
Contribution of the Type II Chaperonin, TRiC/CCT, to Oncogenesis

Abstract
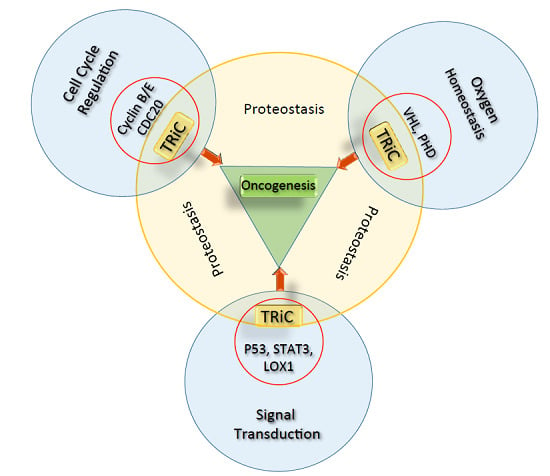
:
{kind=link}
{kind=link}
{kind=link}
1. Molecular Chaperones and Oncogenesis
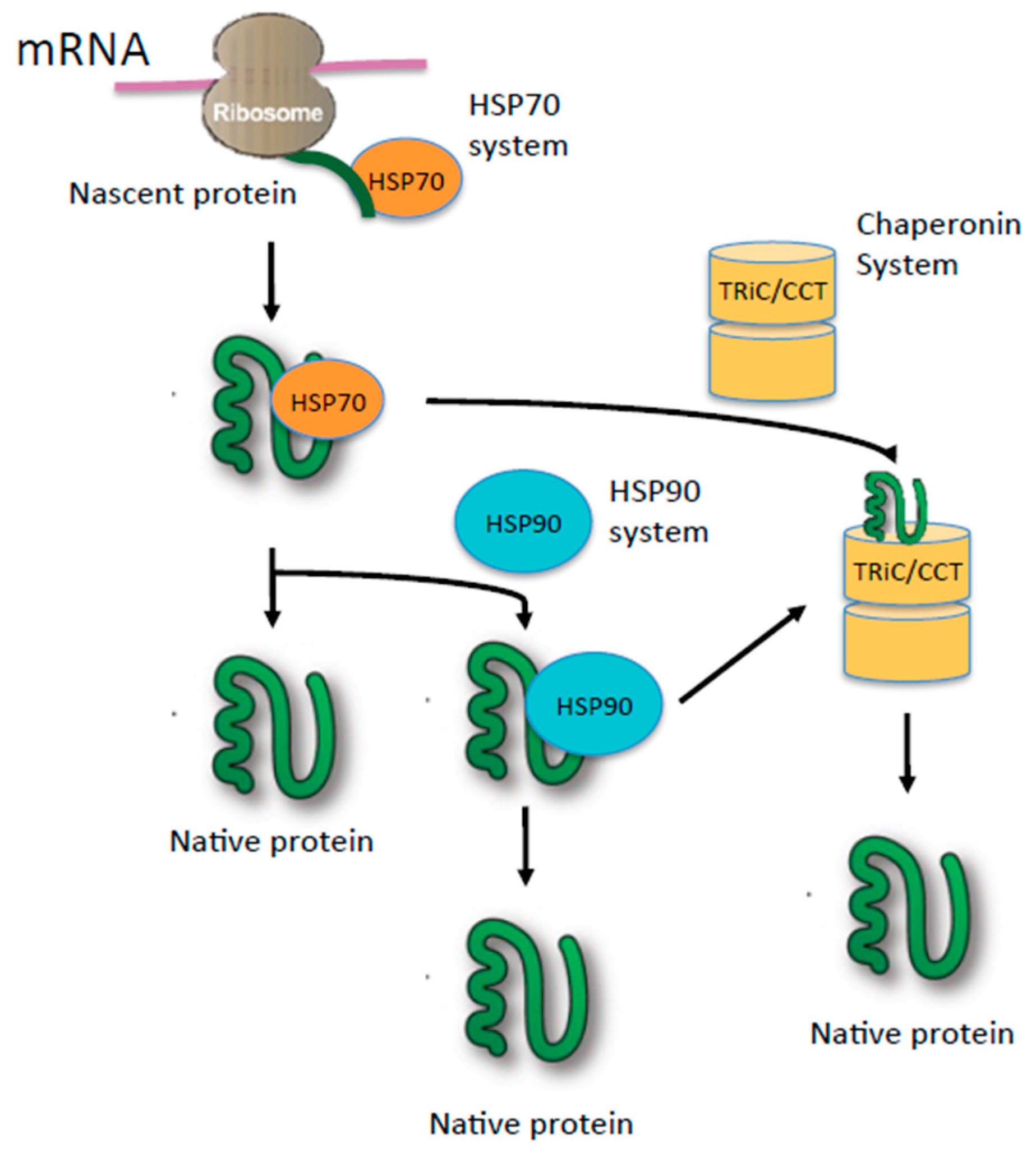
2. Role of Heat Shock Proteins (HSPs) in Oncogenic Signaling
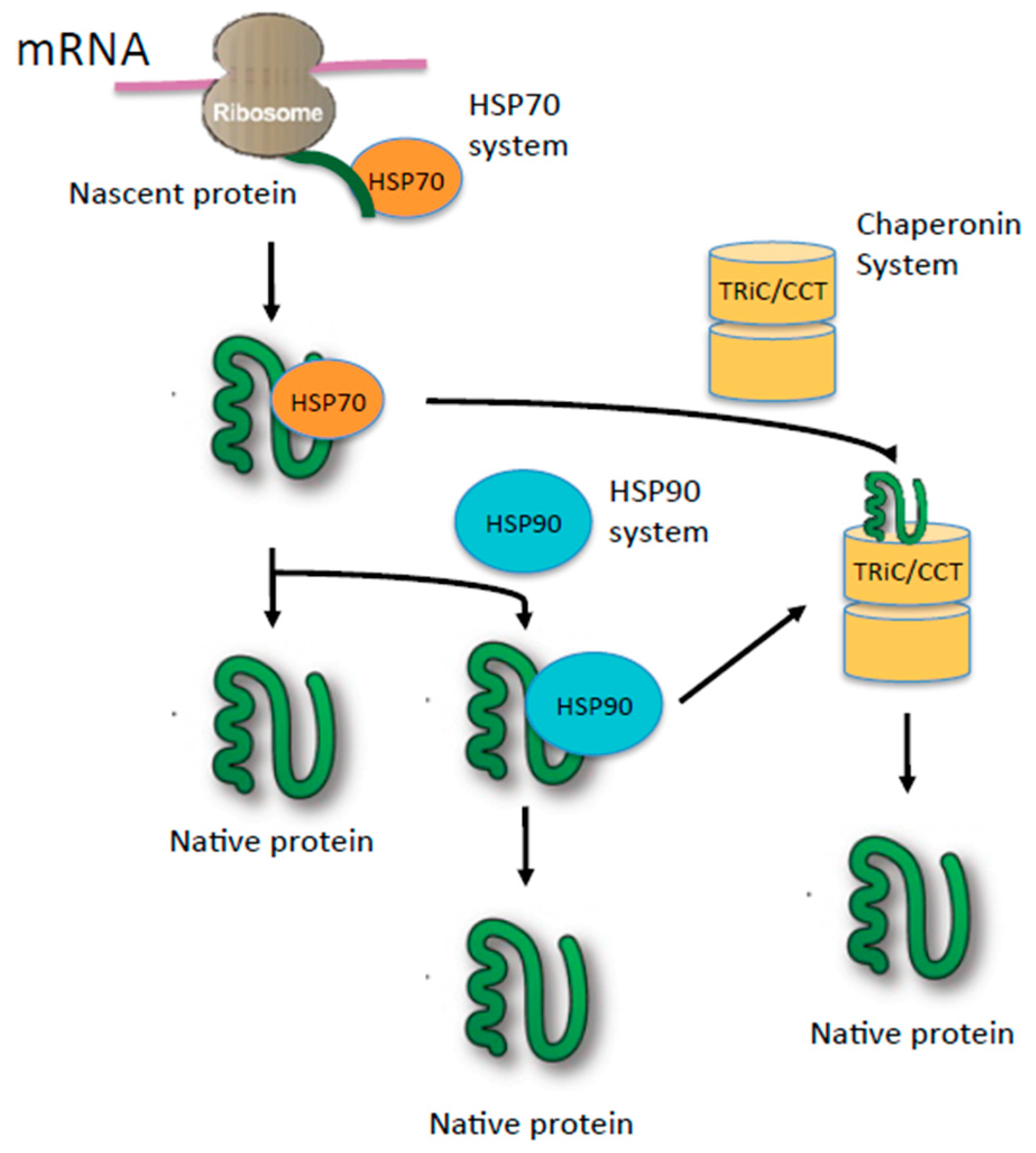
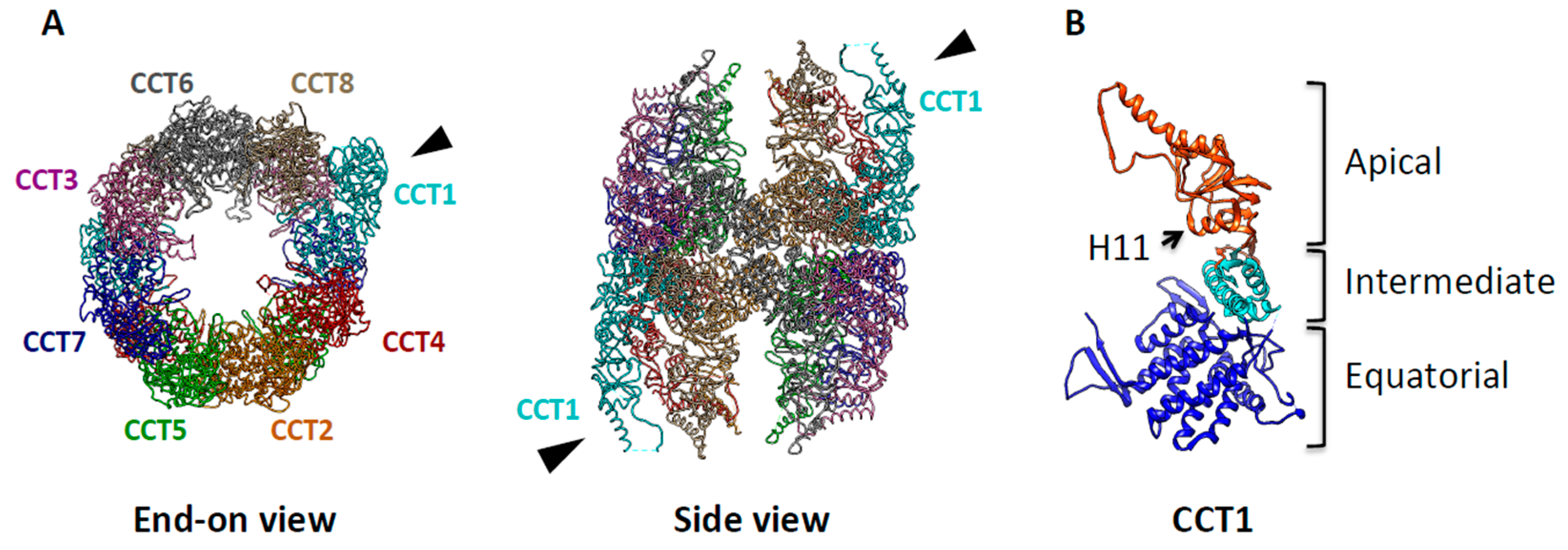
3. TRiC: The Protein-Folding Machine in Eukaryotes
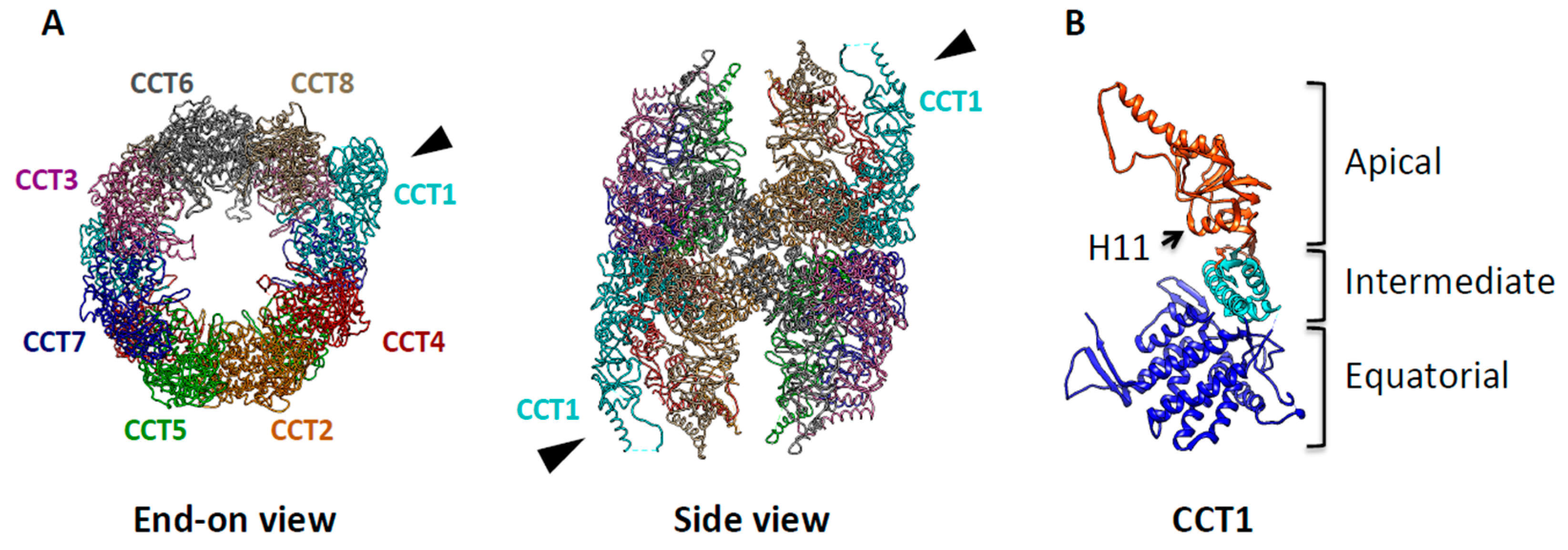
4. TRiC Binds to and Modulates Cancer Related Proteins
4.1. Chaperonin TRiC Works as an Assembly Station for the Tumor Suppressor, VHL (Von Hippel-Lindau)
4.2. TRiC Contributes to STAT Protein Folding and Function
4.3. Interaction of TRiC with p53 Promotes the Protein Folding and Activity of p53
4.4. TRiC Modulates Cell Cycle Regulatory Proteins
4.5. Contribution of LOX-1, a Newly Identified TRiC Substrate, to Inflammation and Oncogenesis
5. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S.; et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, M.; Lau, W.C.; Roh, S.H.; Eckols, T.K.; Frydman, J.; Chiu, W.; Tweardy, D.J. Modulation of STAT3 folding and function by TRiC/CCT chaperonin. PLoS Biol. 2014, 12, e1001844. [Google Scholar] [CrossRef] [PubMed]
- Won, K.-A.; Schumacher, R.J.; Farr, G.W.; Horwich, A.L.; Reed, S.I. Maturation of Human Cyclin E Requires the Function of Eukaryotic Chaperonin CCT. Mol. Cell. Biol. 1998, 18, 7584–7589. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, A.G.; Muller, P.A.; Cuellar, J.; Klejnot, M.; Nobis, M.; Valpuesta, J.M.; Vousden, K.H. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol. Cell 2013, 50, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Shuin, T.; Kondo, K.; Torigoe, S.; Kishida, T.; Kubota, Y.; Hosaka, M.; Nagashima, Y.; Kitamura, H.; Latif, F.; Zbar, B.; et al. Frequent somatic mutations and loss of heterozygosity of the von Hippel-Lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res. 1994, 54, 2852–2855. [Google Scholar] [PubMed]
- Melville, M.W.; McClellan, A.J.; Meyer, A.S.; Darveau, A.; Frydman, J. The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Mol. Cell. Biol. 2003, 23, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Guest, S.T.; Kratche, Z.R.; Bollig-Fischer, A.; Haddad, R.; Ethier, S.P. Two members of the TRiC chaperonin complex, CCT2 and TCP1 are essential for survival of breast cancer cells and are linked to driving oncogenes. Exp. Cell Res. 2015, 332, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, O.A.; Chen, B.; Haase-Pettingell, C.; Ludtke, S.J.; Chiu, W.; King, J.A. Human CCT4 and CCT5 chaperonin subunits expressed in escherichia coli form biologically active homo-oligomers. J. Biol. Chem. 2013, 288, 17734–17744. [Google Scholar] [CrossRef] [PubMed]
- Boudiaf-Benmammar, C.; Cresteil, T.; Melki, R. The cytosolic chaperonin CCT/TRiC and cancer cell proliferation. PLoS ONE 2013, 8, e60895. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Marchion, D.C.; Basso, A.D.; Rosen, N. Degradation of HER2 by ansamycins induces growth arrest and apoptosis in cells with HER2 overexpression via a HER3, phosphatidylinositol 3′-kinase-AKT-dependent pathway. Cancer Res. 2002, 62, 3132–3137. [Google Scholar] [PubMed]
- Zhou, J.; Schmid, T.; Frank, R.; Brune, B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1α from pVHL-independent degradation. J. Biol. Chem. 2004, 279, 13506–13513. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Powers, M.V. Chaperoning cell death: A critical dual role for Hsp90 in small-cell lung cancer. Nat. Chem. Biol. 2007, 3, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Sawarkar, R.; Sievers, C.; Paro, R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell 2012, 149, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Nussbaumer, U.; Chen, Y.; Beisel, C.; Paro, R. Trithorax requires Hsp90 for maintenance of active chromatin at sites of gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1α-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Spiess, C.; Howard, D.E.; Frydman, J. Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol. Cell 2003, 12, 213–224. [Google Scholar] [CrossRef]
- Lindquist, S. Protein folding sculpting evolutionary change. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, D.F.; Lindquist, S. Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science 2010, 330, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Tarafa, G.; Tuck, D.; Ladner, D.; Topazian, M.; Brand, R.; Deters, C.; Moreno, V.; Capella, G.; Lynch, H.; Lizardi, P.; Costa, J. Mutational load distribution analysis yields metrics reflecting genetic instability during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 4306–4311. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Glazer, P.M. Mutagenesis induced by the tumor microenvironment. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1998, 400, 439–446. [Google Scholar] [CrossRef]
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 569, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Frydman, J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Ann. Rev. Biochem. 2001, 70, 603–647. [Google Scholar] [CrossRef] [PubMed]
- Lewis, V.A.; Hynes, G.M.; Zheng, D.; Saibil, H.; Willison, K. T-complex polypeptide-1 is a subunit of a heteromeric particle in the eukaryotic cytosol. Nature 1992, 358, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Hynes, G.; Carne, A.; Ashworth, A.; Willison, K. Identification of six Tcp-1-related genes encoding divergent subunits of the TCP-1-containing chaperonin. Curr. Biol. 1994, 4, 89–99. [Google Scholar] [CrossRef]
- Leitner, A.; Joachimiak, L.A.; Bracher, A.; Monkemeyer, L.; Walzthoeni, T.; Chen, B.; Pechmann, S.; Holmes, S.; Cong, Y.; Ma, B.; et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 2012, 20, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Kalisman, N.; Adams, C.M.; Levitt, M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. USA 2012, 109, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Miller, E.J.; McClellan, A.J.; Frydman, J. Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol. Cell 2006, 24, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Dekker, C.; Roe, S.M.; McCormack, E.A.; Beuron, F.; Pearl, L.H.; Willison, K.R. The crystal structure of yeast CCT reveals intrinsic asymmetry of eukaryotic cytosolic chaperonins. EMBO J. 2011, 30, 3078–3090. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.G.; Yebenes, H.; Zhou, M.; Mesa, P.; Serna, M.; Park, A.Y.; Bragado-Nilsson, E.; Beloso, A.; de Carcer, G.; Malumbres, M.; et al. Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat. Struct. Mol. Biol. 2011, 18, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Schroder, G.F.; Meyer, A.S.; Jakana, J.; Ma, B.; Dougherty, M.T.; Schmid, M.F.; Reissmann, S.; Levitt, M.; Ludtke, S.L.; et al. Symmetry-free cryo-EM structures of the chaperonin TRiC along its ATPase-driven conformational cycle. EMBO J. 2012, 31, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Thomas, J.O.; Chow, R.L.; Lee, G.H.; Cowan, N.J. A cytoplasmic chaperonin that catalyzes β-actin folding. Cell 1992, 69, 1043–1050. [Google Scholar] [CrossRef]
- Melki, R.; Batelier, G.; Soulie, S.; Williams, R.C., Jr. Cytoplasmic chaperonin containing TCP-1: Structural and functional characterization. Biochemistry 1997, 36, 5817–5826. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Gillespie, J.R.; Walther, D.; Millet, I.S.; Doniach, S.; Frydman, J. Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell 2003, 113, 369–381. [Google Scholar] [CrossRef]
- Shimon, L.; Hynes, G.M.; McCormack, E.A.; Willison, K.R.; Horovitz, A. ATP-induced allostery in the eukaryotic chaperonin CCT is abolished by the mutation G345D in CCT4 that renders yeast temperature-sensitive for growth. J. Mol. Biol. 2008, 377, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, R.; Yoshida, T.; Ishii, N.; Zako, T.; Takahashi, K.; Maki, K.; Inobe, T.; Kuwajima, K.; Yohda, M. Characterization of archaeal group II chaperonin-ADP-metal fluoride complexes: Implications that group II chaperonins operate as a “two-stroke engine”. J. Biol. Chem. 2005, 280, 40375–40383. [Google Scholar] [CrossRef] [PubMed]
- Rivenzon-Segal, D.; Wolf, S.G.; Shimon, L.; Willison, K.R.; Horovitz, A. Sequential ATP-induced allosteric transitions of the cytoplasmic chaperonin containing TCP-1 revealed by EM analysis. Nat. Struct. Mol. Biol. 2005, 12, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S.; Parnot, C.; Booth, C.R.; Chiu, W.; Frydman, J. Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat. Struct. Mol. Biol. 2007, 14, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Joachimiak, L.A.; Walzthoeni, T.; Liu, C.W.; Aebersold, R.; Frydman, J. The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 2014, 159, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Yam, A.Y.; Xia, Y.; Lin, H.T.; Burlingame, A.; Gerstein, M.; Frydman, J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 2008, 15, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Kakihara, Y.; Krogan, N.; Greenblatt, J.; Emili, A.; Zhang, Z.; Houry, W.A. An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: Implications to protein folding pathways in the cell. Mol. Syst. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Dekker, C.; Stirling, P.C.; McCormack, E.A.; Filmore, H.; Paul, A.; Brost, R.L.; Costanzo, M.; Boone, C.; Leroux, M.R.; Willison, K.R. The interaction network of the chaperonin CCT. EMBO J. 2008, 27, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Zhong, F.L.; Venteicher, A.S.; Meng, Z.; Veenstra, T.D.; Frydman, J.; Artandi, S.E. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 2014, 159, 1389–1403. [Google Scholar] [PubMed]
- Russmann, F.; Stemp, M.J.; Monkemeyer, L.; Etchells, S.A.; Bracher, A.; Hartl, F.U. Folding of large multidomain proteins by partial encapsulation in the chaperonin TRiC/CCT. Proc. Natl. Acad. Sci. USA 2012, 109, 21208–21215. [Google Scholar] [CrossRef] [PubMed]
- Kalisman, N.; Levitt, M. Insights into the intra-ring subunit order of TRiC/CCT: A structural and evolutionary analysis. Pac. Symp. Biocomput. 2010, 252–259. [Google Scholar]
- Kalisman, N.; Schroder, G.F.; Levitt, M. The crystal structures of the eukaryotic chaperonin CCT reveal its functional partitioning. Structure 2013, 21, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Weisberg, S.J.; Nadler-Holly, M.; McCormack, E.A.; Feldmesser, E.; Kaganovich, D.; Willison, K.R.; Horovitz, A. Equivalent mutations in the eight subunits of the chaperonin CCT produce dramatically different cellular and gene expression phenotypes. J. Mol. Biol. 2010, 401, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S.; Joachimiak, L.A.; Chen, B.; Meyer, A.S.; Nguyen, A.; Frydman, J. A gradient of ATP affinities generates an asymmetric power stroke driving the chaperonin TRIC/CCT folding cycle. Cell Rep. 2012, 2, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Dalton, K.; Frydman, J. The Mechanism and Function of Group II Chaperonins. J. Mol. Biol. 2015, 427, 2919–2930. [Google Scholar] [CrossRef] [PubMed]
- Saegusa, K.; Sato, M.; Sato, K.; Nakajima-Shimada, J.; Harada, A.; Sato, K. Caenorhabditis elegans chaperonin CCT/TRiC is required for actin and tubulin biogenesis and microvillus formation in intestinal epithelial cells. Mol. Biol. Cell 2014, 25, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Scott, M.D.; Frydman, J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 2005, 121, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Katz, S.; Doody, M.; Sheffer, M.; Horesh, S.; Molchadsky, A.; Koifman, G.; Shetzer, Y.; Goldfinger, N.; Rotter, V.; et al. Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc. Natl. Acad. Sci. USA 2014, 111, 7006–7011. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Barneo, J.; Pardal, R.; Ortega-Saenz, P. Cellular mechanism of oxygen sensing. Annu. Rev. Physiol. 2001, 63, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Kishida, T.; Yao, M.; Hustad, T.; Glavac, D.; Dean, M.; Gnarra, J.R.; Orcutt, M.L.; Duh, F.M.; Glenn, G.; et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: Correlations with phenotype. Hum. Mutat. 1995, 5, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Aso, T.; Lane, W.S.; Conaway, J.W.; Conaway, R.C. Elongin (SIII): A multisubunit regulator of elongation by RNA polymerase II. Science 1995, 269, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.R.; Pause, A.; Burgess, W.H.; Aso, T.; Chen, D.Y.; Garrett, K.P.; Conaway, R.C.; Conaway, J.W.; Linehan, W.M.; Klausner, R.D. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science 1995, 269, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Sen, P.; Hofmann, K.; Ma, L.; Goebl, M.; Harper, J.W.; Elledge, S.J. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 1996, 86, 263–274. [Google Scholar] [CrossRef]
- Blasberg, B.; Stool, S.; Oka, S. Choanal atresia-a cryptic congenital anomaly. Cleft Palate J. 1975, 12, 409–416. [Google Scholar] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [PubMed]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1α ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, H.; Gazit, E. The von Hippel-Lindau tumor suppressor protein is a molten globule under native conditions: Implications for its physiological activities. J. Biol. Chem. 2004, 279, 17190–17196. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Thulasiraman, V.; Ferreyra, R.G.; Frydman, J. Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol. Cell 1999, 4, 1051–1061. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Appelhoff, R.J.; Tuckerman, J.R.; Tian, Y.M.; Demol, H.; Puype, M.; Vandekerckhove, J.; Ratcliffe, P.J.; Pugh, C.W. The HIF prolyl hydroxylase PHD3 is a potential substrate of the TRiC chaperonin. FEBS Lett. 2004, 570, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Zaitoun, A.; Fareed, K.R.; Turley, H.; Aloysius, M.; Lobo, D.N.; Harris, A.L.; Madhusudan, S. Expression of key hypoxia sensing prolyl-hydroxylases PHD1, -2 and -3 in pancreaticobiliary cancer. Histopathology 2010, 56, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Loos, M.; Giese, N.; Hines, O.J.; Diebold, I.; Gorlach, A.; Metzen, E.; Pastorekova, S.; Friess, H.; Buchler, P. PHD3 regulates differentiation, tumour growth and angiogenesis in pancreatic cancer. Br. J. Cancer 2010, 103, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Zhu, L.; Schmidt, C.; Tucker, P.W. Hsp90-from signal transduction to cell transformation. Biochem. Biophys. Res. Commun. 2007, 363, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, C.E.; Kasembeli, M.M.; Roh, S.-H.; Tweardy, D.J. Contribution of chaperones to STAT pathway signaling. JAK-STAT 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Jain, S.; Stuhmer, T.; Andrulis, M.; Ungethum, U.; Kuban, R.J.; Lorentz, H.; Bommert, K.; Topp, M.; Kramer, D.; et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90α and β in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; Vu, L.P.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011, 7, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment: JAK-STAT3 signaling. Jakstat 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Hainaut, P. Massively regulated genes: The example of TP53. J. Pathol. 2010, 220, 164–173. [Google Scholar] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 2007, 97, 1–23. [Google Scholar] [PubMed]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Olszewski, M.B.; Gutkowska, M.; Helwak, A.; Zylicz, M.; Zylicz, A. Hsp70 molecular chaperones are required to support p53 tumor suppressor activity under stress conditions. Oncogene 2009, 28, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yanagi, H.; Yura, T.; Kubota, H. Cytosolic chaperonin is up-regulated during cell growth. Preferential expression and binding to tubulin at G(1)/S transition through early S phase. J. Biol. Chem. 1999, 274, 37070–37078. [Google Scholar] [CrossRef] [PubMed]
- Kimata, Y.; Baxter, J.E.; Fry, A.M.; Yamano, H. A role for the Fizzy/Cdc20 family of proteins in activation of the APC/C distinct from substrate recruitment. Mol. Cell 2008, 32, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Yu, H. Cdc20: A WD40 activator for a cell cycle degradation machine. Mol. Cell 2007, 27, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. Embo J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Tan, M.; Yang, J.; Inuzuka, H.; Dai, X.; Wu, T.; Liu, J.; Shaik, S.; Chen, G.; Deng, J.; et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev. Cell 2014, 29, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wan, L.; Zhong, J.; Inuzuka, H.; Liu, P.; Sarkar, F.H.; Wei, W. Cdc20: A potential novel therapeutic target for cancer treatment. Curr. Pharm. Des. 2013, 19, 3210–3214. [Google Scholar] [CrossRef] [PubMed]
- Manchado, E.; Guillamot, M.; de Carcer, G.; Eguren, M.; Trickey, M.; Garcia-Higuera, I.; Moreno, S.; Yamano, H.; Canamero, M.; Malumbres, M. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55α,Δ phosphatase. Cancer Cell 2010, 18, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Sigoillot, F.; Gaur, S.; Choi, S.; Pfaff, K.L.; Oh, D.C.; Hathaway, N.; Dimova, N.; Cuny, G.D.; King, R.W. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010, 18, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lee, E.H.; Han, S.H.; Chung, H.J.; Jeong, J.H.; Kwon, J.; Kim, H. Degradation of human RAP80 is cell cycle regulated by Cdc20 and Cdh1 ubiquitin ligases. Mol. Cancer Res. 2012, 10, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Dimova, N.V.; Tan, M.K.; Sigoillot, F.D.; King, R.W.; Shi, Y. The G2/M regulator histone demethylase PHF8 is targeted for degradation by the anaphase-promoting complex containing CDC20. Mol. Cell. Biol. 2013, 33, 4166–4180. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [PubMed]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Joshi, A.; Zhang, Y.; Jaeger, S.A.; Bulyk, M.; Tsichlis, P.N.; Shirley Liu, X.; Struhl, K. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell 2010, 17, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Khaidakov, M.; Mitra, S.; Kang, B.Y.; Wang, X.; Kadlubar, S.; Novelli, G.; Raj, V.; Winters, M.; Carter, W.C.; Mehta, J.L. Oxidized LDL receptor 1 (OLR1) as a possible link between obesity, dyslipidemia and cancer. PLoS ONE 2011, 6, e20277. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L.; Sanada, N.; Hu, C.P.; Chen, J.; Dandapat, A.; Sugawara, F.; Satoh, H.; Inoue, K.; Kawase, Y.; Jishage, K.; et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 2007, 100, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed]
- Bakthavatsalam, D.; Soung, R.H.; Tweardy, D.J.; Chiu, W.; Dixon, R.A.; Woodside, D.G. Chaperonin-containing TCP-1 complex directly binds to the cytoplasmic domain of the LOX-1 receptor. FEBS Lett. 2014, 13, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Yu, J.; Kao, G.D.; Yen, T.J.; Lazar, M.A. Assembly of the SMRT-histone deacetylase 3 repression complex requires the TCP-1 ring complex. Genes Dev. 2002, 16, 3130–3135. [Google Scholar] [CrossRef] [PubMed]
- Pejanovic, N.; Hochrainer, K.; Liu, T.; Aerne, B.L.; Soares, M.P.; Anrather, J. Regulation of nuclear factor kappaB (NF-κB) transcriptional activity via p65 acetylation by the chaperonin containing TCP1 (CCT). PLoS ONE 2012, 7, e42020. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Thompson, R.C.; Gilmore, T.D. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 2015, 6, 184–213. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roh, S.-H.; Kasembeli, M.; Bakthavatsalam, D.; Chiu, W.; Tweardy, D.J. Contribution of the Type II Chaperonin, TRiC/CCT, to Oncogenesis. Int. J. Mol. Sci. 2015, 16, 26706-26720. https://doi.org/10.3390/ijms161125975
Roh S-H, Kasembeli M, Bakthavatsalam D, Chiu W, Tweardy DJ. Contribution of the Type II Chaperonin, TRiC/CCT, to Oncogenesis. International Journal of Molecular Sciences. 2015; 16(11):26706-26720. https://doi.org/10.3390/ijms161125975
Chicago/Turabian StyleRoh, Soung-Hun, Moses Kasembeli, Deenadayalan Bakthavatsalam, Wah Chiu, and David J. Tweardy. 2015. "Contribution of the Type II Chaperonin, TRiC/CCT, to Oncogenesis" International Journal of Molecular Sciences 16, no. 11: 26706-26720. https://doi.org/10.3390/ijms161125975
APA StyleRoh, S.-H., Kasembeli, M., Bakthavatsalam, D., Chiu, W., & Tweardy, D. J. (2015). Contribution of the Type II Chaperonin, TRiC/CCT, to Oncogenesis. International Journal of Molecular Sciences, 16(11), 26706-26720. https://doi.org/10.3390/ijms161125975