Abstract
In the adult, the source of functionally diverse, mature blood cells are hematopoietic stem cells, a rare population of quiescent cells that reside in the bone marrow niche. Like stem cells in other tissues, hematopoietic stem cells are defined by their ability to self-renew, in order to maintain the stem cell population for the lifetime of the organism, and to differentiate, in order to give rise to the multiple lineages of the hematopoietic system. In recent years, increasing evidence has suggested a role for the accumulation of reactive oxygen species and DNA damage in the decision for hematopoietic stem cells to exit quiescence and to differentiate. In this review, we will examine recent work supporting the idea that detection of cell stressors, such as oxidative and genetic damage, is an important mediator of cell fate decisions in hematopoietic stem cells. We will explore the benefits of such a system in avoiding the development and progression of malignancies, and in avoiding tissue exhaustion and failure. Additionally, we will discuss new work that examines the accumulation of DNA damage and replication stress in aging hematopoietic stem cells and causes us to rethink ideas of genoprotection in the bone marrow niche.
1. Introduction
Hematopoietic stem cells (HSCs), like other stem cell populations, are defined by their ability to self-renew and to differentiate [1,2]. A phenotypically identified and isolated HSC is capable of contributing to all of the mature hematologic lineages and is capable of replenishing the HSC pool after lethal irradiation [3,4,5]. Additionally, the differentiation hierarchy that progresses from the HSC, to the progenitor, to a functionally mature effector cell is well understood. These features make HSCs the best-characterized adult stem cell population. Thus, the hematopoietic system, including HSCs and hematological malignancies, serves as an essential investigative avenue in stem cell biology [6].
Among cellular components, the genome is particularly susceptible to damage, which can result from spontaneous reactions in the nucleus, oxidative damage due to metabolic byproducts or from extrinsic agents, or replication associated defects [7,8]. Because the genome must be maintained for the life of the cell, and because it can be copied and propagated into daughter cells, damage to DNA brings more severe consequences than damage to replaceable cellular macromolecules. Of course, accumulated DNA damage is essential in the development of malignancies, and DNA damage accumulated in HSCs and progenitors is responsible, in part, for hematological malignancies [9,10]. Stem cells, including HSCs, may be particularly susceptible to DNA damage due to their longevity [11].
HSCs replenish themselves and provide mature blood cells, and so, HSCs are thought to be responsible for safeguarding the genomic integrity of the hematopoietic system [12]. An unrepaired genetic lesion maintained in an HSC is capable of being spread throughout the HSC pool, through self-renewing divisions, as well as propagating to all of the hematologic compartments, through differentiation [11]. It is thought that HSCs remain quiescent and reside in the hypoxic bone marrow niche in order to avoid reactive oxygen species (ROS) and other stresses to the genome [13,14,15,16,17]. One might hypothesize that HSCs also exhibit uniquely robust DNA damage repair mechanisms, in order to further fortify their genome. However, much evidence suggests an opposing hypothesis—the consequences of erroneous DNA damage repair in an HSC could be so severe that it is preferable for an HSC faced with oxidative or other genotoxic stress to differentiate, removing it from the stem cell pool and preventing the dissemination of deleterious mutations. The pathways that underlie this decision have been coined the “ROS rheostat [18,19,20,21,22,23,24,25,26]”.
We have previously reviewed the effects of cellular metabolism and oxidative stress, and of the DNA damage response on HSC maintenance [20,26]. In this review, we will investigate three recent papers that have led us to reexamine the contributions of DNA damage repair to the development of malignancy, and also to reconsider the notion that HSCs are privileged to avoid genotoxic stress and the accumulation of DNA damage [27,28,29]. In the first, Santos, et al., the authors explored a role for the DNA damage response and DNA damage resistance in promoting the development of hematological malignancy [27]. This work is the culmination of accumulated evidence that has suggested a mechanism through which HSCs differentiate when faced with genotoxic stressors, likely to avoid the propagation of mutations in a population capable of self-renewal [18,19,20,21,22,23,24,25,26]. The second publication, Beerman, et al., describes the accumulation of DNA damage with age in quiescent HSCs due to an inefficient response to DNA damage [28]. The accumulation of damage and inefficient damage repair seems to be unique to quiescent HSCs in the hematopoietic system, is abrogated upon cell cycling, and may contribute to declining HSC function with age. Finally, in Flach, et al., the authors have identified accumulated DNA damage markers associated with replication stress in old HSCs that is distinct from DNA strand breaks [29]. In this publication, the Passegué group identifies downregulation of Minichromosome maintenance (Mcm) family genes as a mediator of replication stress in old HSCs, which contributes to the reduced function of aged HSCs. Together, these publications contribute greatly to our understanding of the role of DNA damage in aging and malignancy, and identify a number of pathways worthy of further investigation for their potential clinical importance.
2. The DNA Damage Response as a Potent Oncogenic Driver
Recently, the Nussenzweig group presented strong evidence in support of the hypothesis that DNA damage response (DDR) pathways might be cancer protecting in some developing malignancies [27]. This work presents an interesting contrast to other studies that demonstrate a cancer-protective role for DDR-associated genes, such as those associated with the ATM-CHK2-p53 (Ataxia telangiectasia mutated, Checkpoint kinase 2, and p53, respectively) pathway, and complicates our understanding of the role of DNA damage in malignancy [30,31]. In their paper, Santos, et al. identified mixed lineage leukemia 4 (MLL4) as a positive regulator of genes responsible for safeguarding cells against damaging ROS, and observed increased differentiation in Mll4−/− HSCs, consistent with previously reported observations of the effects of accumulated oxidative damage on HSCs [19,23,25,32]. Retroviral expression of MLL1-AF9 (ALL1-fused gene from chromosome 9, or MLLT3) in several hematopoietic compartments results in the generation of acute myeloid leukemia (AML) upon transplantation in mice, and has been used as a model to study AML and leukemia stem cells (LSCs, also known as leukemia-initiating cells) [33,34,35,36,37,38]. The Nussenzweig group demonstrated that Mll4-deficient colonies that express the MLL1-AF9 fusion oncogene contain mature cells, rather than undifferentiated blasts, and are unable to generate AML in mice upon transplantation. Further, they demonstrated that the Mll4-deficient cells expressing MLL1-AF9 displayed higher levels of ROS, as measured by CellROX staining, and higher levels of DNA damage, as indicated by increased Kap-1 staining and γH2AX foci. Treating Mll4-deficient MLL1-AF9 transformed cells with antioxidants (N-acetyl-l-cysteine, or NAC) partially rescues the leukemic phenotype. Additionally, ectopic expression of forkhead box O3 (FoxO3) rescues the leukemic phenotype, suggesting that differentiation in transformed MLL4-deficient cells occurs through target genes for FoxO family of transcription factors, which are responsible for mediating oxidative stress and have been studied in the context of maintaining HSC quiescence (Figure 1) [23,25,32]. Similarly, the Nussenzweig group demonstrated that oxidative stress contributes to differentiation of MLL1-AF9 transformed cells in the context of hydrogen peroxide treatment. Further, DNA damage alone, outside of the context of oxidative stress, is cytoprotective against the effects of the MLL1-AF9 fusion oncogene. Using a Brca1-deficient mouse model, which exhibits DNA damage independent of oxidative stress, and, separately, in a model of DNA double strand breaks using an ectopically expressed restriction endonuclease, Santos, et al. demonstrated that DNA damage alone can also lead to differentiation and exhaustion of MLL1-AF9 transformed leukemia. When DNA damage persists and is detected by cell-cycle checkpoint machinery, leukemic cells enter a differentiation program and lose some of their malignant potential. In their model of MLL1-AF9 transformation, differentiation that results from accumulated DNA damage is dependent on the cell cycle checkpoint protein p21Cip (Cdkn1a) [27]. When p21 is lost in the context of MLL1-AF9, cells are resistant to DNA damage associated growth inhibition and differentiation, consistent with previous reports that cell cycle elongation contributes to differentiation [39,40].
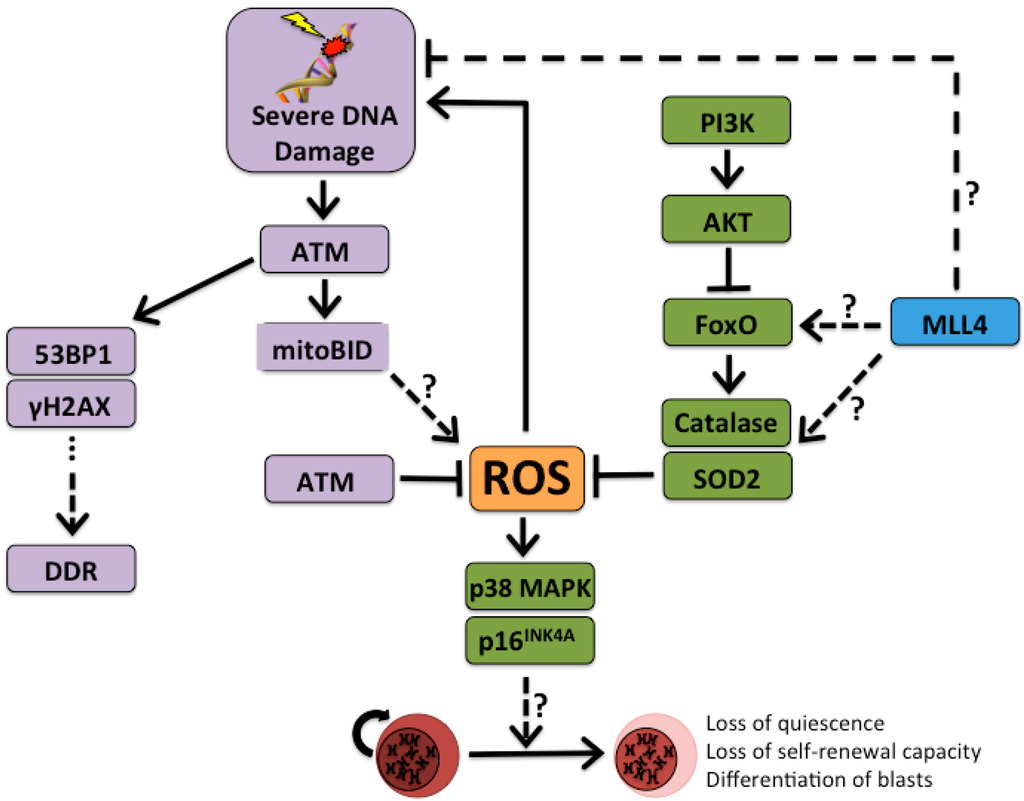
Figure 1.
The ROS rheostat of hematopoietic stem cell (HSC) maintenance. Accumulation of DNA damage and genotoxic oxidative stress contributes to a common pathway that leads to loss of self-renewal capacity of HSCs and leads HSCs to exit their quiescent state. This contributes to the gradual decline of functional HSCs in the bone marrow. Mixed lineage leukemia 4 (MLL4) activates forkhead box O (FoxO) targets through an unknown mechanism, and MLL4 expression is shown to be protective in the MLL1-AF9 (ALL1-fused gene from chromosome 9, or MLLT3) of AML by reducing the accumulation of ROS and, thus, DNA damage. Mll4-deficiency may also contribute to DNA damage through a ROS-independent mechanism. DNA damage results in the activation of ATM and, subsequently, DDR. Accumulation of γH2AX and co-localization with 53BP1 serve as markers of DDR, as in Flach, et al. Under normal conditions, ATM helps to maintain ROS at low levels. However, in the face of severe DNA damage ATM contributes to the accumulation of ROS and loss of quiescence in HSCs. ATM, ataxia telangiectasia mutated; FoxO, forkhead box O; DDR, DNA damage response; γH2AX, phosphorylated histone H2AX; MLL4, mixed-lineage leukemia 4; mitoBID, mitochondrial BH3 interacting-domain death agonist; MLL4, mixed-lineage leukemia 4; p38 MAPK, p38 mitogen-activated protein kinases; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; SOD2, superoxide dismutase 2; TP53BP1, tumor suppressor p53-binding protein 1. p16INK4A, cyclin dependent kinase inhibitor 2A; AKT, protein kinase 3. Solid arrows represent known mechanisms; dashed arrows labeled with question marks represent unknown mechanisms.
The demonstration that pathways that work to maintain genomic integrity are protective in this model of AML presents some interesting prospects for the treatment of these malignancies, namely through inhibition of the DNA damage repair initiators ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR). Treatment with these inhibitors contributes to an accumulation of mature cells and a loss of blasts in the context of MLL1-AF9 transformed cells, and MLL1-AF9 transformed Atm−/− bone marrow is incapable of maintaining leukemic self-renewal without differentiation [27]. In fact, an earlier study utilizing a model of hypomorphic ATR and ATR inhibitors supports the Nussenzweig group’s findings [41]. Additionally, Santos, et al. represents an advance in our understanding of the roles of ROS, DNA damage sensing, and cell-cycle checkpoints in differentiation and cell fate decisions in leukemia and in HSCs. There is much evidence supporting the idea that HSCs, when faced with DNA damage or genotoxic stress, differentiate to lineage-committed progenitors, and this may serve as a method to escape propagating damaged genetic information throughout the HSC pool and the hematopoietic system. Described another way, hematologic malignancies thrive on the failure of this escape mechanism, choosing DNA repair over differentiation, in order to maintain their self-renewal.
3. Sensing Stress and Quitting Quiescence
As previously mentioned, HSCs are particularly susceptible to DNA damage because of their longevity. Additionally, DNA damage in HSCs can be propagated throughout the HSC pool or to mature effector cells through self-renewing and differentiation divisions, respectively. In the face of genotoxic stress the accumulation of ROS serves as a rheostat in the differentiation decision, integrating information from a number of pathways (Figure 1).
Intracellular ROS are byproducts of aerobic metabolism in mitochondria, and may also originate from other organelles [42,43]. DNA is highly susceptible to oxidative damage, which can result in single and double strand breaks (SSBs and DSBs), base and sugar-moiety oxidation, strand crosslinks and the generation of abasic sites [7,8,17,20,44,45]. The initial steps in detection of strand breaks do not require discussion here. Phosphatidylinositol 3 kinase-related kinase (PIKK) family members, the checkpoint kinases ATM and ATR, are recruited to the site of the damage and activated. These enzymes phosphorylate a number of targets initiating signaling cascades that mediate cell cycle arrest and DDR [46,47]. ATM can also be activated to induce DDR in the context of oxidative stress, thus serving as sensor of reactive oxygen species. ATM itself can be oxidized, yielding a disulfide-crosslinked ATM dimer that activates DDR in the absence of DSBs, in contrast to the active monomer generated in the context of DSBs [48]. ATR activation by SSBs, which result from oxidative stress-induced abasic sites, utilizes a unique Apurinic/apyrimidinic (AP) endonuclease 2 (APE2)-dependent mechanism [49]. APE2 participates in the removal of the damaged 3' terminus, generating a 3'–5' SSB end resection and allowing for the recruitment of ATR. Among the downstream targets of ATM are Histone H2AX (H2A histone family, member X), which is phosphorylated at serine 139 to become phosphorylated histone H2AX (γH2AX), and tumor suppressor p53 binding protein 1 (TP53BP1). Due to these interactions, γH2AX staining is commonly used as an indirect marker of DNA damage, and co-localization of TP53BP1 can sometimes be detected in γH2AX foci [27,29]. Studies have demonstrated that ATM is also a powerful mediator of ROS homeostasis, which contributes to its ability to mediate the differentiation decision in response to genotoxic stress. Atm−/− mice suffer from an accumulation of ROS that is mediated by treatment with the antioxidant NAC [50,51,52,53]. The mechanism through which ATM suppresses the accumulation of ROS is unclear. Under normal conditions, phosphorylation of BH3-interacting domain death agonist (BID) by ATM restricts BID to the nucleus and is essential in maintaining HSC quiescence, possibly through and ROS dependent mechanism [21,22]. In the context of severe DNA damage, BID and phosphorylated BID translocate to the mitochondria and result in the accumulation of ROS. Additionally, Atm−/− mice have a reduced reconstitution capacity of HSCs, and elevated levels of the Rb and p53 activators p16INK4A (cyclin-dependent kinase inhibitor 2A) and p19ARF (alternate reading frame tumor suppressor), which contribute to the loss of HSCs—both observations can be rescued with NAC treatment, demonstrating a role for ROS deregulation in the development of this phenotype [18,54] In the context of elevated ROS, p16INK4A levels seem to regulated by the intermediary p38 mitogen-activated protein kinases (p38 MAPK), and p38 MAPK inhibition rescues the hematopoietic phenotype observed in Atm−/− mice [19].
The family of forkhead box binding O (FoxO) proteins is one of the earliest families of transcription factors that have been implicated in sensing and responding to oxidative stress in HSCs. In 2007, both the Gilliland group, using a triple conditional knockout of FoxO1, FoxO3 and FoxO4, and the Hirao and Suda groups, using FoxO3a-deficient mice, reported on the necessity of FoxOs in the maintenance of HSCs [23,25]. The FoxO transcription factors are responsible for positively regulating the expression of oxidative stress mitigating proteins, including superoxide dismutase and catalase enzymes [23,24,25]. FoxO-deficient mice exhibit a decrease in HSC number, a loss of HSC quiescence, and HSCs from FoxO-deficient mice are incapable of reconstituting bone marrow upon transplantation [25]. These defects can be rescued through treatment with NAC and are the result of reduced expression of ROS neutralizing enzymes [25]. Similarly, FoxO3a−/− mice show reduced levels of the ROS scavenging enzymes Superoxide dismutase 2 (SOD2) and catalase, which are positively regulated by FoxO3a, and show loss of HSC function [23,32]. FoxO family proteins are inhibited by exclusion from the nucleus as a result of phosphorylation by AKT, downstream of the PI3K-AKT phosphorylation cascade [55]. The negative effects to HSC number and function that are observed when FoxOs are inhibited strongly suggest a role for accumulated ROS in the decision for an HSC to differentiate.
4. Replication: A Source for Rejuvenation or Stress in Aged Hematopoietic Stem Cells (HSCs)
In the past year, the Rossi and Passegué groups both published dogma-challenging work on the genoprotection of aging HSCs in the bone marrow niche [28,29]. In mice, HSC aging contributes to reduced engraftment upon transplantation, and impaired lymphoid differentiation upon transplantation and in steady state hematopoiesis [56]. In humans, HSC aging is thought to predispose HSCs to the accumulation of malignancy driving mutations. Through studies examining γH2AX, a marker of DNA damage sensing, and examining genetic models of DDR deficiency, it has already been suggested that DNA damage accumulates in old HSCs and contributes to their reduced function with age [57,58,59]. In their recent work, the Rossi group took advantage of the single-cell alkaline comet assay, an older technique which provides a direct measure of DNA single and double strand breaks (Figure 2), in order to demonstrate increased levels of accumulated damage in aged HSCs [60,61,62]. In this assay, isolated cells are mixed with low-temperature gelling agarose, deposited on a glass slide, lysed under alkaline conditions, and subjected to a brief electrophoresis [60]. Staining the slides with a DNA labeling compound, such as SYBR Green, allows for DNA to be visualized microscopically and analyzed with specifically developed software—the tail of the comet in this assay consists of leading strands of DNA that result from single and double strand breaks, whereas unbroken DNA fails to migrate due to its high molecular weight [60]. In Beerman, et al., the authors demonstrate that there are more cells with accumulated moderate-to-severe DNA damage in HSCs from aged mice (24–26 months) as compared to HSCs from young mice (3–4 months), though moderate to severe damage can be detected in young HSCs and undamaged HSCs can be identified in the aged cohort [28]. While hematopoietic progenitors from aged mice also show evidence of accumulated DNA damage, the damage is clearly more severe in the HSC compartment, as evidence by a higher percentage of moderate-to-severe damaged HSCs, suggesting that HSCs are especially susceptible to accumulating DNA damage [28]. Accumulated DNA damage in old HSCs is repaired when the HSCs are caused to cycle. Old HSCs that are cultured in cytokine rich medium for 24 h resemble similarly treated young HSCs, and even display less DNA damage compared to young HSCs at steady state, suggesting that cell cycle entry represents a potent driver of DNA damage repair, which was absent or reduced in quiescent HSCs [28]. Of note, however, is that aged HSCs display a reduced proliferative potential in long-term culture, presumably after DNA damage has been repaired, as compared to young HSCs. HSCs in aged mice treated with 5-fluorouracil, as well as aged HSCs allowed to engraft upon bone marrow transplantation, display significantly reduced DNA damage [28]. Here too, despite damage repair, aged HSCs have reduced reconstitution capacity with a myeloid bias upon transplantation, similar to those observations made in in vitro experiments and consistent with a reduction of HSC function with age. Thus, the DNA damage repair observed upon cell-cycle reentry is not sufficient to completely restore hematopoietic stem cell function.
The Rossi group demonstrated that hematopoietic progenitors, as compared to stem cells, exhibit increased expression of DDR associated genes, which is supported by previous evidence that DDR is tightly linked with cell cycle entry and that progenitors are actively cycling as compared to the largely quiescent HSCs [13,14,63,64]. Fetal liver HSCs, which are actively cycling, as well as young and aged HSCs stimulated to cycle in cytokine-rich medium, demonstrated increased expression of DDR associated genes [28,65]. Interestingly, genes associated with non-homologous end joining (NHEJ), an error-prone mechanism of DNA damage repair, are expressed at similar levels between HSCs and progenitors [28,66]. The authors suggest that NHEJ must not be active in quiescent HSCs, as active NHEJ would be inconsistent with the observed level of DSBs. Instead, they propose a hypothesis that quiescent HSCs are primed to undergo NHEJ upon cell cycle reentry. The authors conclude that DDR pathways are reduced in quiescent HSCs, contributing to the increased DNA damage in young and aged HSCs, as compared to progenitors, observed by the comet assay. Because HSCs remain quiescent for most of adult life they accumulate mutations that limit HSC function with age, despite the repair mechanisms that are initiated upon cell cycle reentry [67].
The persistence of DNA damage and inability to completely restore hematopoietic function, despite activation of DNA repair mechanisms at cell-cycle reentry may be reflected in reports of the accumulation of leukemogenic mutations in HSCs that contribute to the development of “pre-leukemic HSCs” observed by the Majeti group [68,69]. Jan, et al. used a previously described strategy to prospectively isolate residual HSCs from AML samples using T-cell immunoglobulin mucin 3 (TIM3) and other surface markers [70,71]. Exome sequencing and transcriptome analysis of AML cells and residual HSCs revealed genetic mutations that are frequently associated with AML in residual HSCs, strongly supporting the existence of pre-leukemic HSCs in the phenotypically normal HSC population [68]. Further, sequencing of colonies formed from single residual HSCs allowed the Majeti group to observe the sequential accumulation of frequently occurring AML mutations in residual HSCs. This work provides strong evidence in support of a model of sequential accumulation of leukemogenic mutations in self-renewing HSCs contributing to the generation of pre-leukemic HSCs and eventually frank leukemia, however no experimental evidence showing that the accumulation of additional mutations in residual HSCs/pre-leukemic HSCs leads to leukemia is provided in this study. While the recent work from the Rossi group does not provide a clinical avenue to avoid or reduce the accumulation of DNA damage in aging HSCs, it does support the hypothesis that leukemogenic mutations accumulate in HSCs in a stepwise fashion. Treatments that target early mutations found in pre-leukemic HSCs would likely result in increased disease free survival in AML and in other hematologic malignancies.
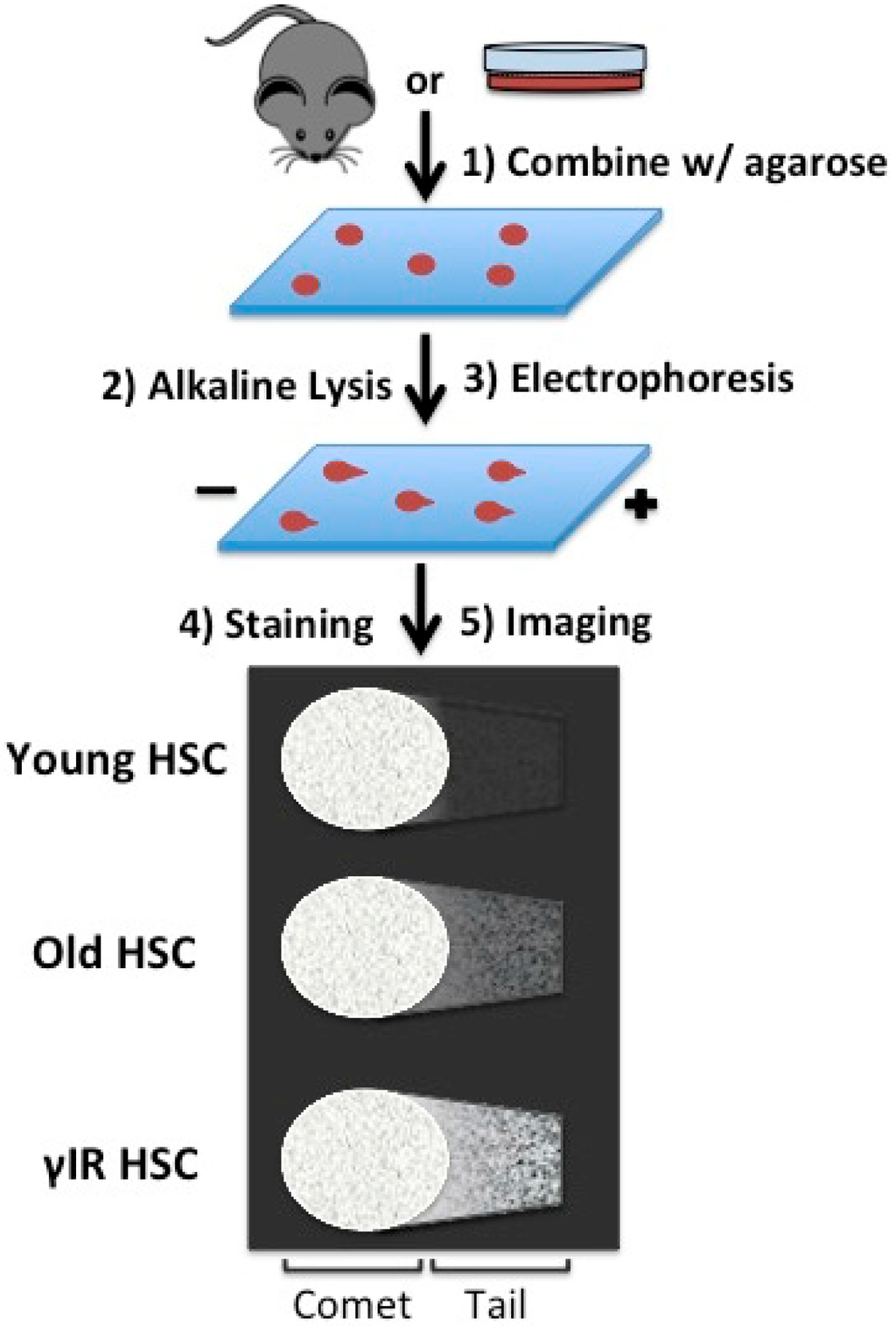
Figure 2.
The alkaline comet assay. The alkaline comet assay allows for the direct microscopic measurement of DNA damage. Isolated cells are mixed with low-temperature gelling agarose and applied to a glass slide. Cells are lysed under alkaline conditions in order to detect single and double strand breaks, though a number of lysis procedures have been described for other purposes. After a brief electrophoresis and staining, DNA damage can be visualized microscopically. High molecular weight DNA, reflecting undamaged DNA, remains in the comet, whereas damaged DNA is susceptible to the electrophoretic field and is found in the tail. A number of methods for quantifying and describing the tail to comet relationship have been described. In Beerman, et al. the authors describe a higher frequency of HSCs with moderate-to-severe DNA damage in aged mice as compared to younger mice [28]. Damage is also more severe among aged hematopoietic stem cells as compared to aged hematopoietic progenitors.
A publication from the Passegué group, also from the past year, suggests that aged HSCs are more susceptible to DNA damage associated with replication stress [29]. This finding draws in to question the strong reparative effects of cell cycling observed in Beerman, et al., and deserves a closer examination.
In Flach, et al., the authors explore the ability of HSCs from aged mice (22–30 months old, identified here as “aged/old HSCs”) to undergo DNA damage repair, as compared to HSCs from young mice (6–12 weeks old, identified here as “young HSCs”) [29]. The authors confirmed that γH2AX is accumulated in aged HSCs, but were unable to observe a significantly elevated tail moment upon alkaline comet assay, as was observed by the Rossi group [27,29]. However, the authors observed that cultured old and young HSCs were similarly capable to clear γH2AX foci after irradiation, albeit old HSCs exhibit an ~8 h delay, and express homologous repair (HR) and NHEJ genes at similar levels to young HSCs [29]. If old HSCs display a similar ability to cope with DNA damage as young HSCs, this raises the question of what is responsible for the accumulation of γH2AX observed by many groups in aged HSCs? Further investigation would clarify this point.
ATR and γH2AX foci have also been reported to accumulate at replication forks, in a DNA damage independent mechanism (Table 1) [45,72]. Consistently, the Passegué group observed signs of increased single-stranded DNA in aged HSCs, namely increased staining for the single-stranded DNA binding proteins ATR interacting protein (ATRIP) and replication protein A (RPA). These findings are suggestive of replication stress in aged HSCs that would explain the increased γH2AX foci, and explain the delayed cell division kinetics observed in EdU/BrdU (5-Ethynyl-2'-Deoxyuridine and 5-Bromo-2'-Deoxyuridine, respectively) incorporation experiments due to delayed onset of S phase and elongated S phase [29,73]. Aged HSCs were also observed to have delayed first and second divisions in culture. Additionally, the Passegué group reported the accumulation of chromosomal gaps and breaks in the daughter cells of cultured old HSCs. Interestingly, they found no evidence of NHEJ, specifically no chromosomal deletions or translocations, in expanded cultures of old HSCs, in contrast of what one might expect of DDR in HSCs, which are typically quiescent [66,74].

Table 1.
DNA damage and DDR associated molecules. Flach, et al. utilized a variety of antigens to examine DNA damage, DDR, replication stress and ribosome biosynthesis by immunofluorescence [29]. A number of these markers are reproduced and described here.
| Antigen | Abbreviation | Indication | Notes | References | |
|---|---|---|---|---|---|
| Gamma-H2AX | γH2AX | DSB single stranded DNA | Target of ATM/ATR may serve as repressive epigenetic mark in quiescent aged HSCs | [29,75,76,77,78,79] | |
| Tumor suppressor p53-binding protein 1 | TP53BP1 | DSB | Target of ATM/ATR | [80,81,82] | |
| Phosphorylated checkpoint kinase 1 | pCHK1 | DNA damage repair | Target of ATR mediates cell cycle arrest | [83,84,85,86,87] | |
| Phosphorylated ataxia-telangiectasia mutated | pATM | DNA damage DSBs | Mediates DDR mediates redox homeostasis in HSCs | [18,22,30,88,89,90,91] | |
| Poly (ADP-ribose) | PAR | Single stranded DNA break | Signals for single strand break repair synthesized by PARP | [92,93] | |
| Replication protein A | RPA | Binds single stranded DNA | Prevents formation of secondary structures during replication | [94,95] | |
| ATR interacting protein | ATRIP | Binds RPA coated single stranded DNA | Associates with ATR, leading to its accumulation at intranuclear DNA damage foci | [96,97,98] | |
| Fibrillarin | FBL | Ribosome biosynthesis | Fibrillarin component of SnRNPs | [99,100,101] | |
| Upstream binding factor | UBF | Ribosome biosynthesis | Upstream binding factor transcription factor of rRNAs | [102] | |
| Nucleolin | NCL | Ribosome biosynthesis | Nucleolin invlolved in ribosome synthesis | [103,104] | |
| Nuclear serine/threonine protein phosphatase 4 catalytic subunit | nPP4c | γH2AX phosphatase | [105,106] | ||
The minichromosome maintenance (MCM) family of genes encodes the subunits of the hexameric MCM DNA helicase, a member of the CDC45–MCM–GINS (cell division control protein 45, minichromosome maintenance, Go-Ichi-Ni-San, respectively) pre-replication complex that is responsible for unwinding DNA at replication forks during the replication initiation [107]. Examination of differentially expressed genes in old HSCs identified the Mcm2–7 as down-regulated, whereas no other members of the pre-replication complex were deregulated [29]. Treating old HSCs with aphidicolin, a DNA polymerase inhibitor, resulted in dramatic accumulation of γH2AX foci and impaired stem cell function as compared to young HSCs treated with aphidicolin, consistent with observations of replication stress in HeLa cells subjected to MCM knockdown by siRNA [108]. Additionally, knockdown of MCM components in young HSCs replicates the stem cell defects observed in old HSCs in culture and upon transplantation [29]. In fact, treatment of young HSCs with aphidicolin, but without modifying MCM levels, also reduces the reconstitution capacity, suggesting a powerful role for replication stress in HSC function.
In quiescent aged HSCs, in which γH2AX foci cannot be explained by replication stress, the Passegué group identified γH2AX accumulation at nucleoli, the sites of ribosome biogenesis, by fluorescent in situ hybridization (FISH) for rDNA genes. The authors suggest that γH2AX scars likely originate from replication stress at rRNA encoding regions, which are difficult to replicate, and may persist because of localization γH2AX phosphatase PP4c in the cytoplasm of quiescent old HSCs [109]. When old quiescent HSCs reenter the cell cycle, nucleolar γH2AX are lost. However, many months after transplantation, when HSCs may have returned to quiescence, nucleolar γH2AX foci can be observed. Interestingly, though no gene regulatory role for γH2AX has been previously identified, γH2AX accumulation at rDNA was associated with decreased expression of ribosomal components, which the authors propose is a non-canonical function of γH2AX to repress transcription in regions undergoing active DNA repair. Decreased ribosomal synthesis in quiescent older HSCs may contribute to decreasing HSC function with age.
This study suggests that identifying a therapeutic strategy to restore MCM expression and activity may prove useful in slowing stem cell decline and rejuvenating an aged HSC compartment. Further, reduction of γH2AX in quiescent aged HSCs might contribute to improved HSC function with age, however additional studies of the role of ribosomal biogenesis stress on quiescent HSCs are needed.
5. Conclusions
DNA damage to HSCs that persists in the stem cell compartment can be deleterious to hematopoietic function and can promote malignancy. Because HSCs have the ability to self-renew and to differentiate, they are thought to be responsible for maintaining genomic integrity of the hematological system, and it had been proposed that HSCs were protected from genomic damage by their hypoxic microenvironment and possibly by cell intrinsic factors. Mounting evidence, including the recent works discussed here, suggests that HSCs, due to their longevity and quiescence, are highly susceptible to accumulating DNA damage. Rather than allow damage to persist in the stem cell compartment where damage can be propagated throughout the hematopoietic system, HSCs faced with DNA damage and genotoxic stress, such as ROS, differentiate. This would serve to restrict the ability of damage to move through the hematopoietic population. To that end, DNA damage repair, commonly the error prone NHEJ in HSCs, might be deleterious to the hematopoietic system and can contribute to the development of malignancy. This is supported by Santos, et al., which identifies mechanisms that reduce oxidative stress and repair DNA damage as protective in the MLL1-AF9 model of AML. When DNA repair mechanisms are inhibited, MLL1-AF9 transformed leukemia is pushed towards differentiation and blasts are lost. With supporting clinical evidence that indicates that malignancy is more sensitive to loss of DDR mechanisms than non-malignant tissues, inhibition of DDR may prove to be a valuable therapeutic option in the treatment of AML that could work similarly to all-trans retinoic acid (ATRA) in the treatment of acute promyelocytic leukemia (APL or AML M3), which pushes leukemic cells towards differentiation.
The Rossi and Passegué groups used different measures of genomic integrity to investigate the role of DNA damage in aging. In Beerman, et al., the authors measured DNA damage directly using the alkaline comet assay and concluded that DNA damage accumulates in quiescent aged HSCs and is repaired upon cell cycle reentry. Upon cell cycle reentry, DNA damage profiles by alkaline comet assay in aged HSCs resemble those of young HSCs, however stem cell function is somewhat decreased in aged HSCs. The persistence of DNA damage in HSCs, perhaps a result of the propensity for HSCs to utilize error-prone NHEJ, may contribute to the stepwise accumulation of leukemogenic mutations and the establishment of pre-leukemic HSCs. In Flach, et al., accumulated γH2AX foci in aged HSCs are attributed to replication stress that results from downregulation of replication helicase machinery in aged HSCs. Interestingly, these two publications include some different findings, which will surely be rectified with continued investigation. Further, the Passegué group identified γH2AX scars at rDNA in quiescent aged HSCs. In this context, γH2AX appears to play a previously undescribed epigenetic role by repressing ribosome synthesis. Further investigations may reveal that this ribosome biogenesis stress may contribute to declining HSC function with aging.
Despite their importance in maintaining the genomic integrity of the hematopoietic system, HSCs are uniquely sensitive to DNA damage and the accumulation of mutation. This is due to their longevity and due to decreased DNA damage repair associated with quiescence. Stepwise accumulation of mutations, as well as replication stress is associated with the functional decline of HSCs with age. Accumulated mutations in HSCs sometimes persist, despite mechanisms that work to remove genotoxically stressed HSCs from the stem cell pool through loss of quiescence and self-renewal. For this reason, DDR mechanisms appear to be oncogenic in HSCs. These publications provide new insights into the accumulation of DNA damage in HSCs and the role of DNA damage and replication stress in the functional decline of hematopoiesis, but also suggest additional questions and areas that require further examination.
Acknowledgments
We are thankful to the Ito lab members for comments and discussion on the effect of reactive oxygen species in stem cells. Keisuke Ito is supported by grants from the National Institutes of Health (NIH) (R00CA139009, R01DK98263, R01DK100689). Cary N. Weiss is supported by an NIH Medical Scientist Training Program (MSTP) training grant (T32GM007288). Many original articles were omitted due to space limitations; for this, we apologize.
Author Contributions
Cary N. Weiss wrote and edited the paper, and generated the figures. Keisuke Ito edited the paper and initiated discussions essential in the conception and creation of this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Hanada, K.; Hamada, H.; Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996, 273, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. Slam family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.H.; Kiel, M.J.; Morrison, S.J. Slam family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood 2006, 107, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in BLAST-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hirao, A. Maintenance of genomic integrity in hematopoietic stem cells. Int. J. Hematol. 2011, 93, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Biol. 2013, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by atm is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Oberkovitz, G.; Niv, H.; Vorobiyov, L.; Zaltsman, Y.; Brenner, O.; Lapidot, T.; Jung, S.; Gross, A. The Atm-bid pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 2012, 14, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. FoxO3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Gilliland, D.G. FoxO transcription factors and stem cell homeostasis: Insights from the hematopoietic system. Cell Stem Cell 2007, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.N.; Ito, K. DNA damage response, redox status and hematopoiesis. Blood Cells Mol. Dis. 2014, 52, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Bakkenist, C.J.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Sehested, M.; Lukas, J.; Kastan, M.B.; Bartek, J. Atm activation in normal human tissues and testicular cancer. Cell Cycle 2005, 4, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FoxO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.; King, G.; Rabbitts, T.H. An MLL-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Cozzio, A.; Passegue, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef] [PubMed]
- Ugale, A.; Norddahl, G.L.; Wahlestedt, M.; Sawen, P.; Jaako, P.; Pronk, C.J.; Soneji, S.; Cammenga, J.; Bryder, D. Hematopoietic stem cells are intrinsically protected against MLL-ENL-mediated transformation. Cell Rep. 2014, 9, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Figueroa, M.E.; Sinha, A.U.; Stubbs, M.C.; Feng, Z.; Valk, P.J.; Delwel, R.; Dohner, K.; Bullinger, L.; Kung, A.L.; et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 2013, 27, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Q.; Morita, Y.; Jiang, H.; Gross, A.; Lechel, A.; Hildner, K.; Guachalla, L.M.; Gompf, A.; Hartmann, D.; et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012, 148, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Aoto, T.; Binh, N.T.; Okamoto, N.; Tanimura, S.; Wakayama, T.; Iseki, S.; Hara, E.; Masunaga, T.; Shimizu, H.; et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 2009, 137, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ROS crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. Atm activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-CHK1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef] [PubMed]
- Takao, N.; Li, Y.; Yamamoto, K. Protective roles for atm in cellular response to oxidative stress. FEBS Lett. 2000, 472, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Kamsler, A.; Daily, D.; Hochman, A.; Stern, N.; Shiloh, Y.; Rotman, G.; Barzilai, A. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from atm-deficient mice. Cancer Res. 2001, 61, 1849–1854. [Google Scholar] [PubMed]
- Schubert, R.; Reichenbach, J.; Royer, N.; Pichler, M.; Zielen, S. Spontaneous and oxidative stress-induced programmed cell death in lymphocytes from patients with ataxia telangiectasia (AT). Clin. Exp. Immunol. 2000, 119, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Takao, N.; Kato, H.; Mori, R.; Morrison, C.; Sonada, E.; Sun, X.; Shimizu, H.; Yoshioka, K.; Takeda, S.; Yamamoto, K. Disruption of atm in p53-Null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene 1999, 18, 7002–7009. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H.; Iwama, A.; Morita, Y.; Kamijo, T.; van Lohuizen, M.; Nakauchi, H. Differential impact of INK4A and ARF on hematopoietic stem cells and their bone marrow microenvironment in BMI1-deficient mice. J. Exp. Med. 2006, 203, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.; Morita, Y.; Eto, K.; Ema, H.; Nakauchi, H. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006, 25, 3515–3523. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Jamieson, C.H.M.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Fricke, A.; Widmann, T.A.; Furst, T.; Madry, H.; Pfreundschuh, M.; Rube, C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One 2011, 6, e17487. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ostling, O.; Johanson, K.J. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem. Biophys. Res. Commun. 1984, 123, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Foudi, A.; Hochedlinger, K.; Van Buren, D.; Schindler, J.W.; Jaenisch, R.; Carey, V.; Hock, H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 2009, 27, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Passegue, E.; Wagers, A.J.; Giuriato, S.; Anderson, W.C.; Weissman, I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005, 202, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Bowie, M.B.; McKnight, K.D.; Kent, D.G.; McCaffrey, L.; Hoodless, P.A.; Eaves, C.J. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Investig. 2006, 116, 2808–2816. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of tim3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA 2011, 108, 5009–5014. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. Atm phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi, T.; Wang, B.; Liu, Q.; Nenoi, M.; Onoda, M. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem. 2014, 156, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. P53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Henderson, C.; Adachi, Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 2001, 21, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Gatei, M.; O'Connell, M.J.; Khanna, K.K.; Bugg, S.J.; Hogg, A.; Scott, S.P.; Hobson, K.; Lavin, M.F. Chk1 complements the G2/M checkpoint defect and radiosensitivity of ataxia-telangiectasia cells. Oncogene 1999, 18, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Walworth, N.C.; Bernards, R. Rad-dependent response of the CHK1-encoded protein kinase at the DNA damage checkpoint. Science 1996, 271, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Flaggs, G.; Plug, A.W.; Dunks, K.M.; Mundt, K.E.; Ford, J.C.; Quiggle, M.R.; Taylor, E.M.; Westphal, C.H.; Ashley, T.; Hoekstra, M.F.; et al. ATM-dependent interactions of a mammalian CHK1 homolog with meiotic chromosomes. Curr. Biol. 1997, 7, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.H.; Walworth, N.C. Association of CHK1 with 14–3-3 proteins is stimulated by DNA damage. Genes Dev. 1999, 13, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Sloper, K.; Sorensen, C.; Syljuasen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.B.; Bartek, J.; et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of CHK1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Kamer, I.; Sarig, R.; Zaltsman, Y.; Niv, H.; Oberkovitz, G.; Regev, L.; Haimovich, G.; Lerenthal, Y.; Marcellus, R.C.; Gross, A. Proapoptotic bid is an ATM effector in the DNA-damage response. Cell 2005, 122, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Takubo, K.; Arai, F.; Satoh, H.; Matsuoka, S.; Ohmura, M.; Naka, K.; Azuma, M.; Miyamoto, K.; Hosokawa, K.; et al. Regulation of reactive oxygen species by atm is essential for proper response to DNA double-strand breaks in lymphocytes. J. Immunol. 2007, 178, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Malanga, M.; Pleschke, J.M.; Kleczkowska, H.E.; Althaus, F.R. Poly(adp-ribose) binds to specific domains of p53 and alters its DNA binding functions. J. Biol. Chem. 1998, 273, 11839–11843. [Google Scholar] [CrossRef] [PubMed]
- Kawamitsu, H.; Hoshino, H.; Okada, H.; Miwa, M.; Momoi, H.; Sugimura, T. Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry 1984, 23, 3771–3777. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Rao, M.R.; Erdile, L.F.; Kelly, T.J.; Kolodner, R.D. An essential saccharomyces cerevisiae single-stranded DNA binding protein is homologous to the large subunit of human RP-A. EMBO J. 1990, 9, 2321–2329. [Google Scholar] [PubMed]
- Wold, M.S.; Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-SSDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Kacmaz, K.; Sancar, A. Quaternary structure of atr and effects of atrip and replication protein a on its DNA binding and kinase activities. Mol. Cell. Biol. 2004, 24, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Bomgarden, R.D.; Yean, D.; Yee, M.C.; Cimprich, K.A. A novel protein activity mediates DNA binding of an ATR-ATRIP complex. J. Biol. Chem. 2004, 279, 13346–13353. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.P.; Hurt, E.C.; Kern, H.; Lehtonen, H.; Carmo-Fonseca, M.; Lapeyre, B.; Tollervey, D. Evolutionary conservation of the human nucleolar protein fibrillarin and its functional expression in yeast. J. Cell Biol. 1991, 113, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, D.; Lehtonen, H.; Carmo-Fonseca, M.; Hurt, E.C. The small nucleolar RNP protein NOP1 (fibrillarin) is required for pre-rrna processing in yeast. EMBO J. 1991, 10, 573–583. [Google Scholar] [PubMed]
- Politz, J.C.; Lewandowski, L.B.; Pederson, T. Signal recognition particle rna localization within the nucleolus differs from the classical sites of ribosome synthesis. J. Cell Biol. 2002, 159, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Learned, R.M.; Learned, T.K.; Haltiner, M.M.; Tjian, R.T. Human rRNA transcription is modulated by the coordinate binding of two factors to an upstream control element. Cell 1986, 45, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Bugler, B.; Caizergues-Ferrer, M.; Bouche, G.; Bourbon, H.; Amalric, F. Detection and localization of a class of proteins immunologically related to a 100-kDa nucleolar protein. FEBS J. 1982, 128, 475–480. [Google Scholar] [CrossRef]
- Borer, R.A.; Lehner, C.F.; Eppenberger, H.M.; Nigg, E.A. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989, 56, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Keogh, M.C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. γ-H2AX dephosphorylation by protein phosphatase 2a facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Chen, G.I.; Gingras, A.C.; Durocher, D. PP4 is a γH2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008, 9, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Guillou, E.; Coloma, J.; Montoya, G.; Mendez, J. The human gins complex associates with CDC45 and MCM and is essential for DNA replication. Nucleic Acids Res. 2009, 37, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Mendez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Ann. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).