
Low T3 State Is Correlated with Cardiac Mitochondrial Impairments after Ischemia Reperfusion Injury: Evidence from a Proteomic Approach



 ,
, 
Abstract
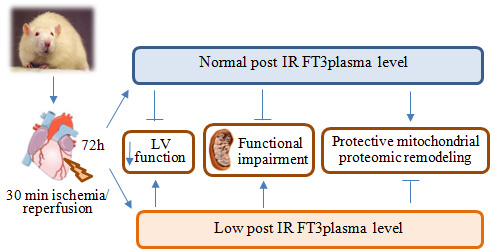
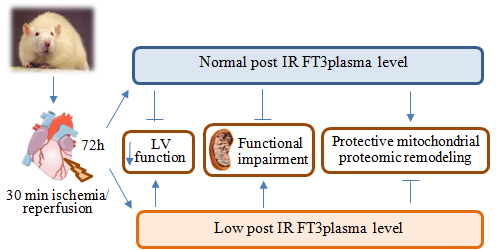
:
1. Introduction
2. Results
2.1. Validation of the Post IR Low-T3 State (LT3S) Model
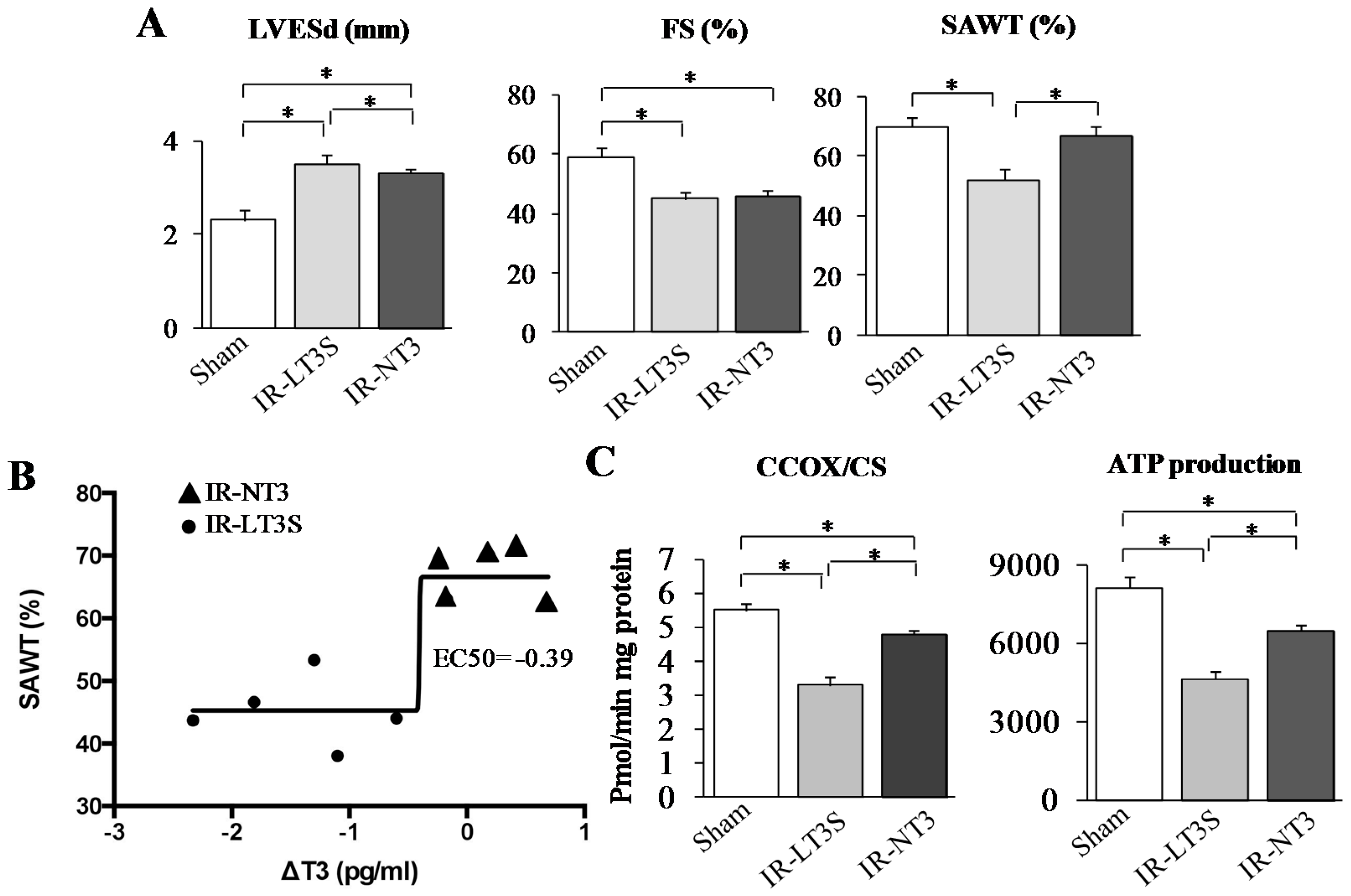
2.2. Post IR Myocardial Functional Parameters and Mitochondrial Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
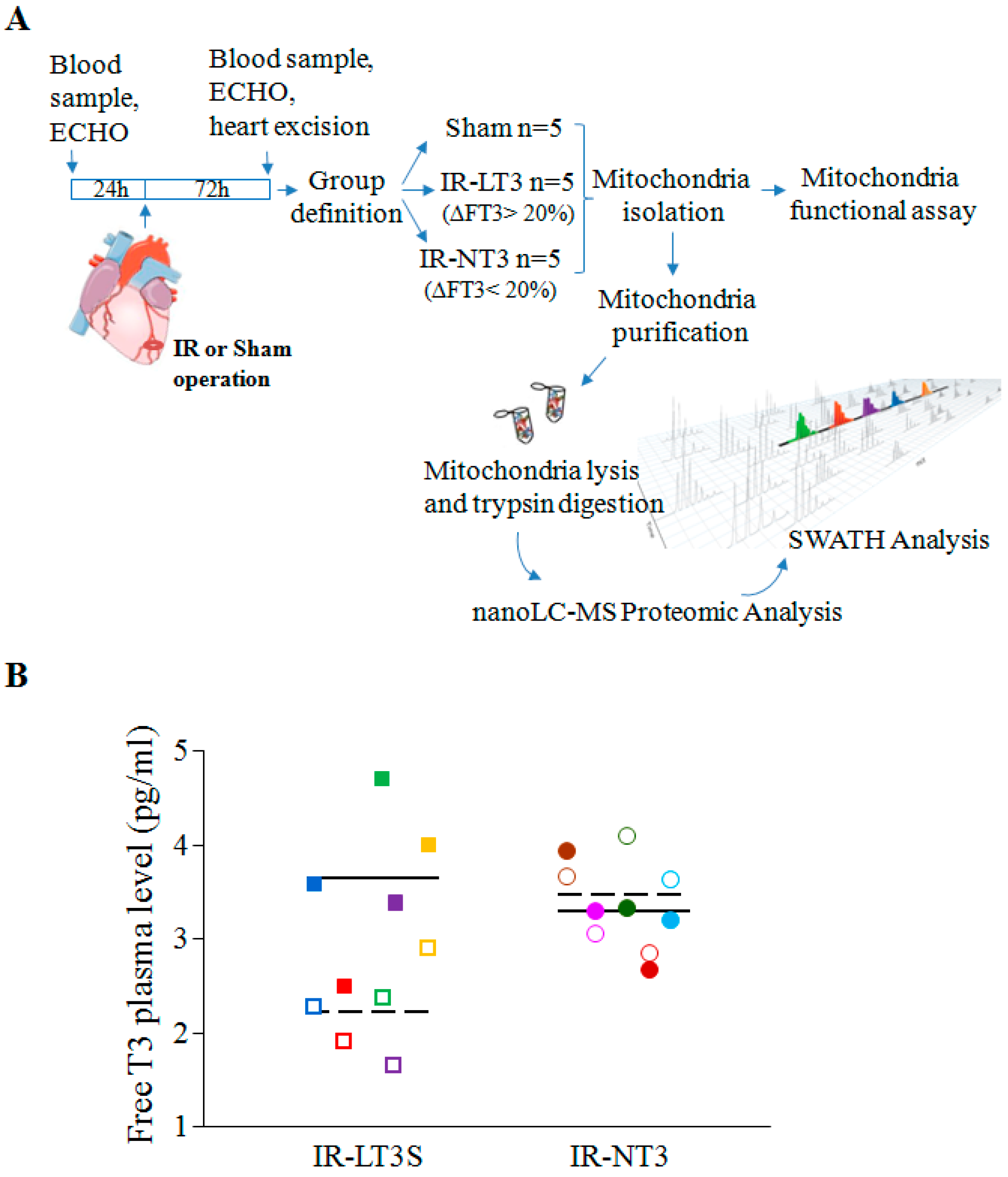{kind=link}
| TH Level (pg/mL) | Sham | IR-NT3 | IR-LT3S | |||
|---|---|---|---|---|---|---|
| Mean ± SEM | Median (IQR) | Mean ± SEM | Median (IQR) | Mean ± SEM | Median (IQR) | |
| FT3 basal | 3.2 ± 0.3 | 3.6 (2.7–3.8) | 3.2 ± 0.3 | 3.2 (2.6–3.9) | 3.5 ± 0.3 | 3.4 (3.3–3.6) |
| FT3 final | 3.5 ± 0.2 | 3.8 (3.1–3.8) | 3.3 ± 0.3 | 3.6 (2.6–3.6) | 2.2 ± 0.3 *,#,† | 2.3 (1.9–2.4) §,&,+ |
| FT4 basal | 12.4 ± 0.6 | 13.4 (11.6–13.7) | 13.0 ± 0.7 | 13.3 (12.9–13.7) | 12.1 ± 1.0 | 13.7 (13.5–14.3) |
| FT4 final | 12.1 ± 1.5 | 12.6 (10.9–13.8) | 14.3 ± 1.8 | 14.3 (12.7–16.3) | 13.3 ± 1.6 | 10.4 (10.1–10.8) |
| Damage Index | Sham | IR-NT3 | IR-LT3S | |||
| – | Mean ± SEM | Mean ± SEM | ||||
| Arrhythmic severity score | NA | 3.3 ± 0.4 | 3.4 ± 0.3 | |||
| Area at risk (% of LV) | NA | 48 ± 3 | 47 ± 5 | |||
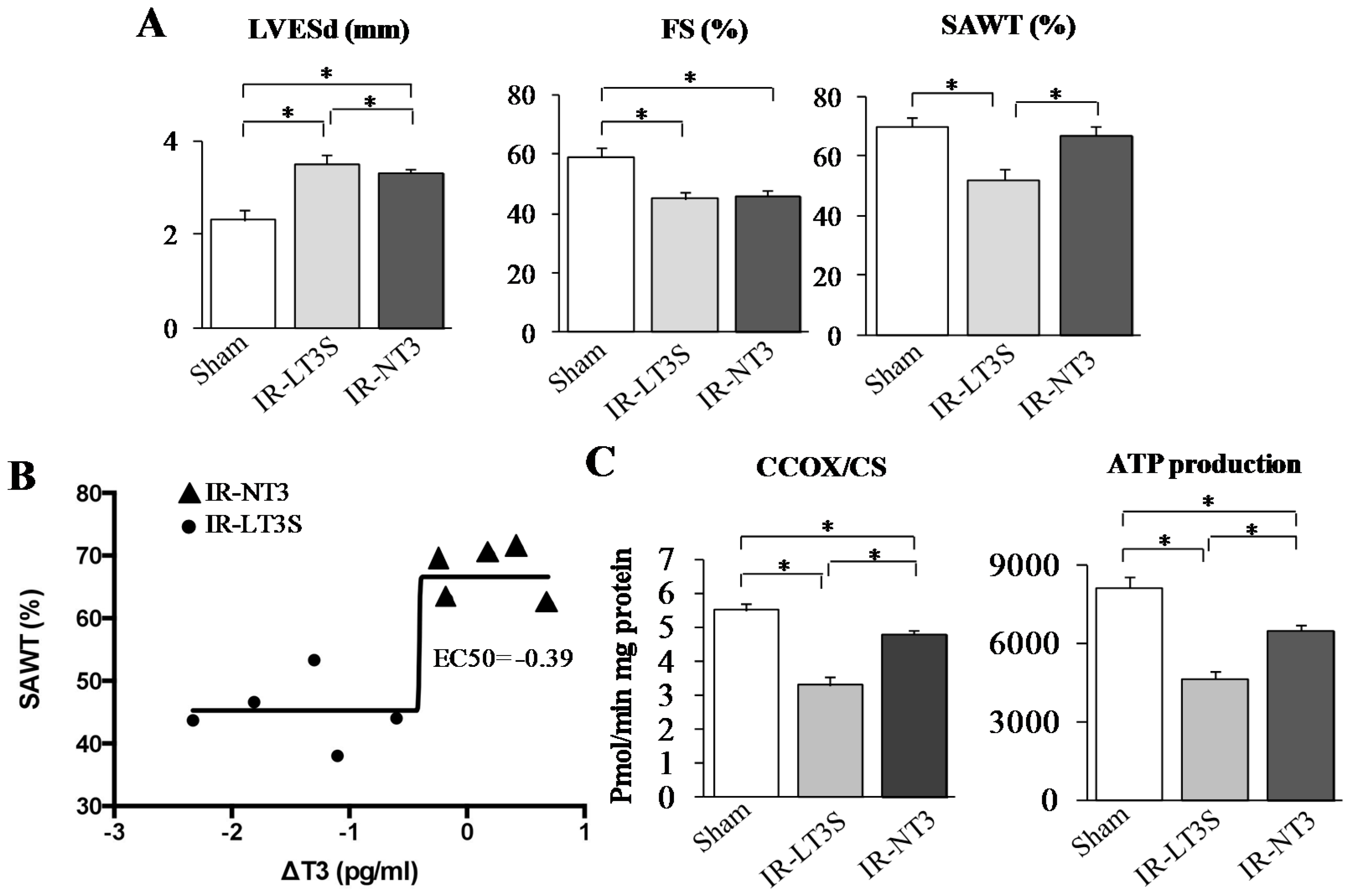
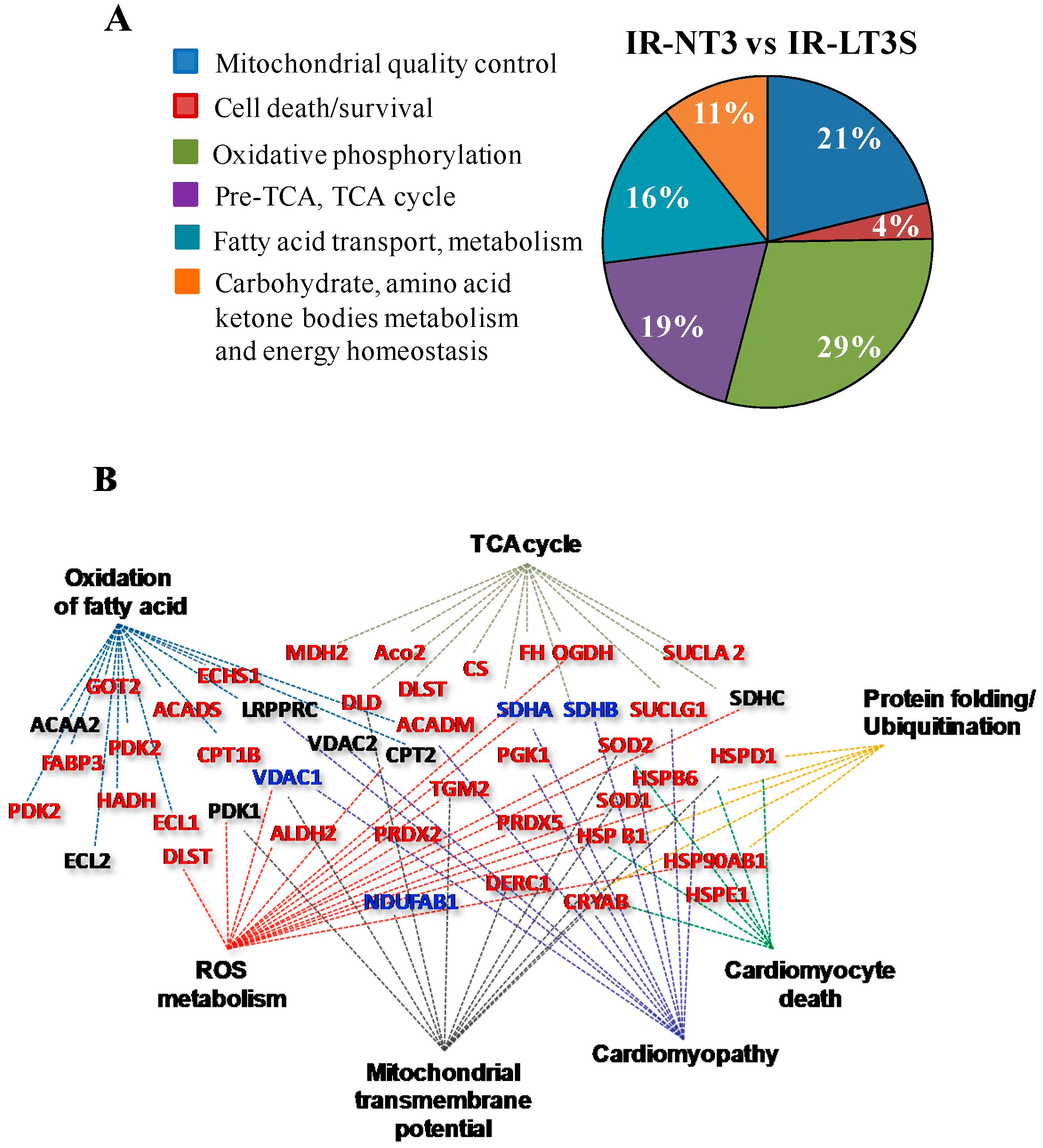
2.3. Mitochondrial Proteome
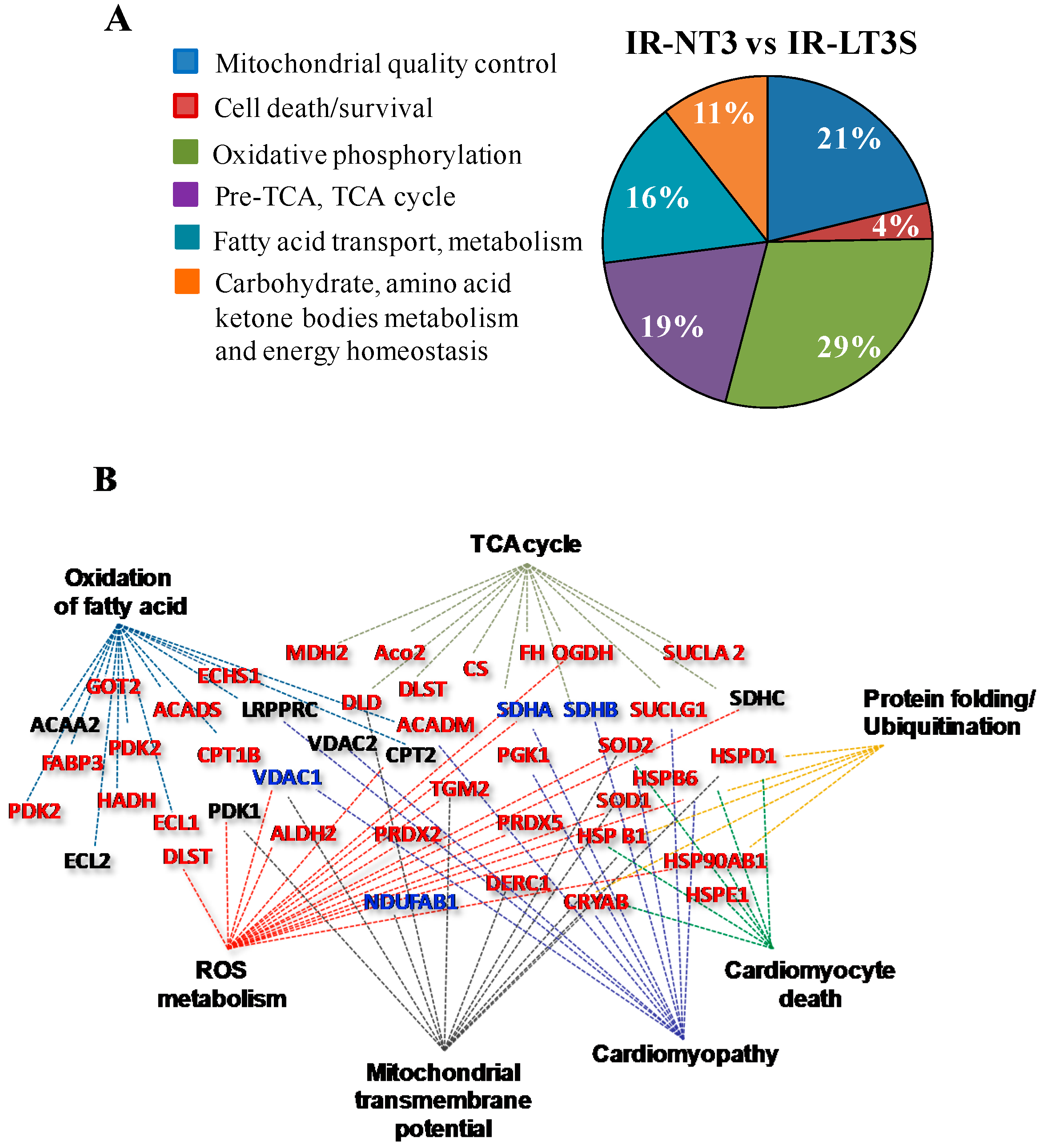
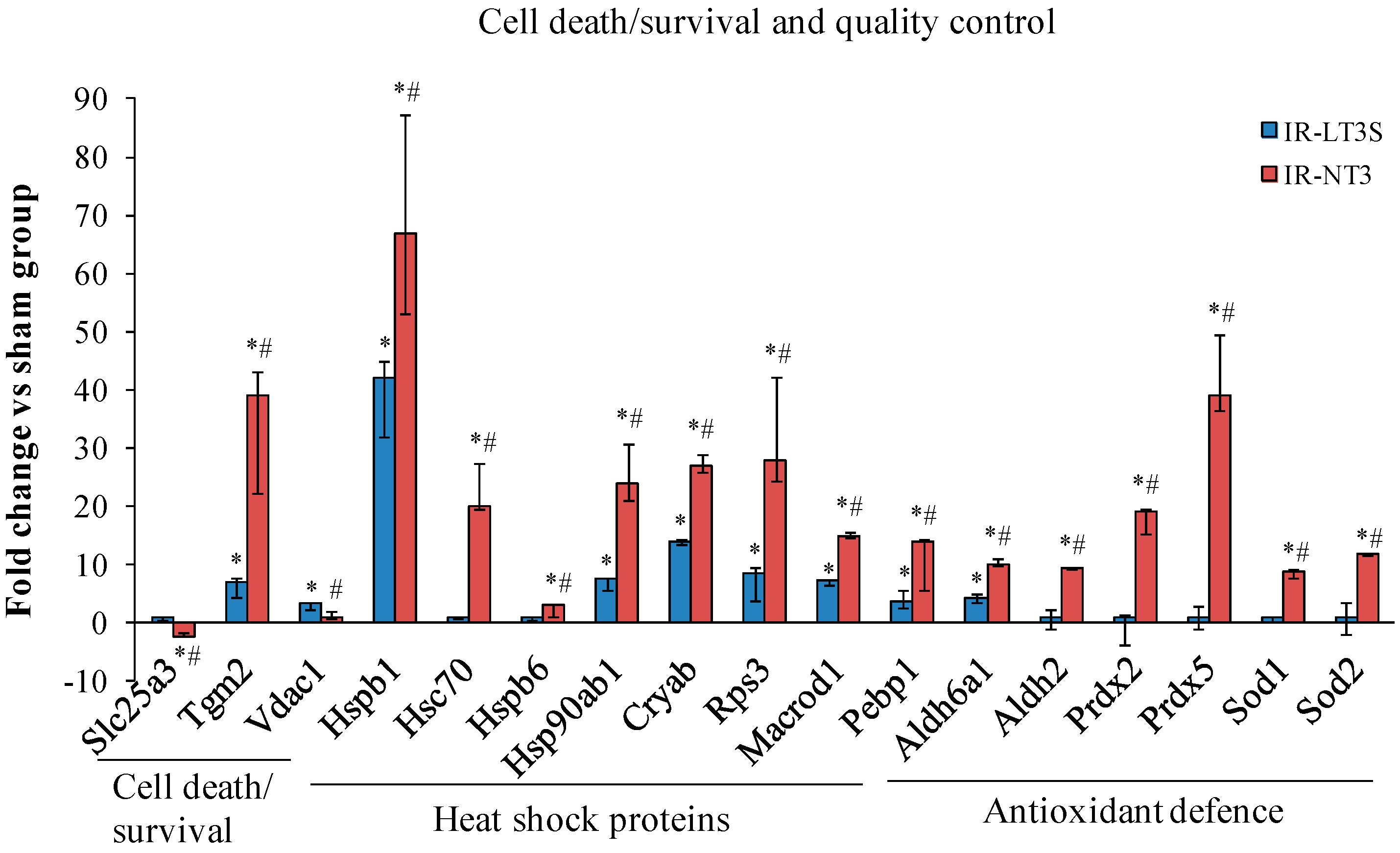
2.4. Mitochondrial Quality Control and Cell Death

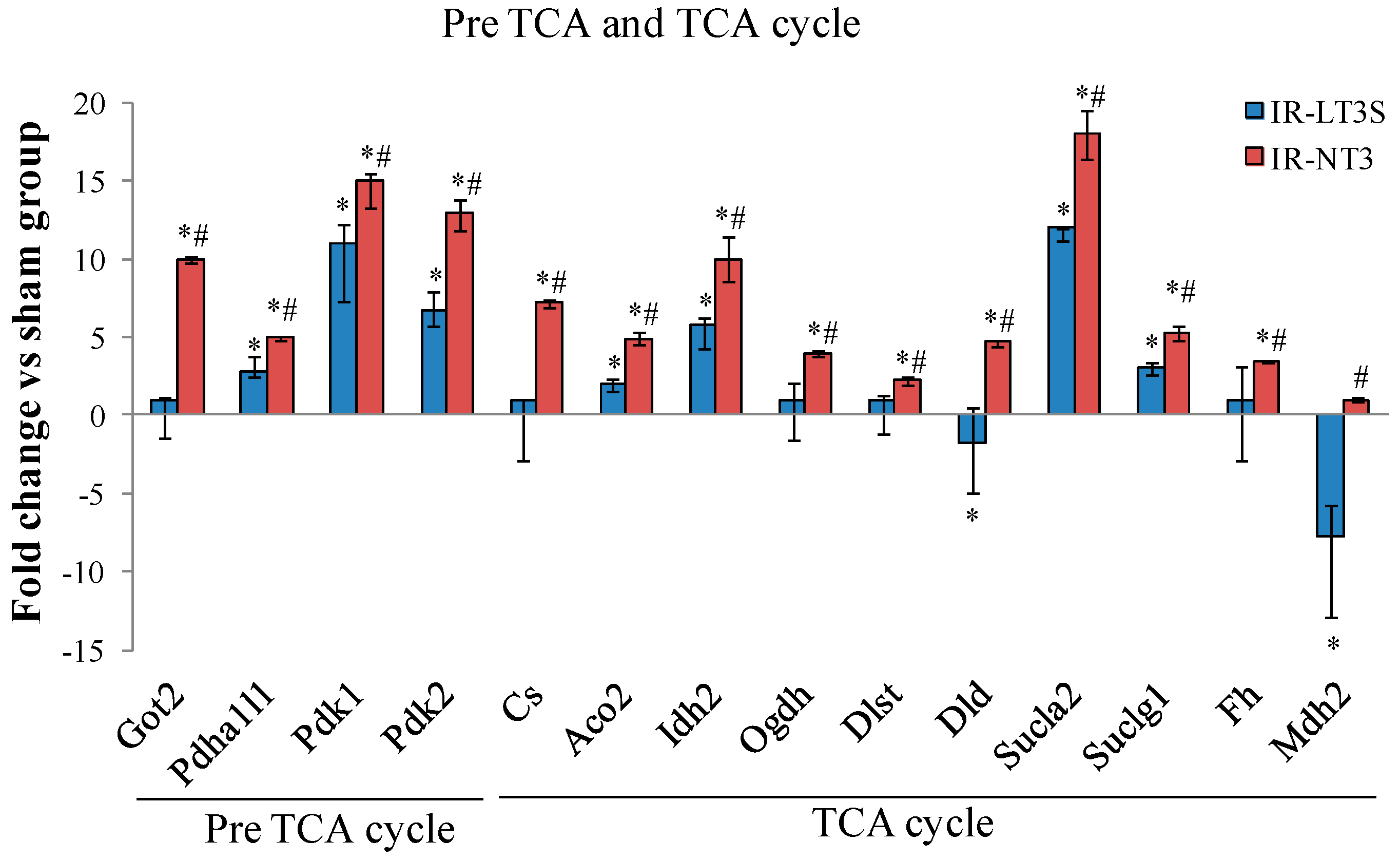
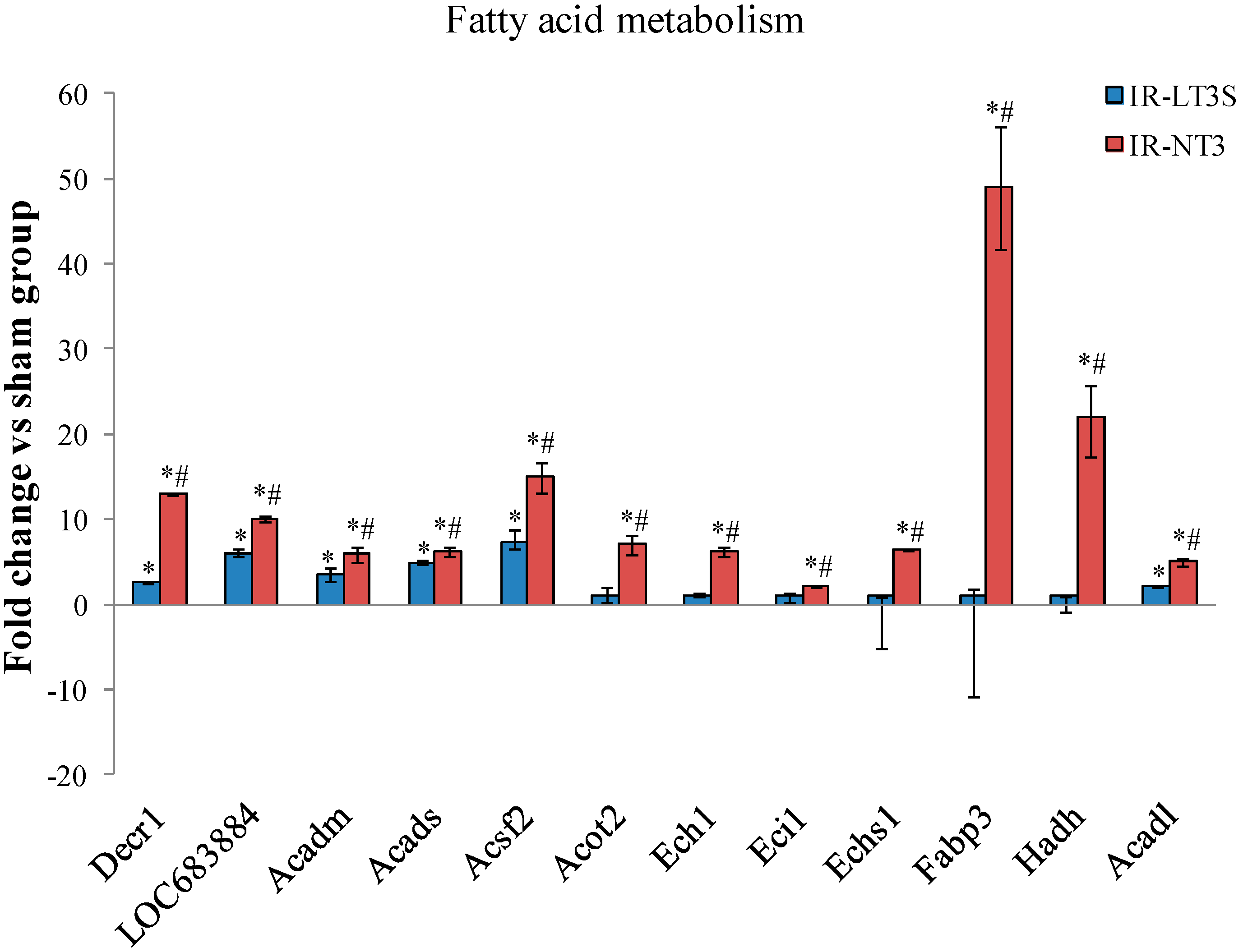
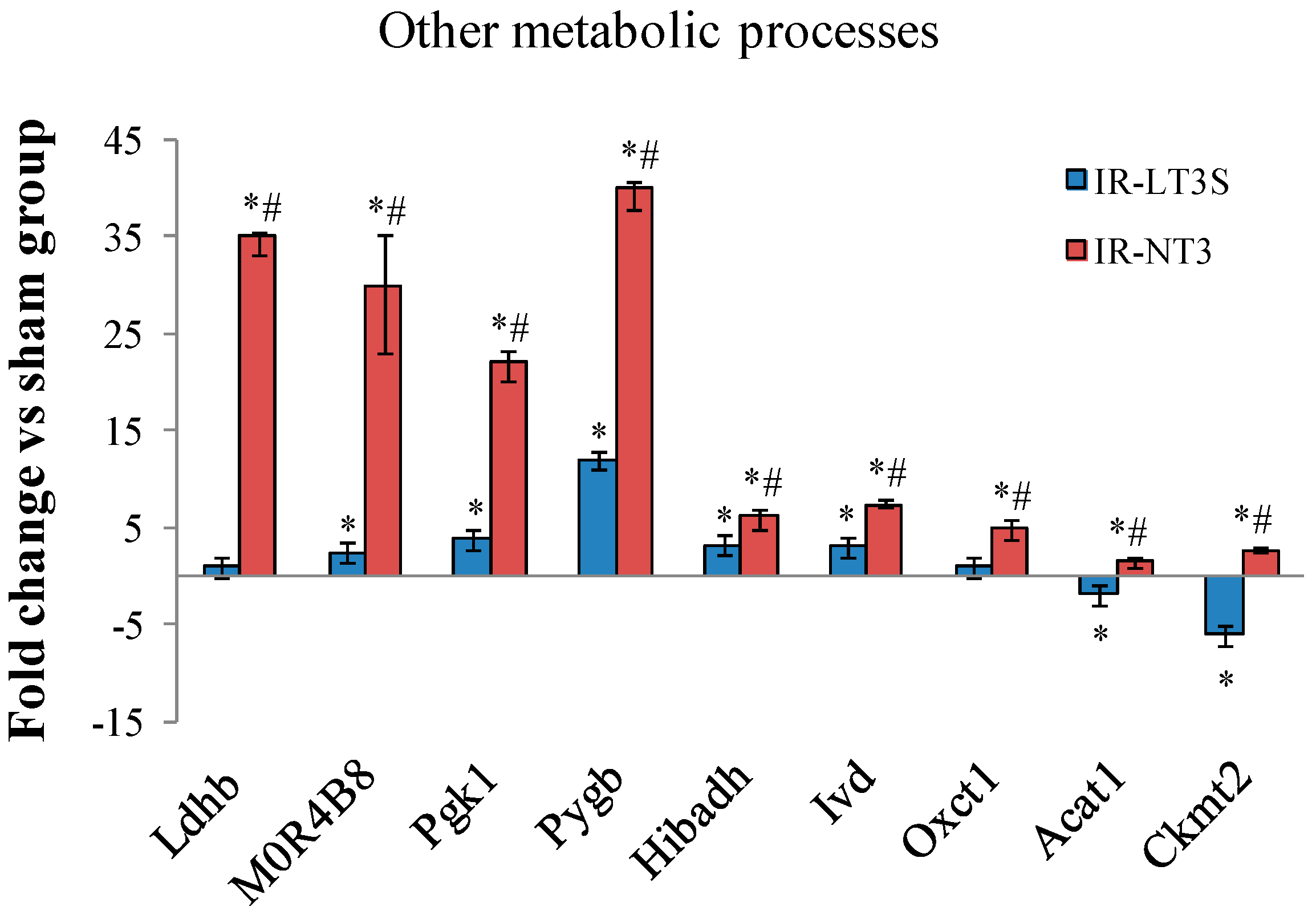
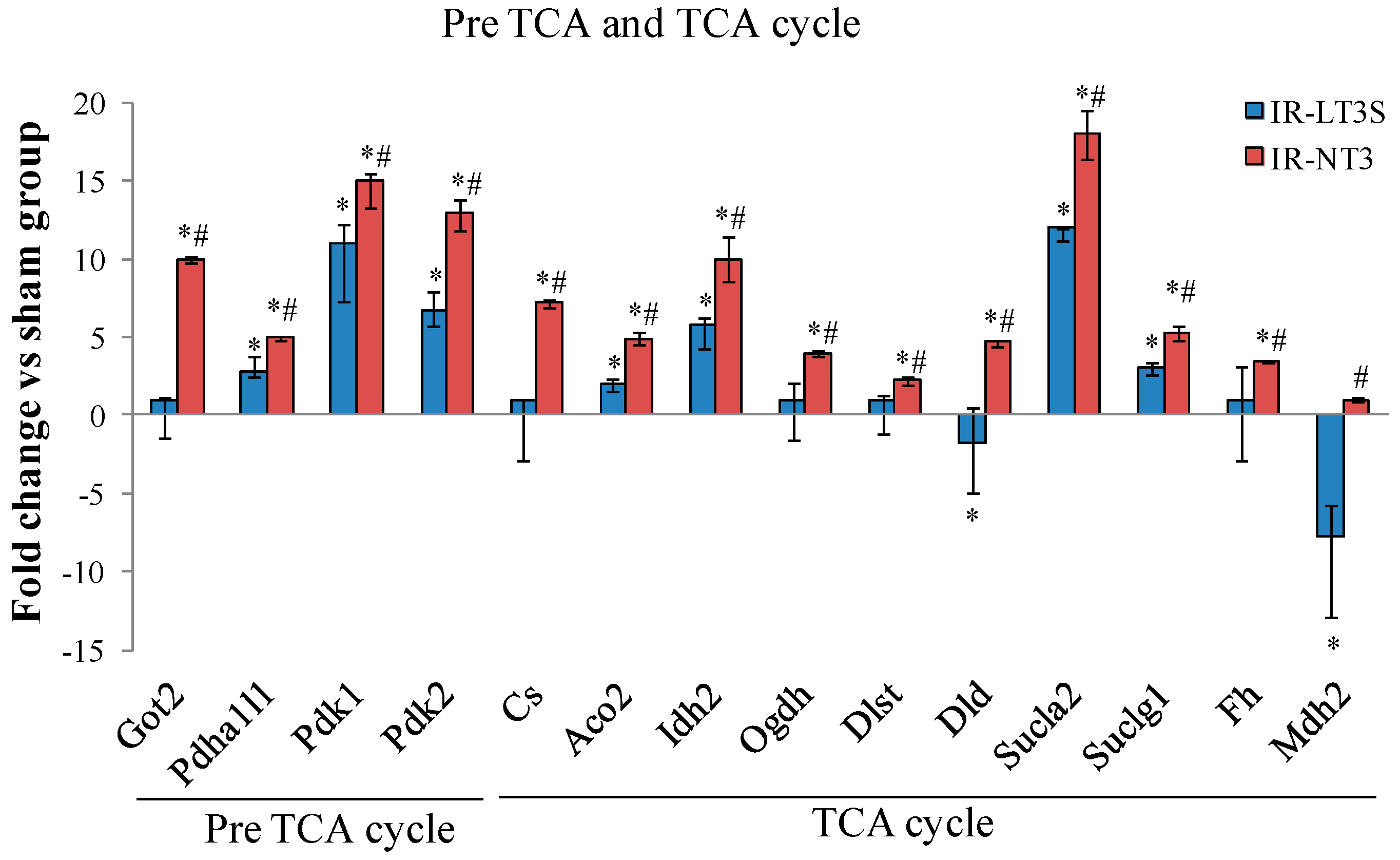
2.5. Cellular Energy Metabolism
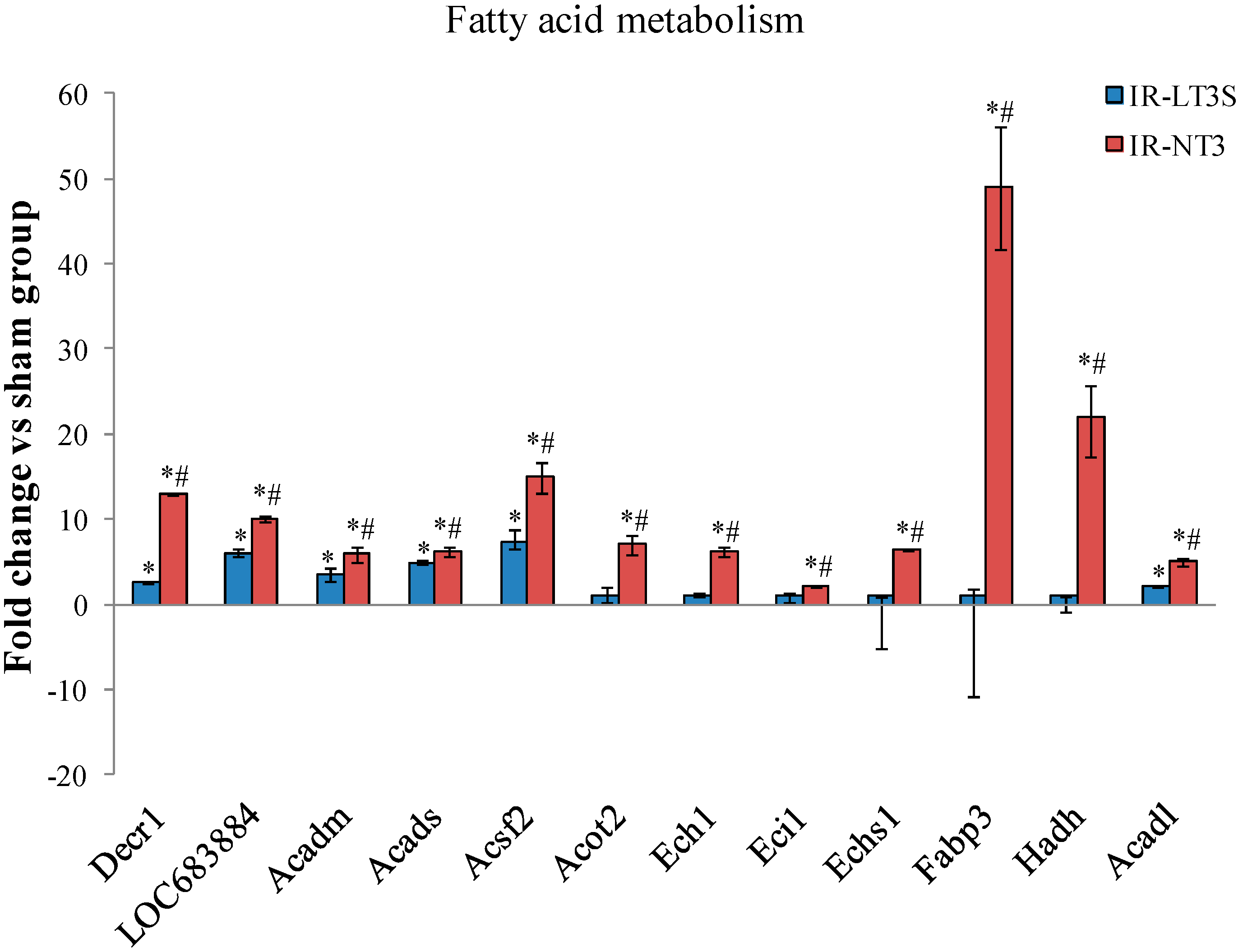
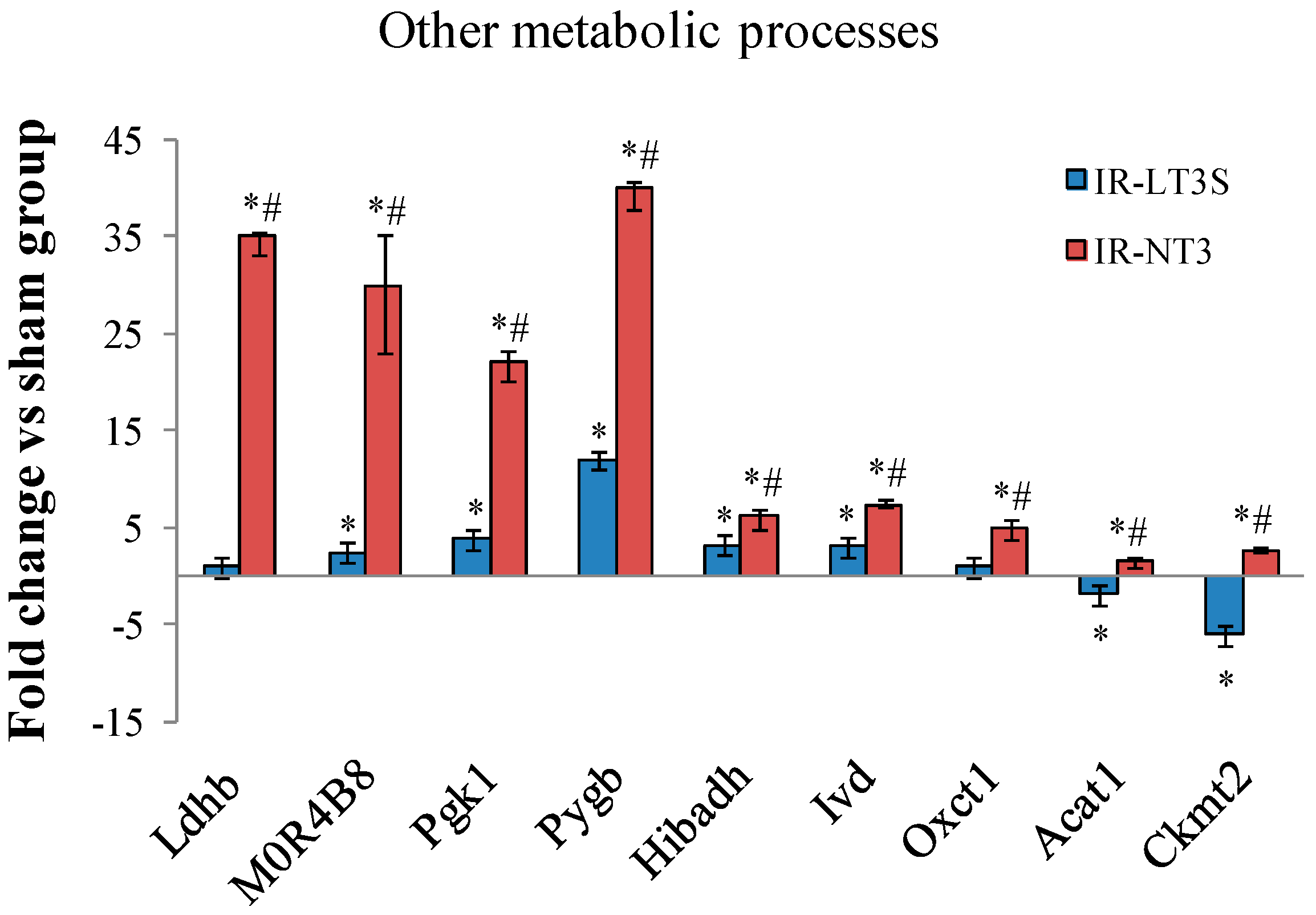
3. Discussion
3.1. Mitochondrial Quality Control and Mitochondrial-Mediated Cell Death
3.1.1. Mitochondrial Quality Control
3.1.2. Cell Fate
3.2. Oxidative Phosphorylation
3.3. Pre TCA, TCA Cycle
3.4. Fatty Acids Metabolism
3.5. Study Limitations and Concluding Remarks
4. Material and Methods
4.1. Animal Procedure
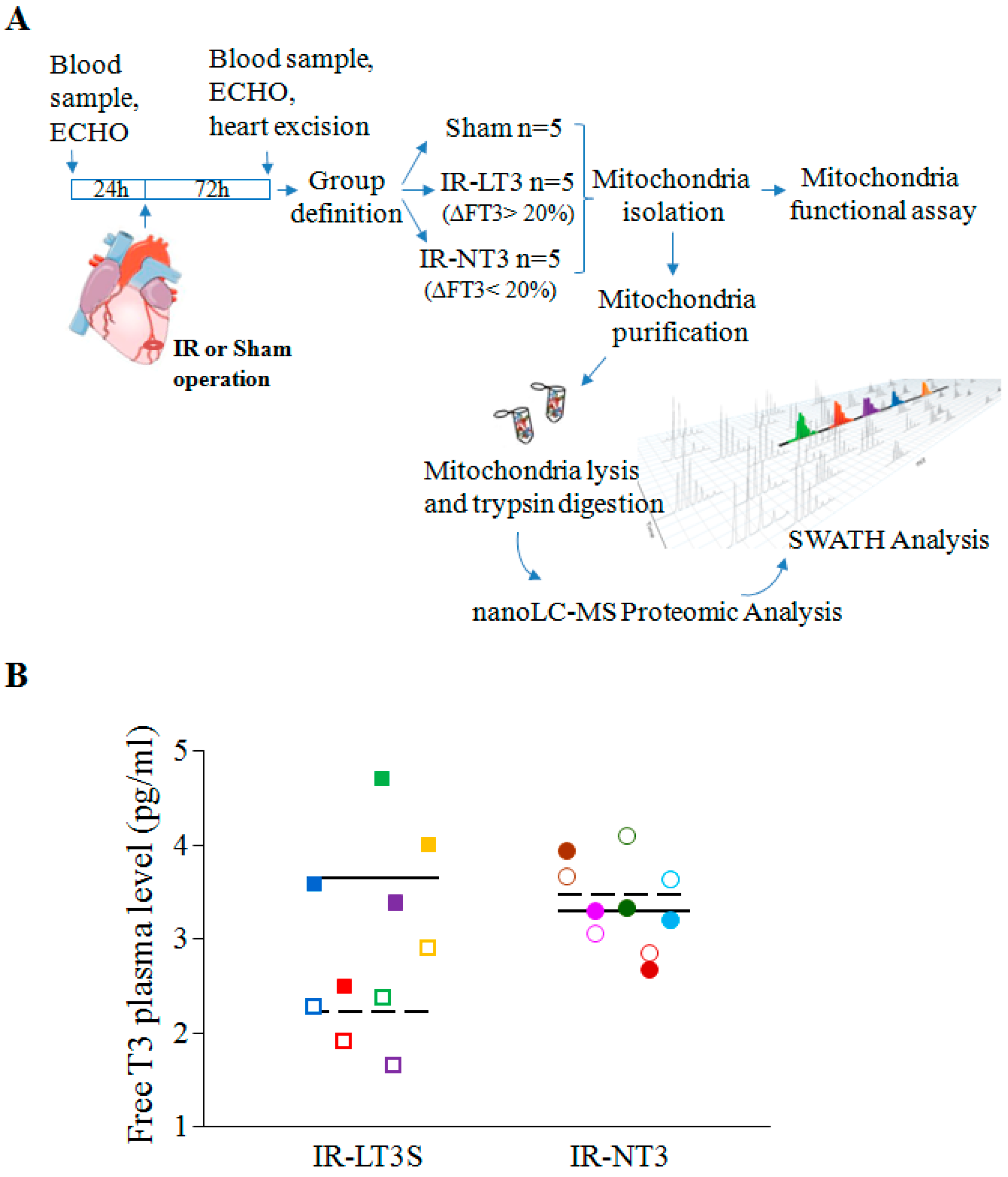
4.2. Echocardiography Study
4.3. Morphometric Analysis to Determine the Area at Risk
4.4. Serum and Tissue Thyroid Hormone Levels
4.5. Mitochondria Isolation
4.6. Mitochondrial Enzyme Activity Assays
4.7. Measurements of ATP Production in Isolated Mitochondria
4.8. Proteomics Sample Pre-Processing
4.9. nanoLC-MS/MS SWATH-Based Analysis
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Acaa2 = 3-ketoacyl-CoA thiolase. |
| Acadl = Long-chain specific acyl-CoA dehydrogenase. |
| Acadm = Acetyl-Coenzyme A dehydrogenase, medium chain. |
| Acads = Acetyl-Coenzyme A dehydrogenase, short chain. |
| Acat1 = Acetyl-CoA acetyltransferase. |
| Aco2 = Aconitate hydratase. |
| Acot2 = Acyl-CoA thioesterase 2. |
| Acsf2 = Acyl-CoA synthetase family member 2. |
| Aldh2 = Aldehyde dehydrogenase. |
| Aldh6a1 = Aldehyde dehydrogenase family 6, isoform CRA. |
| Ckmt2 = Creatine kinase mitochondrial. |
| Cpt1b = Carnitine O-palmitoyltransferase 1. |
| Cryab = α-crystallin B chain. |
| Cs = Citrate synthase. |
| Decr1 = 2,4-dienoyl CoA reductase 1. |
| Dld = Dihydrolipoyl dehydrogenase. |
| Dlst = Dihydrolipoamide S-succinyltransferase. |
| Ech1 = δ(3,5)-δ(2,4)-dienoyl-CoA isomerase. |
| Echs1 = Enoyl-CoA hydratase. |
| Eci1 = Dodecenoyl-Coenzyme A δ isomerase (3,2 trans-enoyl-Coenzyme A isomerase). |
| Ecl1 = Extender of the chronological lifespan protein 1. |
| Ecl2 = Extender of the chronological lifespan protein 2. |
| Fabp3 = Fatty acid-binding protein. |
| Fh = Fumarate hydratase 1. |
| Got2 = Aspartate aminotransferase. |
| Hadh = Hydroxyacyl-coenzyme A dehydrogenase. |
| Hibadh = 3-hydroxyisobutyrate dehydrogenase. |
| Hsc70 = Heat shock cognate 71 kDa. |
| Hspb6 = Heat shock protein α-crystallin-related-B6. |
| Hsp90ab1 = Heat shock protein HSP 90-α class B, member 1. |
| Hspb1 = Heat shock 27 kDa protein 1. |
| Hspd1 = Heat shock protein 60 kDa. |
| Hspe1 = Heat shock 10kDa protein 1. |
| Idh2 = Isocitrate dehydrogenase (NADP). |
| Ivd = Isovaleryl-CoA dehydrogenase. |
| Ldhb = l-lactate dehydrogenase B chain. |
| LOC683884 = Protein Acot13. |
| Lrpprc = Leucine-rich PPR. |
| Ndufab1 = NADH dehydrogenase (ubiquinone) 1, α/β subcomplex, 1. |
| Macrod1 = O-acetyl-ADP-ribose deacetylase MACROD1. |
| Mdh2 = Malate dehydrogenase. |
| MOR4B8 = Pyruvate kinase. |
| Ogdh = 2-oxoglutarate dehydrogenase. |
| Oxct1 = Succinyl-CoA:3-ketoacid coenzyme A transferase 1. |
| Pdha1/1 = Protein Pdha1/1. |
| Pdk1 = Pyruvate dehydrogenase kinase 1. |
| Pdk2 = Pyruvate dehydrogenase kinase 1. |
| Pepb1 = Phosphatidylethanolamine-binding protein 1. |
| Pgk1 = Phosphoglycerate kinase 1. |
| Prdx2 = Peroxiredoxin-2. |
| Prdx5 = Peroxiredoxin-5. |
| Pygb = Glycogen phosphorylase. |
| Rps3 = 40 S ribosomal protein S3. |
| Sdha = Succinate dehydrogenase (ubiquinone) flavoprotein subunit. |
| Sdhb = Succinate dehydrogenase (ubiquinone) iron-sulfur subunit. |
| Sdhc = Succinate dehydrogenase complex, subunit C, integral membrane protein. |
| Slc25a3 = Phosphate carrier protein. |
| Sod1 = Superoxide dismutase [Cu-Zn]. |
| Sod2 = Superoxide dismutase [Cu-Zn]. |
| Sucla2 = succinyl-CoA synthetase. |
| Suclg1 = Succinyl-CoA synthetase (ADP/GDP-forming) subunit α. |
| Tgm2 = transglutaminase 2. |
| Vdac1 = Voltage-dependent anion-selective channel protein 1. |
| Vdac2 = Voltage-dependent anion-selective channel protein 2. |
References
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease mechanisms and significance. Annu. Rev. Phys. 2010, 72, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The cardiac mitochondrion: Nexus of stress. Annu. Rev. Phys. 2010, 72, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. Mitochondrial centrality in heart failure. Heart Fail. Rev. 2008, 13, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 2008, 294, C460–C466. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Baracca, A.; Barbero, G.; Bergamini, C.; Dalmonte, M.E.; del Sole, M.; Faccioli, M.; Falasca, A.; Fato, R.; Genova, M.L.; et al. Mitochondrial respiratory chain super-complex I–III in physiology and pathology. Biochim. Biophys. Acta 2010, 1797, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Genova, M.L. Structure and organization of mitochondrial respiratory complexes: A new understanding of an old subject. Antioxid. Redox Signal. 2010, 12, 961–1008. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, G.; Zweier, J.L.; Duilio, C.; Kuppusamy, P.; Santoro, G.; Elia, P.P.; Tritto, I.; Cirillo, P.; Condorelli, M.; Chiariello, M.; et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 1993, 268, 18532–18541. [Google Scholar] [PubMed]
- Turer, A.T.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; van der Westhuizen, J.; Mathew, J.P.; Schwinn, D.A.; Glower, D.D.; Newgard, C.B.; Podgoreanu, M.V. Metabolomic profiling reveals distinct patterns of myocardial substrate use in humans withcoronary artery disease or left ventricular dysfunction during surgical ischemia/reperfusion. Circulation 2009, 119, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., III; Taegtmeyer, H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J. Clin. Investig. 1991, 87, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Schwarzer, M.; Chen, D.; Schrepper, A.; Amorim, P.A.; Schoepe, M.; Nguyen, T.D.; Mohr, F.W.; Khalimonchuk, O.; Weimer, B.C. Doenst Proteomic remodeling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc. Res. 2010, 85, 376–384. [Google Scholar] [PubMed]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef]
- Birner, C.; Dietl, A.; Deutzmann, R.; Schröder, J.; Schmid, P.; Jungbauer, C.; Resch, M.; Endemann, D.; Stark, K.; Riegger, G.; et al. Proteomic profiling implies mitochondrial dysfunction in tachycardia-induced heart failure. J. Card. Fail. 2012, 18, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Ouzounian, M.; Lee, D.S.; Gramolini, A.O.; Emili, A.; Fukuoka, M.; Liu, P.P. Predict, prevent and personalize: Genomic and proteomic approaches to cardiovascular medicine. Can. J. Cardiol. 2007, 23, 28A–33A. [Google Scholar] [CrossRef]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Goldenthal, M.J.; Ananthakrishnan, R.; Marín-García, J. Nuclear-mitochondrial cross-talk in cardiomyocyte T3 signaling: A time-course analysis. J. Mol. Cell. Cardiol. 2005, 39, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J. Thyroid hormone and myocardial mitochondrial biogenesis. Vascul. Pharmacol. 2010, 52, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.A.; Stevenson, L.W.; Luu, M.; Walden, J.A. Altered thyroid hormone metabolism in advanced heart failure. J. Am. Coll. Cardiol. 1990, 16, 91–95. [Google Scholar] [CrossRef]
- Wiersinga, W.M.; Lie, K.I.; Touber, J.L. Thyroid hormones in acute myocardial infarction. Clin. Endocrinol. (Oxf.) 1981, 14, 367–374. [Google Scholar] [PubMed]
- Friberg, L.; Drvota, V.; Bjelak, A.H.; Eggertsen, G.; Ahnve, S. Association between increased levels of reverse triiodothyronine and mortality after acute myocardial infarction. Am. J. Med. 2001, 111, 699–703. [Google Scholar] [CrossRef]
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Galli, E.; Barison, A.; Iervasi, A.; Scarlattini, M.; Nucci, D.; L’abbate, A.; Mariotti, R.; Iervasi, G. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2008, 93, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I.; Markakis, K.; Tsagoulis, N.; Panagiotou, M.; Cokkinos, D.V. Long term thyroid hormone administration reshapes left ventricular chamber and improves cardiac function after myocardial infarction in rats. Basic Res. Cardiol. 2008, 103, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Kobayashi, S.; Chen, J.; Redetzke, R.A.; Said, S.; Liang, Q.; Gerdes, A.M. Short term triiodo-lthyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J. Mol. Cell. Cardiol. 2008, 44, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Lionetti, V.; Ardehali, H.; Pucci, A.; Cecchetti, F.; Ghanefar, M.; Nicolini, G.; Ichikawa, Y.; Nannipieri, M.; Recchia, F.A.; et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell. Mol. Med. 2011, 5, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Kusmic, C.; Nicolini, G.; Mariani, L.; Zucchi, R.; Matteucci, M.; Iervasi, G.; Pitto, L. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014, 155, 4581–4590. [Google Scholar] [CrossRef] [PubMed]
- Saba, A.; Donzelli, R.; Colligiani, D.; Raffaelli, A.; Nannipieri, M.; Kusmic, C.; dos Remedios, C.G.; Simonides, W.S.; Iervasi, G.; Zucchi, R. Quantification of thyroxine and 3,5,3′-triiodo-thyronine in human and animal hearts by a novel liquid chromatography-tandem mass spectrometry method. Horm. Metab. Res. 2014, 46, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as Key Targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312–6336. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Benjamin, I.J. Protective responses in the ischemic myocardium. HSPs play an important role in the defense mechanism against IR injury. J. Clin. Investig. 2000, 106, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Mestril, R.; HilalDandan, R.; Brunton, L.L.; Dillmann, W.H. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation 1997, 96, 4343–4348. [Google Scholar] [CrossRef] [PubMed]
- Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Sun, L.; Mochly-Rosen, D. Mitochondrial import of PKCepsilon is mediated byHSO90: A role in cardioprotection from ischaemia andreperfusion injury. Cardiovasc. Res. 2010, 88, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W.H. Heat shock proteins and protection against ischemic injury. Infect. Dis. Obstet. Gynecol. 1999, 7, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Kraeft, S.K.; KurtJones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14 dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [PubMed]
- Xiao, X.; Benjamin, I.J. Stress response proteins in cardiovascular disease. Am. J. Hum. Genet. 1999, 64, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Vander Heide, R.S. Increased expression of HSP27 protects canine myocytes from simulated ischemia reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2000, 282, H935–H941. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.D.; Kim, J. Cytoplasmic ribosomal protein S3 (rpS3) plays a pivotal role in mitochondrial DNA damage surveillance. Biochim. Biophys. Acta 2013, 1833, 2943–2952. [Google Scholar] [CrossRef] [PubMed]
- Jankevicius, G.; Hassler, M.; Golia, B.; Rybin, V.; Zacharias, M.; Timinszky, G.; Ladurner, A.G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013, 20, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, F.; Feijs, K.L.; Frugier, E.; Bonalli, M.; Forst, A.H.; Imhof, R.; Winkler, H.C.; Fischer, D.; Caflisch, A.; Hassa, P.O.; et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013, 20, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Fan, G.C.; Zhang, Z.G.; Bandyopadhyay, A.; Zhou, X.; Kranias, E.G. Protection of peroxiredoxine II on oxidative stress-induced cardiomyocyte death and apoptosis. Basic Res. Cardiol. 2009, 104, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kok, K.H.; Chun, A.C.; Wong, C.M.; Wu, H.W.; Lin, M.C.; Fung, P.C.; Kung, H.; Jin, D.Y. Mouse peroxiredoxin V is a thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochem. Biophys. Res. Commun. 2000, 268, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Oba, D.; Dai, S.; Keith, R.; Dimova, N.; Kingery, J.; Zheng, Y.T.; Zweier, J.; Velayutham, M.; Prabhu, S.D.; Li, Q.; et al. Cardiomyocyte restricted overexpression of extracellular superoxide dismutase increases nitric oxide bioavailability and reduces infarct size after ischemia/reperfusion. Basic Res. Cardiol. 2012, 107, 305. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sawa, Y.; Ichikawa, H.; Kaneda, Y.; Matsuda, H. Myocardial protection with endogenous overexpression of manganese superoxide dismutase. Ann. Thorac. Surg. 1999, 68, 1266–1271. [Google Scholar] [CrossRef]
- Shanmuganathan, S.; Hausenloy, D.J.; Duchen, M.R.; Yellon, D.M. Mitochondrial permeability transition pore as a target for cardioprotection in the human heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H237–H242. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Mastroberardino, P.G.; Váradi, J.; Farrace, M.G.; Nagy, N.; Bak, I.; Viti, I.; Wieckowski, M.R.; Melino, G.; Rizzuto, R.; et al. Tissue transglutaminase (TG2) protects cardiomyocytes against ischemia/reperfusion injury by regulating ATP. Cell Death Differ. 2006, 13, 1827–1829. [Google Scholar] [CrossRef] [PubMed]
- Rossin, F.; D’Eletto, M.; Falasca, L.; Sepe, S.; Cocco, S.; Fimia, G.M.; Campanella, M.; Mastroberardino, P.G.; Farrace, M.G.; Piacentini, M. Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic. Cell Death Differ. 2015, 22, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 28, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Brosel, S.; Yang, H.; Schon, E.A.; Manfredi, G. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol. Med. 2009, 1, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Brunati, A.M.; Toninello, A. Tyrosine phosphorylation in mitochondria: A new frontier in mitochondrial signaling. Free Radic. Biol. Med. 2005, 38, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, G.; Varughese, A.; Qinqin, J.; Lee, I.; Salomon, A.; Huttemann, M. Role of cytochrome c phosphorylation in regulation of respiration and apoptosis. FASEB J. 2015, 29, 725. [Google Scholar]
- Chinopoulos, C. Which way does the citric acid cycle turn during hypoxia? The critical role of α ketoglutarate dehydrogenase complex. J. Neurosci. Res. 2013, 91, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.; Aponte, A.M.; French, S.A.; Chess, D.J.; Balaban, R.S. Succinyl-CoA synthetase is a phosphate target for the activation of mitochondrial metabolism. Biochemistry 2009, 48, 7140–7149. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; King, L.M.; Jones, B.E. Energy substrate metabolism, myocardial ischemia, and targets for pharmacotherapy. Am. J. Cardiol. 1998, 82, 54K–60K. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Goodwin, G.W.; Doenst, T.; Frazier, O.H. Substrate metabolism as a determinant for postischemic functional recovery of the heart. Am. J. Cardiol. 1997, 80, 3A–10A. [Google Scholar] [CrossRef]
- Olson, A.K.; Bouchard, B.; Ning, X.-H.; Isern, N.; Rosiers, C.D.; Portman, M.A. Triiodothyronine increases myocardial function and pyruvate entry into the citric acid cycle after reperfusion in a model of infant cardiopulmonary bypass. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1086–H1093. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.K.; Hyti, O.M.; Cohen, G.A.; Ning, X.-H.; Sadilek, M.; Isern, N.; Portman, M.A. Superior cardiac function via anaplerotic pyruvate in the immature swine heart after cardiopulmonary bypass and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2315–H2320. [Google Scholar] [CrossRef] [PubMed]
- Kjellman, U.; Bjork, K.; Ekroth, R.; Karlsson, H.; Jagenburg, R.; Nilsson, F.; Svensson, G.; Wernerman, J. α-Ketoglutarate for myocardial protection in heart surgery. Lancet 1995, 345, 552–553. [Google Scholar] [CrossRef]
- Kjellman, U.W.; Bjork, K.; Ekroth, R.; Karlsson, H.; Jagenburg, R.; Nilsson, F.N.; Svensson, G.; Wernerman, J. Addition of alpha-ketoglutarate to blood cardioplegia improves cardioprotection. Ann. Thor. Surg. 1997, 63, 1625–1633. [Google Scholar] [CrossRef]
- Mullis-Jansson, S.L.; Argenziano, M.; Corwin, S.; Homma, S.; Weinberg, A.D.; Williams, M.; Rose, E.A.; Smith, C.R. A randomized double-blind study of the effect of triiodothyronine on cardiac function and morbidity after coronary bypass surgery. J. Thorac. Cardiovasc. Surg. 1999, 117, 1128–1134. [Google Scholar] [CrossRef]
- Bilsen, M.; van Nieuwenhoven, F.A.; van der Vusse, G.J. Metabolic remodelling of the failing heart: Beneficial or detrimental? Cardiovasc. Res. 2009, 81, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A. Reversal of metabolic shift in post-infarct-remodelled hearts: Possible novel therapeutic approach. Cardiovasc. Res. 2013, 1, 195–196. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lou, P.H.; Zhang, L.; Lucchinetti, E.; Heck, M.; Affolter, A.; Gandhi, M.; Kienesberger, P.C.; Hersberger, M.; Clanachan, A.S.; Zaugg, M. Infarct-remodelled hearts with limited oxidative capacity boost fatty acid oxidation after conditioning against ischaemia/reperfusion injury. Cardiovasc. Res. 2013, 97, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Goldenthal, M.J.; Weiss, H.R.; Marín-García, J. Bioenergetic remodeling of heart mitochondria by thyroid hormone. Mol. Cell. Biochem. 2004, 265, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kusmic, C.; Barsanti, C.; Matteucci, M.; Vesentini, N.; Pelosi, G.; Abraham, N.G.; L’Abbate, A. Up-regulation of heme oxygenase-1 after infarct initiation reduces mortality, infarct size and left ventricular remodeling: Experimental evidence and proof of concept. J. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kristián, T.; Hopkins, I.B.; McKenna, M.C.; Fiskum, G. Isolation of mitochondria with high respiratory control from primary cultures of neurons and astrocytes using nitrogen cavitation. J. Neurosci. Methods 2006, 152, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hüttenhain, R.; Collins, B.; Aebersold, R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Rev. Mol. Diagn. 2013, 13, 811–825. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forini, F.; Ucciferri, N.; Kusmic, C.; Nicolini, G.; Cecchettini, A.; Rocchiccioli, S.; Citti, L.; Iervasi, G. Low T3 State Is Correlated with Cardiac Mitochondrial Impairments after Ischemia Reperfusion Injury: Evidence from a Proteomic Approach. Int. J. Mol. Sci. 2015, 16, 26687-26705. https://doi.org/10.3390/ijms161125973
Forini F, Ucciferri N, Kusmic C, Nicolini G, Cecchettini A, Rocchiccioli S, Citti L, Iervasi G. Low T3 State Is Correlated with Cardiac Mitochondrial Impairments after Ischemia Reperfusion Injury: Evidence from a Proteomic Approach. International Journal of Molecular Sciences. 2015; 16(11):26687-26705. https://doi.org/10.3390/ijms161125973
Chicago/Turabian StyleForini, Francesca, Nadia Ucciferri, Claudia Kusmic, Giuseppina Nicolini, Antonella Cecchettini, Silvia Rocchiccioli, Lorenzo Citti, and Giorgio Iervasi. 2015. "Low T3 State Is Correlated with Cardiac Mitochondrial Impairments after Ischemia Reperfusion Injury: Evidence from a Proteomic Approach" International Journal of Molecular Sciences 16, no. 11: 26687-26705. https://doi.org/10.3390/ijms161125973
APA StyleForini, F., Ucciferri, N., Kusmic, C., Nicolini, G., Cecchettini, A., Rocchiccioli, S., Citti, L., & Iervasi, G. (2015). Low T3 State Is Correlated with Cardiac Mitochondrial Impairments after Ischemia Reperfusion Injury: Evidence from a Proteomic Approach. International Journal of Molecular Sciences, 16(11), 26687-26705. https://doi.org/10.3390/ijms161125973