 | RAS-related protein 1 |
| RAPTOR | Regulatory-associated protein of mammalian target of rapamycin |
| RAS | Rat sarcoma |
| RASSF | RAS association family |
| RBD | RAS-binding domain |
| RCE1 | RAS-converting enzyme |
| RGC | Retinal ganglion cell |
| RGL | RAL guanine nucleotide dissociation stimulator-like |
| RHEB | RAS homolog protein enriched in Brain |
| RHO | RAS homologs |
| RHOA | Ras homolog gene family, member A |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| RIN | RAS and RAB interactor |
| RNA | Ribonucleic acid |
| ROCK | RHO-associated protein kinase |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinases |
| S6K1 | p70 S6 Kinase |
| SARAH | Salvador-RASSF-Hippo |
| SFK | SRC family kinase |
| SNAIL | Zinc finger protein SNAI1 |
| SPIONs | Superparamagnetic iron oxide nanoparticles |
| TCTP | Translationally controlled tumor protein |
| TH | Tyrosine hydroxylase |
| TIAM1 | T-lymphoma invasion and metastasis-inducing protein 1 |
| TNFα | Tumor necrosis factor α |
| TRK | Tyrosine receptor kinase |
| TRKB | Tropomyosin receptor kinase B |
| TRP | Transient receptor potential |
| TRPV4 | Transient receptor potential cation channel subfamily V member 4 |
| TSC | Tuberous sclerosis complex |
| TSC1 | Hamartin |
| TSC2 | Tuberin |
| U2OS | Human bone osteosarcoma epithelial cell line |
| ULK1 | Uncoordinated 51-like kinase |
| VPS21 | Vacuolar protein sorting-associated protein 21 |
| WAVE | WASP-family verprolin homologous protein |
| WNT | int/Wingless |
| ZO-1 | Zonula occludens-1 |
References
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.J. An unidentified virus which causes the rapid production of tumours in mice. Nature 1964, 204, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, W.H.; Mayer, L.A. Morphologic responses to a murine erythroblastosis virus. J. Natl. Cancer Inst. 1967, 39, 311–335. [Google Scholar] [PubMed]
- Malumbres, M.; Barbacid, M. Ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Urano, J.; Ellis, C.; Clark, G.J.; Tamanoi, F. Characterization of RHEB functions using yeast and mammalian systems. Methods Enzymol. 2001, 333, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Heard, J.J.; Fong, V.; Bathaie, S.Z.; Tamanoi, F. Recent progress in the study of the RHEB family gtpases. Cell Signal. 2014, 26, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Araki, Y.; Kontani, K.; Nishina, H.; Katada, T. Novel role of the small gtpase RHEB: Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 2005, 137, 423–430. [Google Scholar] [CrossRef]
- Yamagata, K.; Sanders, L.K.; Kaufmann, W.E.; Yee, W.; Barnes, C.A.; Nathans, D.; Worley, P.F. RHEB, a growth factor- and synaptic activity-regulated gene, encodes a novel ras-related protein. J. Biol. Chem. 1994, 269, 16333–16339. [Google Scholar]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef]
- Chen, M.; Peters, A.; Huang, T.; Nan, X. Ras dimer formation as a new signaling mechanism and potential cancer therapeutic target. Mini Rev. Med. Chem. 2016, 16, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Inouye, K.; Mizutani, S.; Koide, H.; Kaziro, Y. Formation of the ras dimer is essential for raf-1 activation. J. Biol. Chem. 2000, 275, 3737–3740. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of ras. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Grosseruschkamp, F.; Stephan, S.; Cui, Q.; Kotting, C.; Xia, F.; Gerwert, K. Specific substates of ras to interact with gaps and effectors: Revealed by theoretical simulations and ftir experiments. J. Phys. Chem. Lett. 2018, 9, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Hall, M.N. MTORC1: Turning off is just as important as turning on. Cell 2014, 156, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Ehrkamp, A.; Herrmann, C.; Stoll, R.; Heumann, R. Ras and RHEB signaling in survival and cell death. Cancers 2013, 5, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.; Hobbs, G.A.; Aghajanian, A.; Campbell, S.L. Redox regulation of ras and rho gtpases: Mechanism and function. Antioxid. Redox Signal. 2013, 18, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Wolfman, A.; Macara, I.G. A cytosolic protein catalyzes the release of gdp from p21ras. Science 1990, 248, 67–69. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. Gefs and gaps: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Rehmann, H.; Bruning, M.; Berghaus, C.; Schwarten, M.; Kohler, K.; Stocker, H.; Stoll, R.; Zwartkruis, F.J.; Wittinghofer, A. Biochemical characterisation of tctp questions its function as a guanine nucleotide exchange factor for RHEB. FEBS Lett. 2008, 582, 3005–3010. [Google Scholar] [CrossRef]
- Schopel, M.; Potheraveedu, V.N.; Al-Harthy, T.; Abdel-Jalil, R.; Heumann, R.; Stoll, R. The small gtpases ras and RHEB studied by multidimensional nmr spectroscopy: Structure and function. Biol. Chem. 2017, 398, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Parton, R.G. Ras plasma membrane signalling platforms. Biochem. J. 2005, 389, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Inoki, K.; Guan, K.-L. Biochemical and functional characterizations of small gtpase RHEB and TSC2 gap activity. Mol. Cell. Biol. 2004, 24, 7965–7975. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Otto, J.C.; Bergo, M.O.; Young, S.G.; Casey, P.J. The c-terminal polylysine region and methylation of k-ras are critical for the interaction between k-ras and microtubules. J. Biol. Chem. 2000, 275, 41251–41257. [Google Scholar] [CrossRef] [PubMed]
- Thissen, J.A.; Gross, J.M.; Subramanian, K.; Meyer, T.; Casey, P.J. Prenylation-dependent association of ki-ras with microtubules. Evidence for a role in subcellular trafficking. J. Biol. Chem. 1997, 272, 30362–30370. [Google Scholar] [CrossRef] [PubMed]
- Schmick, M.; Vartak, N.; Papke, B.; Kovacevic, M.; Truxius, D.C.; Rossmannek, L.; Bastiaens, P.I.H. Kras localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Scotto-Lavino, E.; Sobczyk, A.; Bar-Sagi, D. Differential modification of ras proteins by ubiquitination. Mol. Cell 2006, 21, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of RHEB to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun. 2006, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. Pras40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nakagawa, M.; Young, S.G.; Yamanaka, S. Differential membrane localization of eras and RHEB, two ras-related proteins involved in the phosphatidylinositol 3-kinase/mtor pathway. J. Biol. Chem. 2005, 280, 32768–32774. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Therrien, M. Regulation of raf protein kinases in erk signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Baljuls, A.; Kholodenko, B.N.; Kolch, W. It takes two to tango—Signalling by dimeric raf kinases. Mol. bioSyst. 2013, 9, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hancock, J.F. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 2015, 1853, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.G.; Satoh, T.; Liao, Y.; Song, C.; Gao, X.; Kariya, K.; Hu, C.D.; Kataoka, T. Role of the cdc25 homology domain of phospholipase cepsilon in amplification of rap1-dependent signaling. J. Biol. Chem. 2001, 276, 30301–30307. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mtor. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Peng, H.; Kasada, A.; Ueno, M.; Hoshii, T.; Tadokoro, Y.; Nomura, N.; Ito, C.; Takase, Y.; Vu, H.T.; Kobayashi, M.; et al. Distinct roles of RHEB and raptor in activating mtor complex 1 for the self-renewal of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 2018, 495, 1129–1135. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian tor complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Gβl, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mtor. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. Deptor is an mtor inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.-I.; Natsume, T.; Mizushima, N. Tti1 and tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-I.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (tor), mediates tor action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Mtor interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jenö, P.; Arrieumerlou, C.; Hall, M.N. Pras40 and prr5-like protein are new mtor interactors that regulate apoptosis. PLoS ONE 2007, 2, e1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. Pras40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. RHEB binds and regulates the mtor kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Dos, D.S.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mtor, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. Msin1 is necessary for akt/pkb phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of protor as a novel rictor-binding component of mtor complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. Raft1 phosphorylation of the translational regulators p70 s6 kinase and 4e-bp1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-fkbp specifically blocks growth-dependent activation of and signaling by the 70 kd s6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Kuo, C.J.; Chung, J.; Fiorentino, D.F.; Flanagan, W.M.; Blenis, J.; Crabtree, G.R. Rapamycin selectively inhibits interleukin-2 activation of p70 s6 kinase. Nature 1992, 358, 70. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mtor-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ulk1–atg13–fip200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bickle, M.; Beck, T.; Hall, M.N. The yeast phosphatidylinositol kinase homolog tor2 activates rho1 and rho2 via the exchange factor rom2. Cell 1997, 88, 531–542. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-oh kinase as a direct target of ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Feinstein, E. Ral-gtpases: Good chances for a long-lasting fame. Oncogene 2005, 24, 326. [Google Scholar] [CrossRef]
- Teodoro, R.O.; Pekkurnaz, G.; Nasser, A.; Higashi-Kovtun, M.E.; Balakireva, M.; McLachlan, I.G.; Camonis, J.; Schwarz, T.L. Ral mediates activity-dependent growth of postsynaptic membranes via recruitment of the exocyst. EMBO J. 2013, 32, 2039–2055. [Google Scholar] [CrossRef]
- Rosse, C.; Hatzoglou, A.; Parrini, M.C.; White, M.A.; Chavrier, P.; Camonis, J. Ralb mobilizes the exocyst to drive cell migration. Mol. Cell. Biol. 2006, 26, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Camonis, J.H.; White, M.A. Ral gtpases: Corrupting the exocyst in cancer cells. Trends Cell Biol. 2005, 15, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Feig, L.A. Ral-gtpases: Approaching their 15 minutes of fame. Trends Cell Biol. 2003, 13, 419–425. [Google Scholar] [CrossRef]
- Martin, T.D.; Chen, X.W.; Kaplan, R.E.; Saltiel, A.R.; Walker, C.L.; Reiner, D.J.; Der, C.J. Ral and RHEB gtpase activating proteins integrate mtor and gtpase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 2014, 53, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Lalli, G.; Hall, A. Ral gtpases regulate neurite branching through gap-43 and the exocyst complex. J. Cell Biol. 2005, 171, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates ras activation of rac by a pi(3)k-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, J.; Miyamoto, Y.; Tanoue, A.; Shooter, E.M.; Chan, J.R. Ras activation of a rac1 exchange factor, tiam1, mediates neurotrophin-3-induced schwann cell migration. Proc. Natl. Acad. Sci. USA 2005, 102, 14889–14894. [Google Scholar] [CrossRef]
- Arthur, W.T.; Quilliam, L.A.; Cooper, J.A. Rap1 promotes cell spreading by localizing rac guanine nucleotide exchange factors. J. Cell Biol. 2004, 167, 111–122. [Google Scholar] [CrossRef]
- Demarco, R.S.; Struckhoff, E.C.; Lundquist, E.A. The rac gtp exchange factor tiam-1 acts with cdc-42 and the guidance receptor unc-40/dcc in neuronal protrusion and axon guidance. PLoS Genet. 2012, 8, e1002665. [Google Scholar] [CrossRef]
- Ehler, E.; van Leeuwen, F.; Collard, J.G.; Salinas, P.C. Expression of tiam-1 in the developing brain suggests a role for the tiam-1-rac signaling pathway in cell migration and neurite outgrowth. Mol. Cell. Neurosci. 1997, 9, 1–12. [Google Scholar] [CrossRef]
- Zheng, C.; Diaz-Cuadros, M.; Chalfie, M. Gefs and rac gtpases control directional specificity of neurite extension along the anterior-posterior axis. Proc. Natl. Acad. Sci. USA 2016, 113, 6973–6978. [Google Scholar] [CrossRef] [PubMed]
- Tall, G.G.; Barbieri, M.A.; Stahl, P.D.; Horazdovsky, B.F. Ras-activated endocytosis is mediated by the rab5 guanine nucleotide exchange activity of rin1. Dev. Cell 2001, 1, 73–82. [Google Scholar] [CrossRef]
- Doi, M.; Minematsu, H.; Kubota, Y.; Nishiwaki, K.; Miyamoto, M. The novel rac effector rin-1 regulates neuronal cell migration and axon pathfinding in C. elegans. Development 2013, 140, 3435–3444. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Satoh, T.; Edamatsu, H.; Wu, D.; Tadano, M.; Gao, X.; Kataoka, T. Differential roles of ras and rap1 in growth factor-dependent activation of phospholipase C epsilon. Oncogene 2002, 21, 8105–8113. [Google Scholar] [CrossRef]
- Zhadanov, A.B.; Provance, D.W., Jr.; Speer, C.A.; Coffin, J.D.; Goss, D.; Blixt, J.A.; Reichert, C.M.; Mercer, J.A. Absence of the tight junctional protein af-6 disrupts epithelial cell-cell junctions and cell polarity during mouse development. Curr. Biol. 1999, 9, 880–888. [Google Scholar] [CrossRef]
- Zhang, Z.; Rehmann, H.; Price, L.S.; Riedl, J.; Bos, J.L. Af6 negatively regulates rap1-induced cell adhesion. J. Biol. Chem. 2005, 280, 33200–33205. [Google Scholar] [CrossRef] [PubMed]
- Toker, A. Signaling through protein kinase C. Front. Biosci. 1998, 3, D1134–D1147. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.S. Ras target proteins in eukaryotic cells. FASEB J. 1995, 9, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, M.; Harada, N.; Kuroda, S.; Yamamoto, T.; Nakafuku, M.; Iwamatsu, A.; Yamamoto, D.; Prasad, R.; Croce, C.; Canaani, E.; et al. Identification of af-6 and canoe as putative targets for ras. J. Biol. Chem. 1996, 271, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lynn, B.D.; Nagy, J.I. The effector and scaffolding proteins af6 and mupp1 interact with connexin36 and localize at gap junctions that form electrical synapses in rodent brain. Eur. J. Neurosci. 2012, 35, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Carmena, A.; Speicher, S.; Baylies, M. The pdz protein canoe/af-6 links ras-mapk, notch and wingless/wnt signaling pathways by directly interacting with ras, notch and dishevelled. PLoS ONE 2006, 1, e66. [Google Scholar] [CrossRef]
- Boettner, B.; Harjes, P.; Ishimaru, S.; Heke, M.; Fan, H.Q.; Qin, Y.; Van Aelst, L.; Gaul, U. The af-6 homolog canoe acts as a rap1 effector during dorsal closure of the drosophila embryo. Genetics 2003, 165, 159–169. [Google Scholar] [PubMed]
- Speicher, S.; Fischer, A.; Knoblich, J.; Carmena, A. The pdz protein canoe regulates the asymmetric division of drosophila neuroblasts and muscle progenitors. Curr. Biol. 2008, 18, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Schmidt, M.L.; Mezzanote, J.; Barnoud, T.; Clark, G.J. Ras signaling through rassf proteins. Semin. Cell Dev. Biol. 2016, 58, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J. Ras and downstream raf-mek and PI3K-akt signaling in neuronal development, function and dysfunction. Biol. Chem. 2016, 397, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Potheraveedu, V.N.; Schöpel, M.; Stoll, R.; Heumann, R. RHEB in neuronal degeneration, regeneration, and connectivity. Biol. Chem. 2017, 398, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, M.; Gozdz, A.; Swiech, L.J.; Jaworski, J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J. Biol. Chem. 2012, 287, 30240–30256. [Google Scholar] [CrossRef] [PubMed]
- Fritsche-Guenther, R.; Witzel, F.; Kempa, S.; Brummer, T.; Sers, C.; Blüthgen, N. Effects of raf inhibitors on PI3K/akt signalling depend on mutational status of the ras/raf signalling axis. Oncotarget 2016, 7, 7960–7969. [Google Scholar] [CrossRef] [PubMed]
- Borasio, G.D.; John, J.; Wittinghofer, A.; Barde, Y.A.; Sendtner, M.; Heumann, R. Ras p21 protein promotes survival and fiber outgrowth of cultured embryonic neurons. Neuron 1989, 2, 1087–1096. [Google Scholar] [CrossRef]
- Anderson, C.N.; Tolkovsky, A.M. A role for mapk/erk in sympathetic neuron survival: Protection against a p53-dependent, jnk-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 1999, 19, 664–673. [Google Scholar] [CrossRef]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the ras-mapk signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Xifro, X.; Minano-Molina, A.J.; Saura, C.A.; Rodriguez-Alvarez, J. Ras protein activation is a key event in activity-dependent survival of cerebellar granule neurons. J. Biol. Chem. 2014, 289, 8462–8472. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.A.; Kupzig, S.; Bouyoucef, D.; Davies, L.C.; Tsuboi, T.; Bivona, T.G.; Cozier, G.E.; Lockyer, P.J.; Buckler, A.; Rutter, G.A.; et al. Identification of a ras gtpase-activating protein regulated by receptor-mediated Ca2+ oscillations. EMBO J. 2004, 23, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.A.; Cullen, P.J.; Taylor, J.A.; Lockyer, P.J. Control of ras cycling by Ca2+. FEBS Lett. 2003, 546, 6–10. [Google Scholar] [CrossRef]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhöhl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic activation of ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef]
- Gärtner, U.; Alpár, A.; Seeger, G.; Heumann, R.; Arendt, T. Enhanced ras activity in pyramidal neurons induces cellular hypertrophy and changes in afferent and intrinsic connectivity in synras mice. Int. J. Dev. Neurosci. 2004, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Felderhoff-Mueser, U.; Bittigau, P.; Sifringer, M.; Jarosz, B.; Korobowicz, E.; Mahler, L.; Piening, T.; Moysich, A.; Grune, T.; Thor, F.; et al. Oxygen causes cell death in the developing brain. Neurobiol. Dis. 2004, 17, 273–282. [Google Scholar] [CrossRef]
- Chan, C.B.; Liu, X.; Pradoldej, S.; Hao, C.; An, J.; Yepes, M.; Luo, H.R.; Ye, K. Phosphoinositide 3-kinase enhancer regulates neuronal dendritogenesis and survival in neocortex. J. Neurosci. 2011, 31, 8083–8092. [Google Scholar] [CrossRef]
- Leinninger, G.M.; Backus, C.; Uhler, M.D.; Lentz, S.I.; Feldman, E.L. Phosphatidylinositol 3-kinase and akt effectors mediate insulin-like growth factor-i neuroprotection in dorsal root ganglia neurons. FASEB J. 2004, 18, 1544–1546. [Google Scholar] [CrossRef]
- Anderton, R.S.; Price, L.L.; Turner, B.J.; Meloni, B.P.; Mitrpant, C.; Mastaglia, F.L.; Goh, C.; Wilton, S.D.; Boulos, S. Co-regulation of survival of motor neuron and bcl-xl expression: Implications for neuroprotection in spinal muscular atrophy. Neuroscience 2012, 220, 228–236. [Google Scholar] [CrossRef]
- Markus, A.; Zhong, J.; Snider, W.D. Raf and akt mediate distinct aspects of sensory axon growth. Neuron 2002, 35, 65–76. [Google Scholar] [CrossRef]
- Orike, N.; Middleton, G.; Borthwick, E.; Buchman, V.; Cowen, T.; Davies, A.M. Role of pi 3-kinase, akt and bcl-2-related proteins in sustaining the survival of neurotrophic factor-independent adult sympathetic neurons. J. Cell Biol. 2001, 154, 995–1005. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-akt signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef]
- Chakrabarty, K.; Serchov, T.; Mann, S.A.; Dietzel, I.D.; Heumann, R. Enhancement of dopaminergic properties and protection mediated by neuronal activation of ras in mouse ventral mesencephalic neurones. Eur. J. Neurosci. 2007, 25, 1971–1981. [Google Scholar] [CrossRef]
- Arya, R.; White, K. Cell death in development: Signaling pathways and core mechanisms. Semin. Cell Dev. Biol. 2015, 39, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Gulbins, E.; Brenner, B.; Koppenhoefer, U.; Linderkamp, O.; Lang, F. Fas or ceramide induce apoptosis by ras-regulated phosphoinositide-3-kinase activation. J. Leukoc. Biol. 1998, 63, 253–263. [Google Scholar] [CrossRef]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.C.; Liu, J.P.; Mao, Y.W.; Xiang, H.; Wang, J.; Ma, W.Y.; Dong, Z.G.; Pike, H.M.; Brown, R.E.; Reed, J.C. Calcium-activated raf/mek/erk signaling pathway mediates p53-dependent apoptosis and is abrogated by alpha b-crystallin through inhibition of ras activation. Mol. Biol. Cell 2005, 16, 4437–4453. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Pervaiz, S. Crosstalk between bcl-2 family and ras family small gtpases: Potential cell fate regulation? Front. Oncol. 2012, 2, 206. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The rassf proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.; Baksh, S. Rassf1a: Not a prototypical ras effector. Small GTPases 2011, 2, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Volodko, N.; Gordon, M.; Salla, M.; Ghazaleh, H.A.; Baksh, S. Rassf tumor suppressor gene family: Biological functions and regulation. FEBS Lett. 2014, 588, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Recino, A.; Jeffries, A.; Ward, A.; Chalmers, A.D. The n-terminal rassf family: A new group of ras-association-domain-containing proteins, with emerging links to cancer formation. Biochem. J. 2009, 425, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Vavvas, D.; Li, X.; Avruch, J.; Zhang, X.F. Identification of nore1 as a potential ras effector. J. Biol. Chem. 1998, 273, 5439–5442. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kang, S.I.; Lee, S.Y.; Zhang, X.F.; Kim, M.S.; Beers, L.F.; Lim, D.S.; Avruch, J.; Kim, H.S.; Lee, S.B. Tumor suppressor ras association domain family 5 (rassf5/nore1) mediates death receptor ligand-induced apoptosis. J. Biol. Chem. 2010, 285, 35029–35038. [Google Scholar] [CrossRef] [PubMed]
- Elmetwali, T.; Salman, A.; Palmer, D.H. Nore1a induction by membrane-bound cd40l (mcd40l) contributes to cd40l-induced cell death and g1 growth arrest in p21-mediated mechanism. Cell Death Dis. 2016, 7, e2146. [Google Scholar] [CrossRef]
- Khokhlatchev, A.; Rabizadeh, S.; Xavier, R.; Nedwidek, M.; Chen, T.; Zhang, X.F.; Seed, B.; Avruch, J. Identification of a novel ras-regulated proapoptotic pathway. Curr. Biol. 2002, 12, 253–265. [Google Scholar] [CrossRef]
- Koturenkiene, A.; Makbul, C.; Herrmann, C.; Constantinescu-Aruxandei, D. Kinetic characterization of apoptotic ras signaling through nore1-mst1 complex formation. Biol. Chem. 2017, 398, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fang, Y.J.; Xu, S.B.; Reis, C.; Zhang, J.M. Mammalian sterile20-like kinases: Signalings and roles in central nervous system. Aging Dis. 2018, 9, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Titus, H.E.; Lopez-Juarez, A.; Silbak, S.H.; Rizvi, T.A.; Bogard, M.; Ratner, N. Oligodendrocyte rasg12v expressed in its endogenous locus disrupts myelin structure through increased mapk, nitric oxide, and notch signaling. Glia 2017, 65, 1990–2002. [Google Scholar] [CrossRef] [PubMed]
- Serdar, M.; Herz, J.; Kempe, K.; Winterhager, E.; Jastrow, H.; Heumann, R.; Felderhoff-Muser, U.; Bendix, I. Protection of oligodendrocytes through neuronal overexpression of the small gtpase ras in hyperoxia-induced neonatal brain injury. Front. Neurol. 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Niihori, T.; Kawame, H.; Kurosawa, K.; Ohashi, H.; Tanaka, Y.; Filocamo, M.; Kato, K.; Suzuki, Y.; Kure, S.; et al. Germline mutations in hras proto-oncogene cause costello syndrome. Nat. Genet. 2005, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag gtpases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef]
- Patel, P.H.; Tamanoi, F. Increased RHEB-tor signaling enhances sensitivity of the whole organism to oxidative stress. J. Cell Sci. 2006, 119, 4285–4292. [Google Scholar] [CrossRef]
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.-L.; Inoki, K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-RHEB gtpase pathway. J. Biol. Chem. 2011, 286, 32651–32660. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Kramvis, I.; Cho, N.; Sadowski, A.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mtor-dependent manner. J. Neurosci. 2009, 29, 5926–5937. [Google Scholar] [CrossRef]
- Lee, H.; Paik, S.G. Regulation of BNIP3 in normal and cancer cells. Mol. Cells 2006, 21, 1–6. [Google Scholar] [PubMed]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.-Y.; Swanson, J.; You, M.; Guan, K.-L. BNIP3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with RHEB. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Wu, Y.P.; Sugaya, S.; Moriya, T.; Nomura, J.; Takahashi, S.; Yamamori, H.; Nakajima, N.; Suzuki, N. Search for UV-responsive genes in human cells by differential mRNA display: Involvement of human ras-related GTP-binding protein, RHEB, in UV susceptibility. Biochem. Biophys. Res. Commun. 2000, 274, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Tan, X.; Jin, W.; Zheng, H.; Xu, W.; Rui, Y.; Li, L.; Cao, J.; Wu, X.; Cui, G.; et al. Upregulation of ras homolog enriched in the brain (RHEB) in lipopolysaccharide-induced neuroinflammation. Neurochem. Int. 2013, 62, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Norsted Gregory, E.; Codeluppi, S.; Gregory, J.A.; Steinauer, J.; Svensson, C.I. Mammalian target of rapamycin in spinal cord neurons mediates hypersensitivity induced by peripheral inflammation. Neuroscience 2010, 169, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Karassek, S.; Berghaus, C.; Schwarten, M.; Goemans, C.G.; Ohse, N.; Kock, G.; Jockers, K.; Neumann, S.; Gottfried, S.; Herrmann, C.; et al. Ras homolog enriched in brain (RHEB) enhances apoptotic signaling. J. Biol. Chem. 2010, 285, 33979–33991. [Google Scholar] [CrossRef]
- Soga, M.; Matsuzawa, A.; Ichijo, H. Oxidative stress-induced diseases via the ASK1 signaling pathway. Int. J. Cell Biol. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. Hdac6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Ozcan, L.; Yilmaz, E.; Duvel, K.; Sahin, M.; Manning, B.D.; Hotamisligil, G.S. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 2008, 29, 541–551. [Google Scholar] [CrossRef]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Okamura, M.; Du, S.; Kasai, A.; Takano, Y.; Shitamura, A.; Shimada, T.; Yao, J.; et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J. Biol. Chem. 2008, 283, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Hinerfeld, D.; Traini, M.D.; Weinberger, R.P.; Cochran, B.; Doctrow, S.R.; Harry, J.; Melov, S. Endogenous mitochondrial oxidative stress: Neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J. Neurochem. 2004, 88, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.; Shahani, N.; Gorgen, L.; Ferretti, M.; Pryor, W.; Chen, P.Y.; Swarnkar, S.; Worley, P.F.; Karbstein, K.; Snyder, S.H.; et al. RHEB inhibits protein synthesis by activating the PERK-eIF2alpha signaling cascade. Cell Rep. 2015, 10, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORC1 signaling in the pathogenesis of huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef]
- Shahani, N.; Pryor, W.; Swarnkar, S.; Kholodilov, N.; Thinakaran, G.; Burke, R.E.; Subramaniam, S. RHEB gtpase regulates beta-secretase levels and amyloid beta generation. J. Biol. Chem. 2014, 289, 5799–5808. [Google Scholar] [CrossRef]
- Gallo, G.; Letourneau, P.C. Axon guidance: Gtpases help axons reach their targets. Curr. Biol. 1998, 8, R80–R82. [Google Scholar] [CrossRef]
- Flynn, K.C. The cytoskeleton and neurite initiation. Bioarchitecture 2013, 3, 86–109. [Google Scholar] [CrossRef] [PubMed]
- Bradke, F.; Dotti, C.G. The role of local actin instability in axon formation. Science 1999, 283, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Guo, W.; Liang, X.; Rao, Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: Critical roles of GSK-3β and its upstream regulators. Cell 2005, 120, 123–135. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Z.; Chen, Y.-K.; Chai, Z.; Zhou, C.; Zhang, Y. Neurons with multiple axons have functional axon initial segments. Neurosci. Bull. 2017, 33, 641–652. [Google Scholar] [CrossRef]
- Lange-Carter, C.; Johnson, G. Ras-dependent growth factor regulation of mek kinase in PC12 cells. Science 1994, 265, 1458–1461. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Moodie, S.A.; Wolfman, A.; Landreth, G.E. The mitogen-activated protein kinase cascade is activated by b-raf in response to nerve growth factor through interaction with p21ras. Mol. Cell. Biol. 1994, 14, 6944–6953. [Google Scholar] [CrossRef]
- Sarah, T.; Philip, C. Identification of a latent map kinase kinase kinase in PC12 cells as b-raf. FEBS Lett. 1994, 350, 13–18. [Google Scholar] [CrossRef]
- Vaillancourt, R.R.; Gardner, A.M.; Johnson, G.L. B-raf-dependent regulation of the mek-1/mitogen-activated protein kinase pathway in PC12 cells and regulation by cyclic amp. Mol. Cell. Biol. 1994, 14, 6522–6530. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Feramisco, J.R. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell 1985, 42, 841–848. [Google Scholar] [CrossRef]
- Robinson, M.J.; Stippec, S.A.; Goldsmith, E.; White, M.A.; Cobb, M.H. A constitutively active and nuclear form of the map kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr. Biol. 1998, 8, 1141–1152. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Lampe, P.D.; Kurata, W.E.; Kanemitsu, M.Y.; Loo, L.W.M.; Eckhart, W.; Lau, A.F. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J. Biol. Chem. 1996, 271, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, M.Y.; Lau, A.F. Epidermal growth factor stimulates the disruption of gap junctional communication and connexin43 phosphorylation independent of 12-0-tetradecanoylphorbol 13-acetate-sensitive protein kinase C: The possible involvement of mitogen-activated protein kinase. Mol. Biol. Cell 1993, 4, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Mamoru, S. Chromatographic resolution and characterization of a nerve growth factor-dependent kinase that phosphorylates microtubule-associated proteins 1 and 2 in PC12 cells. J. Neurochem. 1992, 59, 1263–1272. [Google Scholar] [CrossRef]
- Sturgill, T.W.; Ray, L.B. Muscle proteins related to microtubule associated protein-2 are substrates for an insulin-stimulatable kinase. Biochem. Biophys. Res. Commun. 1986, 134, 565–571. [Google Scholar] [CrossRef]
- Drewes, G.; Lichtenberg-Kraag, B.; Döring, F.; Mandelkow, E.M.; Biernat, J.; Goris, J.; Dorée, M.; Mandelkow, E. Mitogen activated protein (MAP) kinase transforms tau protein into an alzheimer-like state. EMBO J. 1992, 11, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Mitsushima, M.; Suwa, A.; Amachi, T.; Ueda, K.; Kioka, N. Extracellular signal-regulated kinase activated by epidermal growth factor and cell adhesion interacts with and phosphorylates vinexin. J. Biol. Chem. 2004, 279, 34570–34577. [Google Scholar] [CrossRef]
- Atwal, J.K.; Massie, B.; Miller, F.D.; Kaplan, D.R. The trkb-shc site signals neuronal survival and local axon growth via mek and pi3-kinase. Neuron 2000, 27, 265–277. [Google Scholar] [CrossRef]
- Mills, J.; Digicaylioglu, M.; Legg, A.T.; Young, C.E.; Young, S.S.; Barr, A.M.; Fletcher, L.; O’Connor, T.P.; Dedhar, S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J. Neurosci. 2003, 23, 1638–1648. [Google Scholar] [CrossRef]
- Zhou, F.-Q.; Zhou, J.; Dedhar, S.; Wu, Y.-H.; Snider, W.D. Ngf-induced axon growth is mediated by localized inactivation of GSK-3β and functions of the microtubule plus end binding protein apc. Neuron 2004, 42, 897–912. [Google Scholar] [CrossRef]
- Nishimura, T.; Fukata, Y.; Kato, K.; Yamaguchi, T.; Matsuura, Y.; Kamiguchi, H.; Kaibuchi, K. Crmp-2 regulates polarized numb-mediated endocytosis for axon growth. Nat. Cell Biol. 2003, 5, 819. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Menager, C.; Fukata, Y.; Kaibuchi, K. Role of crmp-2 in neuronal polarity. J. Neurobiol. 2004, 58, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho gtpases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Bregman, B.; Goldberger, M. Anatomical plasticity and sparing of function after spinal cord damage in neonatal cats. Science 1982, 217, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Kunkel-Bagden, E.; Dai, H.-N.; Bregman, B.S. Recovery of function after spinal cord hemisection in newborn and adult rats: Differential effects on reflex and locomotor function. Exp. Neurol. 1992, 116, 40–51. [Google Scholar] [CrossRef]
- David, S.; Aguayo, A. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science 1981, 214, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W. The paper that restarted modern central nervous system axon regeneration research. Trends Neurosci. 2018, 41, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.T. Recapitulate development to promote axonal regeneration: Good or bad approach? Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1565–1574. [Google Scholar] [CrossRef]
- Lindsay, R.M. Neuron saving schemes. Nature 1995, 373, 289. [Google Scholar] [CrossRef]
- Oppenheim, R.W. Neurotrophic survival molecules for motoneurons: An embarrassment of riches. Neuron 1996, 17, 195–197. [Google Scholar] [CrossRef]
- Namikawa, K.; Honma, M.; Abe, K.; Takeda, M.; Mansur, K.; Obata, T.; Miwa, A.; Okado, H.; Kiyama, H. Akt/protein kinase b prevents injury-induced motoneuron death and accelerates axonal regeneration. J. Neurosci. 2000, 20, 2875–2886. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Dietz, V.; Schwab, M.E. From the rodent spinal cord injury model to human application: Promises and challenges. J. Neurotrauma 2017, 34, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Hollis, E.R.; Jamshidi, P.; Löw, K.; Blesch, A.; Tuszynski, M.H. Induction of corticospinal regeneration by lentiviral trkb-induced erk activation. Proc. Natl. Acad. Sci. USA 2009, 106, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- Sapieha, P.S.; Hauswirth, W.W.; Di Polo, A. Extracellular signal-regulated kinases 1/2 are required for adult retinal ganglion cell axon regeneration induced by fibroblast growth factor-2. J. Neurosci. Res. 2006, 83, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Kermer, P.; Digicaylioglu, M.H.; Kaul, M.; Zapata, J.M.; Krajewska, M.; Stenner-Liewen, F.; Takayama, S.; Krajewski, S.; Lipton, S.A.; Reed, J.C. BAG1 over-expression in brain protects against stroke. Brain Pathol. 2003, 13, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Pernet, V.; Hauswirth, W.W.; Di Polo, A. Extracellular signal-regulated kinase 1/2 mediates survival, but not axon regeneration, of adult injured central nervous system neurons in vivo. J. Neurochem. 2005, 93, 72–83. [Google Scholar] [CrossRef]
- O’Donovan, K.J.; Ma, K.; Guo, H.; Wang, C.; Sun, F.; Han, S.B.; Kim, H.; Wong, J.K.; Charron, J.; Zou, H.; et al. B-raf kinase drives developmental axon growth and promotes axon regeneration in the injured mature CNS. J. Exp. Med. 2014, 211, 801–814. [Google Scholar] [CrossRef]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the pten/mtor pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 2010, 13, 1075. [Google Scholar] [CrossRef]
- Christie, K.J.; Webber, C.A.; Martinez, J.A.; Singh, B.; Zochodne, D.W. Pten inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J. Neurosci. 2010, 30, 9306–9315. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Borson, S.H.; Gambello, M.J.; Wang, F.; Cavalli, V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J. Biol. Chem. 2010, 285, 28034–28043. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.G. Rho gtpases and their regulators in neuronal functions and development. Neurosignals 2007, 15, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Auer, M.; Schweigreiter, R.; Hausott, B.; Thongrong, S.; Höltje, M.; Just, I.; Bandtlow, C.; Klimaschewski, L. Rho-independent stimulation of axon outgrowth and activation of the erk and akt signaling pathways by c3 transferase in sensory neurons. Front. Cell. Neurosci. 2012, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Rishal, I.; Fainzilber, M. Axon–soma communication in neuronal injury. Nat. Rev. Neurosci. 2013, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Qiu, J.; Cao, Z.; McAtee, M.; Bregman, B.S.; Filbin, M.T. Neuronal cyclic amp controls the developmental loss in ability of axons to regenerate. J. Neurosci. 2001, 21, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Deng, K.; Hou, J.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated creb is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Roy, A.; Wu, Z.; Goncharov, A.; Jin, Y.; Chisholm, A.D. Calcium and cyclic amp promote axonal regeneration in Caenorhabditis elegans and require dlk-1 kinase. J. Neurosci. 2010, 30, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Grandpré, T.; Strittmatter, S.M. Nogo: A molecular determinant of axonal growth and regeneration. Neuroscientist 2001, 7, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Brochier, C.; Jones, J.I.; Willis, D.E.; Langley, B. Poly(adp-ribose) polymerase 1 is a novel target to promote axonal regeneration. Proc. Natl. Acad. Sci. USA 2015, 112, 15220–15225. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sekine, Y.; Byrne, A.B.; Cafferty, W.B.J.; Hammarlund, M.; Strittmatter, S.M. Inhibition of poly-ADP-ribosylation fails to increase axonal regeneration or improve functional recovery after adult mammalian CNS injury. Eneuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Kosmaczewski, S.G.; Han, S.M.; Han, B.; Irving Meyer, B.; Baig, H.S.; Athar, W.; Lin-Moore, A.T.; Koelle, M.R.; Hammarlund, M. RNA ligation in neurons by RtcB inhibits axon regeneration. Proc. Natl. Acad. Sci. USA 2015, 112, 8451–8456. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Sretavan, D.; Salegio, E.A.; Berg, J.; Huang, X.; Cheng, T.; Xiong, X.; Meltzer, S.; Han, C.; Nguyen, T.-T.; et al. Regulation of axon regeneration by the RNA repair and splicing pathway. Nat. Neurosci. 2015, 18, 817. [Google Scholar] [CrossRef] [PubMed]
- Imielski, Y.; Schwamborn, J.C.; Lüningschrör, P.; Heimann, P.; Holzberg, M.; Werner, H.; Leske, O.; Püschel, A.W.; Memet, S.; Heumann, R.; et al. Regrowing the adult brain: Nf-κb controls functional circuit formation and tissue homeostasis in the dentate gyrus. PLoS ONE 2012, 7, e30838. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.L.; Goldberg, J.L. Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 2011, 71, 1186–1211. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kareva, T.; Yarygina, O.; Kholodilov, N.; Burke, R.E. AAV transduction of dopamine neurons with constitutively active RHEB protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 2012, 20, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Klaw, M.C.; Connors, T.; Kholodilov, N.; Burke, R.E.; Tom, V.J. Expressing constitutively active RHEB in adult neurons after a complete spinal cord injury enhances axonal regeneration beyond a chondroitinase-treated glial scar. J. Neurosci. 2015, 35, 11068–11080. [Google Scholar] [CrossRef]
- Wu, D.; Klaw, M.C.; Connors, T.; Kholodilov, N.; Burke, R.E.; Côté, M.-P.; Tom, V.J. Combining constitutively active RHEB expression and chondroitinase promotes functional axonal regeneration after cervical spinal cord injury. Mol. Ther. 2017, 25, 2715–2726. [Google Scholar] [CrossRef]
- Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 2015, 18, 1213–1225. [Google Scholar] [CrossRef]
- Liu, H.; Yu, X.; Li, K.; Klejnot, J.; Yang, H.; Lisiero, D.; Lin, C. Photoexcited cry2 interacts with cib1 to regulate transcription and floral initiation in arabidopsis. Science 2008, 322, 1535–1539. [Google Scholar] [CrossRef]
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid blue light induction of protein interactions in living cells. Nat. Methods 2010, 7, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Christie, J.M.; Salomon, M.; Nozue, K.; Wada, M.; Briggs, W.R. Lov (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): Binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. USA 1999, 96, 8779–8783. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Tepperman, J.M.; Quail, P.H. Binding of phytochrome b to its nuclear signalling partner pif3 is reversibly induced by light. Nature 1999, 400, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Sato, S.; Huq, E.; Tepperman, J.M.; Quail, P.H. A light-switchable gene promoter system. Nat. Biotechnol. 2002, 20, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Chung, H.K.; Lam, A.J.; Lin, M.Z. Optical control of protein activity by fluorescent protein domains. Science 2012, 338, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Tischer, D.; Weiner, O.D. Illuminating cell signalling with optogenetic tools. Nat. Rev. Mol. Cell Biol. 2014, 15, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Bugaj, L.J.; Choksi, A.T.; Mesuda, C.K.; Kane, R.S.; Schaffer, D.V. Optogenetic protein clustering and signaling activation in mammalian cells. Nat. Methods 2013, 10, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.I.; Frey, D.; Lungu, O.I.; Jaehrig, A.; Schlichting, I.; Kuhlman, B.; Hahn, K.M. A genetically encoded photoactivatable rac controls the motility of living cells. Nature 2009, 461, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Yang, Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods 2012, 9, 266–269. [Google Scholar] [CrossRef]
- Strickland, D.; Lin, Y.; Wagner, E.; Hope, C.M.; Zayner, J.; Antoniou, C.; Sosnick, T.R.; Weiss, E.L.; Glotzer, M. Tulips: Tunable, light-controlled interacting protein tags for cell biology. Nat. Methods 2012, 9, 379–384. [Google Scholar] [CrossRef]
- Guntas, G.; Hallett, R.A.; Zimmerman, S.P.; Williams, T.; Yumerefendi, H.; Bear, J.E.; Kuhlman, B. Engineering an improved light-induced dimer (ilid) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.P.; Hallett, R.A.; Bourke, A.M.; Bear, J.E.; Kennedy, M.J.; Kuhlman, B. Tuning the binding affinities and reversion kinetics of a light inducible dimer allows control of transmembrane protein localization. Biochemistry 2016, 55, 5264–5271. [Google Scholar] [CrossRef] [PubMed]
- Hallett, R.A.; Zimmerman, S.P.; Yumerefendi, H.; Bear, J.E.; Kuhlman, B. Correlating in vitro and in vivo activities of light-inducible dimers: A cellular optogenetics guide. ACS Synth. Biol. 2016, 5, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 2009, 461, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Gambetta, G.A.; Lagarias, J.C. Genetic engineering of phytochrome biosynthesis in bacteria. Proc. Natl. Acad. Sci. USA 2001, 98, 10566–10571. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Engesser, R.; Timmer, J.; Nagy, F.; Zurbriggen, M.D.; Weber, W. Synthesis of phycocyanobilin in mammalian cells. Chem. Commun. 2013, 49, 8970–8972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Cui, B. Optogenetic control of intracellular signaling pathways. Trends Biotechnol. 2015, 33, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.P.; Asokan, S.B.; Kuhlman, B.; Bear, J.E. Cells lay their own tracks—Optogenetic cdc42 activation stimulates fibronectin deposition supporting directed migration. J. Cell Sci. 2017, 130, 2971–2983. [Google Scholar] [CrossRef] [PubMed]
- Toettcher, J.E.; Weiner, O.D.; Lim, W.A. Using optogenetics to interrogate the dynamic control of signal transmission by the ras/erk module. Cell 2013, 155, 1422–1434. [Google Scholar] [CrossRef]
- Zhang, K.; Duan, L.; Ong, Q.; Lin, Z.; Varman, P.M.; Sung, K.; Cui, B. Light-mediated kinetic control reveals the temporal effect of the raf/mek/erk pathway in PC12 cell neurite outgrowth. PLoS ONE 2014, 9, e92917. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Kumagai, Y.; Sakurai, A.; Komatsu, N.; Fujita, Y.; Shionyu, C.; Matsuda, M. Stochastic erk activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 2013, 52, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Kondo, Y.; Naoki, H.; Hiratsuka, T.; Itoh, R.E.; Matsuda, M. Propagating wave of erk activation orients collective cell migration. Dev. Cell 2017, 43, 305–317.e305. [Google Scholar] [CrossRef] [PubMed]
- Wend, S.; Wagner, H.J.; Muller, K.; Zurbriggen, M.D.; Weber, W.; Radziwill, G. Optogenetic control of protein kinase activity in mammalian cells. ACS Synth. Biol. 2014, 3, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Dine, E.; Gil, A.A.; Uribe, G.; Brangwynne, C.P.; Toettcher, J.E. Protein phase separation provides long-term memory of transient spatial stimuli. Cell Syst. 2018, 6, 655–663.e655. [Google Scholar] [CrossRef] [PubMed]
- Goglia, A.G.; Wilson, M.Z.; DiGiorno, D.B.; Toettcher, J.E. Optogenetic control of ras/erk signaling using the phy-pif system. Methods Mol. Biol. 2017, 1636, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Chatelle, C.V.; Hovermann, D.; Muller, A.; Wagner, H.J.; Weber, W.; Radziwill, G. Optogenetically controlled raf to characterize braf and craf protein kinase inhibitors. Sci. Rep. 2016, 6, 23713. [Google Scholar] [CrossRef]
- Krishnamurthy, V.V.; Khamo, J.S.; Mei, W.; Turgeon, A.J.; Ashraf, H.M.; Mondal, P.; Patel, D.B.; Risner, N.; Cho, E.E.; Yang, J.; et al. Reversible optogenetic control of kinase activity during differentiation and embryonic development. Development 2016, 143, 4085–4094. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Toettcher, J.E. Light-based feedback for controlling intracellular signaling dynamics. Nat. Methods 2011, 8, 837–839. [Google Scholar] [CrossRef]
- Kakumoto, T.; Nakata, T. Optogenetic control of pip3: Pip3 is sufficient to induce the actin-based active part of growth cones and is regulated via endocytosis. PLoS ONE 2013, 8, e70861. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Dickson, E.J.; Hille, B.; Toomre, D.K.; De Camilli, P. Optogenetic control of phosphoinositide metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, E2316–E2323. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, J.; Lee, M.; Kim, C.; Chang, K.-Y.; Heo, W. Spatiotemporal control of fibroblast growth factor receptor signals by blue light. Chem. Biol. 2014, 21, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Grusch, M.; Schelch, K.; Riedler, R.; Reichhart, E.; Differ, C.; Berger, W.; Ingles-Prieto, A.; Janovjak, H. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 2014, 33, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Suzuki, H.; Yamamoto, R.; Sahashi, H.; Takido, Y.; Sawada, M. Optogenetic control of cell differentiation in channelrhodopsin-2-expressing os3, a bipotential glial progenitor cell line. Neurochem. Int. 2017, 104, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Warscheid, B.; Weber, W.; Radziwill, G. Optogenetic clustering of cnk1 reveals mechanistic insights in raf and akt signalling controlling cell fate decisions. Sci. Rep. 2016, 6, 38155. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Sadaghiani, A.M.; Hsueh, B.; Dolmetsch, R.E. Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 2009, 27, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Beyer, H.M.; Naumann, S.; Weber, W.; Radziwill, G. Optogenetic control of signaling in mammalian cells. Biotechnol. J. 2015, 10, 273–283. [Google Scholar] [CrossRef]
- Chen, S.; Weitemier, A.Z.; Zeng, X.; He, L.; Wang, X.; Tao, Y.; Huang, A.J.Y.; Hashimotodani, Y.; Kano, M.; Iwasaki, H.; et al. Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science 2018, 359, 679–684. [Google Scholar] [CrossRef]
- Kanczler, J.M.; Sura, H.S.; Magnay, J.; Green, D.; Oreffo, R.O.; Dobson, J.P.; El Haj, A.J. Controlled differentiation of human bone marrow stromal cells using magnetic nanoparticle technology. Tissue Eng. Part A 2010, 16, 3241–3250. [Google Scholar] [CrossRef]
- Seo, D.; Southard, K.M.; Kim, J.W.; Lee, H.J.; Farlow, J.; Lee, J.U.; Litt, D.B.; Haas, T.; Alivisatos, A.P.; Cheon, J.; et al. A mechanogenetic toolkit for interrogating cell signaling in space and time. Cell 2016, 165, 1507–1518. [Google Scholar] [CrossRef]
- Stanley, S.A.; Sauer, J.; Kane, R.S.; Dordick, J.S.; Friedman, J.M. Remote regulation of glucose homeostasis in mice using genetically encoded nanoparticles. Nat. Med. 2015, 21, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Van Bergeijk, P.; Adrian, M.; Hoogenraad, C.C.; Kapitein, L.C. Optogenetic control of organelle transport and positioning. Nature 2015, 518, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Dhennin-Duthille, I.; Ay, A.S.; Rybarczyk, P.; Korichneva, I.; Ouadid-Ahidouch, H. New insights into pharmacological tools to tr(i)p cancer up. Br. J. Pharmacol. 2014, 171, 2582–2592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, Y.; Chang, Y.; Seyf, H.R.; Henry, A.; Mattheyses, A.L.; Yehl, K.; Zhang, Y.; Huang, Z.; Salaita, K. Nanoscale optomechanical actuators for controlling mechanotransduction in living cells. Nat. Methods 2016, 13, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Monzel, C. Magnetic control of cellular processes using biofunctional nanoparticles. Chem. Sci. 2017, 8, 7330–7338. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, S.; Lohmann, K.J. The physics and neurobiology of magnetoreception. Nat. Rev. Neurosci. 2005, 6, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Ritz, T.; Ahmad, M.; Mouritsen, H.; Wiltschko, R.; Wiltschko, W. Photoreceptor-based magnetoreception: Optimal design of receptor molecules, cells, and neuronal processing. J. R. Soc. Interface 2010, 7 (Suppl. 2), S135–S146. [Google Scholar] [CrossRef]
- Pankhurst, Q.A.; Connolly, J.; Jones, S.; Dobson, J. Applications of magnetic nanoparticles in biomedicine. J. Phys. D Appl. Phys. 2003, 36, R167. [Google Scholar] [CrossRef]
- Gupta, A.K.; Curtis, A.S. Surface modified superparamagnetic nanoparticles for drug delivery: Interaction studies with human fibroblasts in culture. J. Mater. Sci. Mater. Med. 2004, 15, 493–496. [Google Scholar] [CrossRef]
- Griffete, N.; Fresnais, J.; Espinosa, A.; Wilhelm, C.; Bee, A.; Menager, C. Design of magnetic molecularly imprinted polymer nanoparticles for controlled release of doxorubicin under an alternative magnetic field in athermal conditions. Nanoscale 2015, 7, 18891–18896. [Google Scholar] [CrossRef]
- Georgelin, T.; Bombard, S.; Siaugue, J.M.; Cabuil, V. Nanoparticle-mediated delivery of bleomycin. Angew. Chem. 2010, 49, 8897–8901. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J. Magnetic micro- and nano-particle-based targeting for drug and gene delivery. Nanomedicine 2006, 1, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bulte, J.W.; Douglas, T.; Witwer, B.; Zhang, S.C.; Strable, E.; Lewis, B.K.; Zywicke, H.; Miller, B.; van Gelderen, P.; Moskowitz, B.M.; et al. Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat. Biotechnol. 2001, 19, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Hyeon, T. Designed synthesis of uniformly sized iron oxide nanoparticles for efficient magnetic resonance imaging contrast agents. Chem. Soc. Rev. 2012, 41, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J. Remote control of cellular behaviour with magnetic nanoparticles. Nat. Nanotechnol. 2008, 3, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Tajik, A.; Zhang, Y.; Wei, F.; Sun, J.; Jia, Q.; Zhou, W.; Singh, R.; Khanna, N.; Belmont, A.S.; Wang, N. Transcription upregulation via force-induced direct stretching of chromatin. Nature materials 2016, 15, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, J.W.; Levy, M.; Kao, A.; Noh, S.H.; Bozovic, D.; Cheon, J. Magnetic nanoparticles for ultrafast mechanical control of inner ear hair cells. ACS Nano 2014, 8, 6590–6598. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Smith, C.J.; Ottolini, M.; Barker, B.S.; Purohit, A.M.; Grippo, R.M.; Gaykema, R.P.; Spano, A.J.; Beenhakker, M.P.; Kucenas, S.; et al. Genetically targeted magnetic control of the nervous system. Nat. Neurosci. 2016, 19, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Rotherham, M.; El Haj, A.J. Remote activation of the wnt/beta-catenin signalling pathway using functionalised magnetic particles. PLoS ONE 2015, 10, e0121761. [Google Scholar] [CrossRef]
- Tseng, P.; Judy, J.W.; Di Carlo, D. Magnetic nanoparticle-mediated massively-parallel mechanical modulation of single-cell behavior. Nat. Methods 2012, 9, 1113–1119. [Google Scholar] [CrossRef]
- Riedinger, A.; Guardia, P.; Curcio, A.; Garcia, M.A.; Cingolani, R.; Manna, L.; Pellegrino, T. Subnanometer local temperature probing and remotely controlled drug release based on azo-functionalized iron oxide nanoparticles. Nano Lett. 2013, 13, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role for trp channels in thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef]
- Premkumar, L.S.; Abooj, M. Trp channels and analgesia. Life Sci. 2013, 92, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Park, H.; Griffin, R.J. Theoretical and experimental basis of hyperthermia. In Thermotherapy for Neoplasia, Inflammation, and Pain; Springer: Berlin/Heidelberg, Germany, 2001; pp. 394–407. [Google Scholar]
- Huang, H.; Delikanli, S.; Zeng, H.; Ferkey, D.M.; Pralle, A. Remote control of ion channels and neurons through magnetic-field heating of nanoparticles. Nat. Nanotechnol. 2010, 5, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Romero, G.; Christiansen, M.G.; Mohr, A.; Anikeeva, P. Wireless magnetothermal deep brain stimulation. Science 2015, 347, 1477–1480. [Google Scholar] [CrossRef]
- Stanley, S.A.; Gagner, J.E.; Damanpour, S.; Yoshida, M.; Dordick, J.S.; Friedman, J.M. Radio-wave heating of iron oxide nanoparticles can regulate plasma glucose in mice. Science 2012, 336, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Kelly, L.; Latcha, K.N.; Schmidt, S.F.; Yu, X.; Nectow, A.R.; Sauer, J.; Dyke, J.P.; Dordick, J.S.; Friedman, J.M. Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 2016, 531, 647–650. [Google Scholar] [CrossRef]
- Mannix, R.J.; Kumar, S.; Cassiola, F.; Montoya-Zavala, M.; Feinstein, E.; Prentiss, M.; Ingber, D.E. Nanomagnetic actuation of receptor-mediated signal transduction. Nat. Nanotechnol. 2008, 3, 36–40. [Google Scholar] [CrossRef]
- Bharde, A.A.; Palankar, R.; Fritsch, C.; Klaver, A.; Kanger, J.S.; Jovin, T.M.; Arndt-Jovin, D.J. Magnetic nanoparticles as mediators of ligand-free activation of egfr signaling. PLoS ONE 2013, 8, e68879. [Google Scholar] [CrossRef]
- Cho, M.H.; Lee, E.J.; Son, M.; Lee, J.H.; Yoo, D.; Kim, J.W.; Park, S.W.; Shin, J.S.; Cheon, J. A magnetic switch for the control of cell death signalling in in vitro and in vivo systems. Nat. Mater. 2012, 11, 1038–1043. [Google Scholar] [CrossRef]
- Lisse, D.; Monzel, C.; Vicario, C.; Manzi, J.; Maurin, I.; Coppey, M.; Piehler, J.; Dahan, M. Engineered ferritin for magnetogenetic manipulation of proteins and organelles inside living cells. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Etoc, F.; Vicario, C.; Lisse, D.; Siaugue, J.M.; Piehler, J.; Coppey, M.; Dahan, M. Magnetogenetic control of protein gradients inside living cells with high spatial and temporal resolution. Nano Lett. 2015, 15, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; Kolch, W.; Mahmoudi, M. Cell type-specific activation of akt and erk signaling pathways by small negatively-charged magnetic nanoparticles. Sci. Rep. 2012, 2, 868. [Google Scholar] [CrossRef] [PubMed]
- Etoc, F.; Lisse, D.; Bellaiche, Y.; Piehler, J.; Coppey, M.; Dahan, M. Subcellular control of rac-gtpase signalling by magnetogenetic manipulation inside living cells. Nat. Nanotechnol. 2013, 8, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Mazari, E.; Lallet, S.; Le Borgne, R.; Marchi, V.; Gosse, C.; Gueroui, Z. Spatiotemporal control of microtubule nucleation and assembly using magnetic nanoparticles. Nat. Nanotechnol. 2013, 8, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Björklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef]
- Makwana, M.; Serchov, T.; Hristova, M.; Bohatschek, M.; Gschwendtner, A.; Kalla, R.; Liu, Z.; Heumann, R.; Raivich, G. Regulation and function of neuronal gtp-ras in facial motor nerve regeneration. J. Neurochem. 2009, 108, 1453–1463. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human ips cell-derived dopaminergic neurons function in a primate parkinson’s disease model. Nature 2017, 548, 592. [Google Scholar] [CrossRef]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human trials of stem cell-derived dopamine neurons for parkinson’s disease: Dawn of a new era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef]
- Dinca, A.; Chien, W.M.; Chin, M.T. Intracellular delivery of proteins with cell-penetrating peptides for therapeutic uses in human disease. Int. J. Mol. Sci. 2016, 17, 263. [Google Scholar] [CrossRef] [PubMed]
- Paliga, D.; Raudzus, F.; Leppla, S.H.; Heumann, R.; Neumann, S. Lethal factor domain-mediated delivery of nurr1 transcription factor enhances tyrosine hydroxylase activity and protects from neurotoxin-induced degeneration of dopaminergic cells. Mol. Neurobiol. 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schöneborn, H.; Raudzus, F.; Coppey, M.; Neumann, S.; Heumann, R. Perspectives of RAS and RHEB GTPase Signaling Pathways in Regenerating Brain Neurons. Int. J. Mol. Sci. 2018, 19, 4052. https://doi.org/10.3390/ijms19124052
Schöneborn H, Raudzus F, Coppey M, Neumann S, Heumann R. Perspectives of RAS and RHEB GTPase Signaling Pathways in Regenerating Brain Neurons. International Journal of Molecular Sciences. 2018; 19(12):4052. https://doi.org/10.3390/ijms19124052
Chicago/Turabian StyleSchöneborn, Hendrik, Fabian Raudzus, Mathieu Coppey, Sebastian Neumann, and Rolf Heumann. 2018. "Perspectives of RAS and RHEB GTPase Signaling Pathways in Regenerating Brain Neurons" International Journal of Molecular Sciences 19, no. 12: 4052. https://doi.org/10.3390/ijms19124052
APA StyleSchöneborn, H., Raudzus, F., Coppey, M., Neumann, S., & Heumann, R. (2018). Perspectives of RAS and RHEB GTPase Signaling Pathways in Regenerating Brain Neurons. International Journal of Molecular Sciences, 19(12), 4052. https://doi.org/10.3390/ijms19124052