Abstract
Senescent cells are relatively stable, lacking proliferation capacity yet retaining metabolic activity. In contrast, cancer cells are rather invasive and devastating, with uncontrolled proliferative capacity and resistance to cell death signals. Although tumorigenesis and cellular senescence are seemingly opposite pathological events, they are actually driven by a unified mechanism: DNA damage. Integrity of the DNA damage response (DDR) network can impose a tumorigenesis barrier by navigating abnormal cells to cellular senescence. Compromise of DDR, possibly due to the inactivation of DDR components, may prevent cellular senescence but at the expense of tumor formation. Here we provide an overview of the fundamental role of DDR in tumorigenesis and cellular senescence, under the light of the Yin-Yang concept of Chinese philosophy. Emphasis is placed on discussing DDR outcome in the light of in vivo models. This information is critical as it can help make better decisions for clinical treatments of cancer patients.
1. Introduction
1.1. DNA Damage
The integrity and fidelity of DNA is pivotal for accurately passing genetic information from generation to generation. However, over an individual’s lifespan, DNA is constantly exposed to exogenous and endogenous insults. Exogenous sources of damage can come from harmful chemicals, ultraviolet light (UV) and ionizing radiation (IR), whereas endogenous hazards arise from reactive oxygen species (ROS) produced in normal metabolic processes, telomere shortening induced by cell division and “DNA replication stress” imposed by activation of oncogenes or inactivation of tumor suppressor genes. In response to DNA damage, organisms are capable of launching repair mechanisms, predominantly homologous recombination (HR) and non-homologous end joining (NHEJ), to counteract the potential damage. HR is mostly error-free, requiring an intact sister chromatid as a template for repair, by contrast, NHEJ is error-prone due to the lack of an intact template. The molecular mechanisms of DNA damage repair are not the primary focus of our review as this has been comprehensively reviewed by Thompson et al. [1].
1.2. DNA Damage Response (DDR)
In addition to repair mechanisms, individuals have evolved a so called “DNA damage response”, which is responsible for invoking a myriad of cellular events in response to genotoxic stress. DNA damage response is mainly mediated by the activation of ATM (ataxia telangiectasia mutated)-CHK2 (cell cycle the checkpoint kinase 2)-p53 and ATR (ataxia telangiectasia and rad3-related)-CHK1 (cell cycle checkpoint kinase 1)-CDKs (Cyclin-dependent kinases) pathways. Once activated, these signaling cascades can trigger cell cycle arrest (so called “checkpoint”), thereby gaining time for DNA damage repair and preventing the propagation of damaged cells [2–5]. ATM and ATR belong to the same family and share some functional redundancy. However, these proteins are distinct because they respond to different aberrant DNA structures. ATM, in principle, is elicited by double-strand breaks (DSB) and recruited via interaction with DSB sensors, MRN complex (MRE11-RAD50-NBS1) [6–12]. In contrast, ATR is induced by single-strand breaks (SSB) and engaged by its partner protein, ATRIP (ATR interacting protein) through interaction with the SSB sensor RPA (replicative protein A) [13–17]. Consequently, phosphorylated ATM and ATR, acting as transducer proteins, can active the effector kinases CHK1 and CHK2, with the help of the mediator proteins; MDC1 (mediator of DNA damage checkpoint), 53BP1 (p53-binding protein 1), BRCA1 (breast cancer 1) for ATM, and TopBP1 (topoisomerase-binding protein 1) and Claspin for ATR [1,18,19] (Figure 1). Under normal circumstances, p53 is easily degraded and hence rarely detected. Upon stress, p53 is phosphorylated and stabilized following ATM and, to a less extent, ATR stimulation [20–23]. Additionally, p53 stabilization can be achieved through ARF (alternate reading frame) activation imposed by oncogene-induced replication stress [24]. The stimulation of ARF relieves the inhibitory effect of MDM2 (mouse double minute 2 homolog) on p53 [25–27]. Once p53 is stabilized and activated, it can orchestrate a range of cellular stress responses including cell cycle arrest, senescence and apoptosis. The various outcomes are determined by the intensity of stress as well as the tissue and cellular context [28,29].
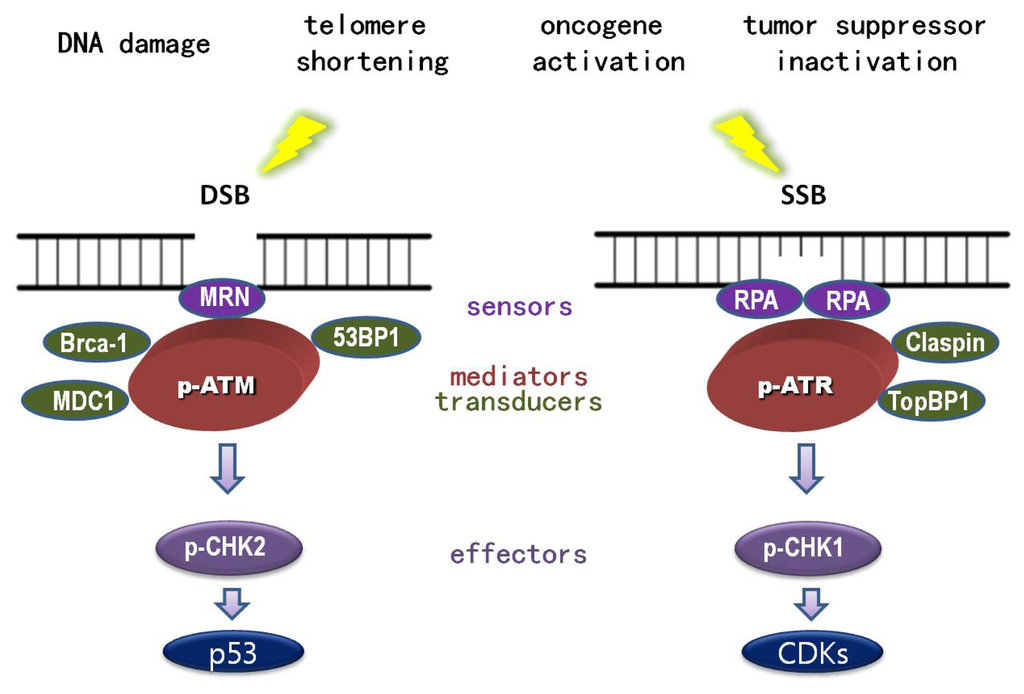
Figure 1.
DNA damage and DNA damage response (DDR). Both external insults and internal hazards can cause DNA damage. DNA damage response is coordinated by various proteins whose functions can be categorized as DNA damage sensors, transducers, mediators, and effectors. Double strand DNA damage (DSB) can be detected by MRN complex (sensor) to recruit and activate transducer ATM (ataxia Telangiectasia mutated) to activate CHK2 (effector), with the help of DDR mediators MDC1 (mediator of DNA damage checkpoint), 53BP1 (p53-binding protein 1), and BRCA1 (breast cancer 1). In contrast, single strand DNA damage (SSB) could be detected by sensor protein, RPA (replicative protein A), to recruit and activate transducer ATR (ataxia telangiectasia- and Rad3-related), to activate CHK1 (effector), with the help of mediators TopBP1 (topoisomerase-binding protein 1) and Claspin. p53 and CDKs are the major downstream substrates in response to DSB and SSB respectively.
2. Pathways of Senescence-Associated Cell Cycle Arrest
The cell cycle, comprised of S phase (DNA synthesis), M phase (mitosis) and two gap phases (G0 and G1), is coordinately regulated by cell cycle proteins (cyclins), cyclin-dependent kinases (CDKs) and cyclin-dependent kinase inhibitors (CDKIs). CDKIs are negative modulators of cell cycle and hence also viewed as tumor suppressor genes. CDKIs can be grouped into two categories, the KIP/CIP family (p21Cip1, p27Kip1 and p57KipII) and the INK4 family (p16Ink4a, p15Ink4b, p18Ink4c and p19Ink4d). As illustrated in Figure 2, through cellular events triggered by genomic stress, stimulated p53 transactivates p21, which, in turn, inhibits CDK2/cyclin E and thereby retains Rb (Retinoblastoma) in an inactive unphosphorylated state. Unphosphorylated Rb suppresses the function of the G1/S phase-promoting, E2F, and as a result, cells are subjected to proliferation arrest and DNA damage repair [30–32]. Compelling evidence also points to a critical role of p16 as one of the central modulators of cell cycle arrest. The p16 can inhibit the CDK4-6/cyclin D complex thereby reducing Rb phosphorylation and subsequent downstream signal transduction pathways. Thus, cells will arrest in G1 phase and fail to complete the cell cycle. Since both pathways engage pRb, it is plausible to speculate proliferation arrest, in response to cellular stress, which is coordinately regulated by p53/p21/pRb/E2F and p16/pRb/E2F signal transduction pathways.
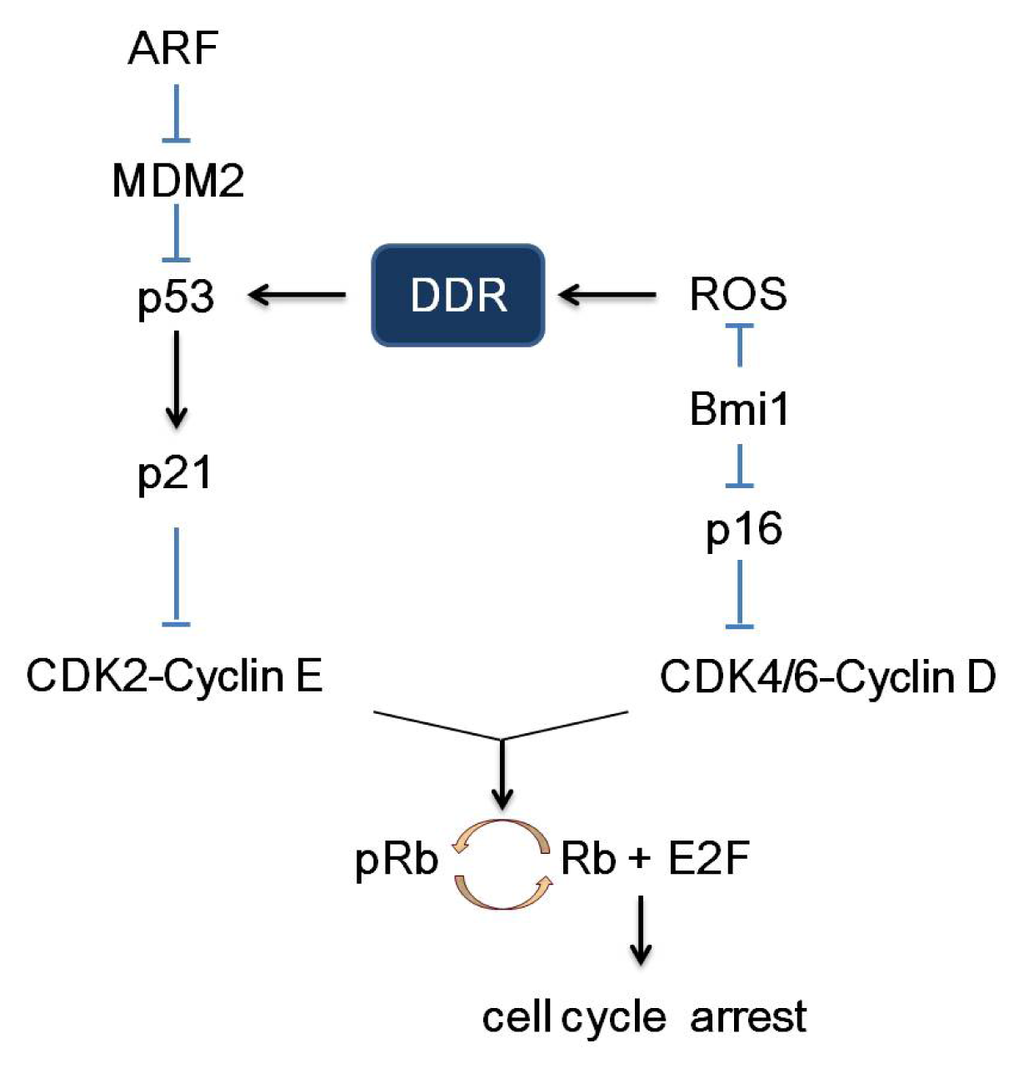
Figure 2.
p53/p21/pRb/E2F and p16/pRb/E2F signaling pathway. The INK4a/ARF locus encodes both ARF (alternate reading frame) and p16 protein. ARF could stimulate p53 through inhibition and degradation of MDM2 (mouse double minute 2 homolog). Activated p53 transactivates p21, which, in turn, inhibits CDK2/cyclin E with the consequent inhibition of CDK2-dependent phosphorylation of Rb. Unphosphorylated Rb suppresses the function of the G1/S phase-promoting factor, E2F, and as a result, cells are subjected to proliferation arrest and DNA damage repair. Similarly, p16 can inhibit the CDK4-6/cyclin D complex thereby reducing Rb phosphorylation and subsequent downstream signal transduction pathways. Thus, cells will arrest in G1 phase and fail to complete the cell cycle. Bmi1, a polycomb group protein, is a transcriptional repressor of p16. Bim1 also has a potential inhibitory role on reactive oxygen species (ROS) production [33].
Senescence is a permanent form of cell-cycle arrest, first discovered in normal human fibroblasts by Hayflick [34]. Unlike normal cells, senescent cells are relatively stable, lacking proliferation capacity but retaining metabolic activity. These cells possess large and flattened morphology, increased intracellular particles, as well as enhanced senescence-associated β-galactosidase (SA-β-gal) activity. Under normal circumstances, as cells cycles, telomere, a special structure at the ends of chromosomes, is gradually shortened. When the length of the telomere reaches a certain limit, cell proliferation is halted and cellular senescence is elicited [35]. Such senescence is known as replicative senescence. In addition to the replicative senescence, senescence can also result from DNA damage aroused from oxidative stress or oncogene activation-induced replication stress, which is termed as premature senescence, or oncogene-induced senescence (OIS) [36–39].
Consistent with the notion that senescence is a permanent form of cell cycle arrest, factors central to checkpoint events, such as p53, p21, p16 and Rb, are also key regulators of the senescence program. In human cells, replication senescence is commonly dependent on p53/p21/pRb/E2F pathway, whereas premature senescence can be mediated through p53/p21/pRb/E2F pathway, p16/pRb/E2F pathway or both [35,40]. Mechanistically, little is known as to how a cell chooses one way over the other, however, some evidence implies that it might be associated with types of stimulus and cell context [41–44]. Given the complexity of cellular responses to various stimuli, the chances are that these pathways could be cooperative and intertwined in stress-induced senescence and associated cell cycle arrest [45,46].
3. DDR Is the Common Link between Tumorigenesis and Senescence
It has been known for decades that constitutive activation of oncogenes, such as Ras, is capable of driving the proliferation of malignant tumor cells [47]. Together with the observations that expression of the same oncogenes in normal cell culture leads to cell senescence rather than cell transformation [36,48,49], it raises an obvious question as to how oncogenes lead to both tumor and senescence in a similar scenario. One explanation to reconcile the paradox is that oncogene-induced DDR and resultant senescence may occur prior to tumorigenesis, imposing an intrinsic barrier to the development of the malignant tumor. In effect, a large body of emerging evidence from both bench and clinical works supports this conception [50–52]. In the early stages of tumor formation, many factors along the DDR pathway, such as ATM, CHK1, CHK 2, p53 and p16, as well as markers of DNA damage foci, such as H2AX and 53BP1, could be detected, mostly in their phosphorylated activated form [50,51,53–58]. Senescent cells and their specific markers, such as SA-β-Gal, were also present in precancerous lesions [53,59–64]. The correlation between markers of activated DDR with those of cellular senescence reinforces the crucial role of DDR signaling in oncogene-induced senescence [50,51,65]. Remarkably, markers for senescence-associated DDR were attenuated or absent in the later stage of cancer [53,59–62], leading to the speculation that senescence impinges selective pressure on hyper-proliferative tumors with mutations of checkpoint genes. Agreeably, cells escaped from OIS by depletion or inhibition of several important DNA damage signaling factors, such as 53BP1, ATM, CHK1, CHK2, and p53, predisposes to cell proliferation and oncogenic transformation [36,50,51,53,54,57,58]. Collectively, based on the results summarized above, the following concepts are emerging: (1) DNA damage is the common driving force for both tumorigenesis and cellular senescence (2) senescence-associated DDR acts as a natural barrier for tumorigenesis, and abrogation of the barrier may rescue defective cell growth and limit cell senescence in the incipient malignant form but at the expense of tumor progression [66–68]. The outcome as to whether cells predispose to tumor or senescence upon DNA damage is largely dependent on the competence of DDR signaling. To achieve the “Yin-Yang balance” of DDR, signaling is critical for preventing both tumors and senescence. The Yin and Yang of DDR in tumors and senescence could be further exemplified in the light of a number of in vivo mice models with genetic deletions of certain genes involved in the DDR pathways.
3.1. p53
Given the critical role of p53 as the “guardian of the genome”, its activity must be tightly regulated. Either too much, or too little p53 activity will have adverse effects, as demonstrated comprehensively in various mouse models using gene manipulation to alter p53 levels [69,70]. The p53 null mice are largely tumor-prone, consistent with the fact that p53 mutations are the most prevalent mutations in human cancers [71–74]. By contrast, mice with high p53 activity, such as p53+/m mice, are less cancer-prone compared to the control mice, but display obvious age-related phenotypes such as tissue atrophy [69,75]. The overall high p53 activity in p53+/m mice was attributed to the stabilization and enhancement of the p53 by the m allele product [69]. These findings clearly demonstrated the existence of a delicate balance between the tumor suppression and age promoting functions of p53. To optimize the outcome of p53-dependent DNA damage response and tip the Yin-Yang balance between tumor suppression and age promoting it is crucial to involve p53 in clinical applications. Cancer protection without negatively affecting aging was observed in mice containing an extra copy of p53 (super-p53 mice) [76,77]. In contrast to the p53+/m mice model, the desired phenotype from super-p53 mice might be attributed to the maintenance of the normal regulation of p53 activity. Nevertheless, super-p53 mice raise great hopes of involving p53 as a potential anti-tumor treatment and further studies are needed to ascertain whether this scenario could be sustainable in humans.
3.2. p21 and p27
It is commonly noted that p53-induced senescence is executed, at least partly, via p21 upregulation [45]. The p21 levels are elevated in prematurely senescing fibroblasts in both humans and mice displaying premature aging syndromes, and p21−/− mice exhibited deficiencies in senescence response in comparison to WT mice after UV damage [78–81]. In support of the notion that p53/p21 is the major determinant in response to telomere-dysfunction-aroused replicative senescence, p21 deficiency could reverse the short lifespan of mice with telomerase deficiency (Terc−/−) [82,83]. By contrast, lacking p53 increases genomic instability and cancer formation in vivo, thereby reducing the lifespan of mice with dysfunctional telomeres [84,85]. In the early age of p21−/− mice, tumor is rarely detected and an increased tumor formation occurs at an average age of 16 months, with the most common tumor types being sarcomas and B cell lymphomas [86,87]. Mice with genetic deletion of DDB2 (damaged DNA binding protein 2), a significant player in recognizing DNA damage in NER (nucleotide excision repair), exhibited reduced senescence response and induced enhanced incidence of UV-induced skin cancers in comparison to wild-type mice [81,88–91]. A stronger inhibition of premature senescence and accelerated tumor formation in the DDB2−/− p21−/− mice compared to the DDB2−/− single knockouts, further support the tumor-suppressive role of p21 [92]. However, taking into consideration the extremely low incidence of p21 mutations in human cancer and the less drastic tumor-prone phenotype in p21-deficient mice in comparison to mice deficient in other tumor suppressors, such as p53 or p16, the tumor suppressive role of p21 is not viewed as crucial. Recently, the tumor suppressive role of p21 was complicated by findings indicating that p21 has an inhibitory role in apoptosis [93–95]. The involvement of p21 in these pathways could confer its oncogenic activities, particularly in lymphomas [96,97].
Similar to p21, p27, another member from the KIP/CIP family, binds and inactivates CDK2-cyclin E and CDK2-cyclin A complex thereby inhibiting cell-cycle progression [98]. The p27 expression is frequently reduced in human epithelial cancers, and is correlated with tumor progression and poor survival [99–103]. Targeted disruption of p27 in the mouse model leads to multi-organ hyperplasia, loss of senescence markers and an increased tumor latency [104–108].
3.3. BRCA1
The tumor suppressor BRCA1 is intimately associated with an increased risk of breast and ovarian cancer [109–114]. BRCA1 is a DNA damage repair protein crucial for maintaining genomic stability. The abrogation of the full-length isoform leads to genomic instability and embryonic death [114–118]. This lethal phenotype is partially due to the excessive activation of the ATM-CHK2-p53 axis which leads to accelerated aging, in response to untimely repaired chromosome breaks in the absence of BRCA1 [119,120]. In support of this notion, loss of p53 prolonged the survival of BRCA1 mutant embryos from E7.5 to E9.5 [121]. Moreover, haploid loss of p53 in BRCA1 null mice (BRCA1Δ11/Δ11 p53+/−) could completely overcome embryonic lethality, but display cancer susceptibility mostly in female mice, and premature aging, mainly in male mice [119,122]. The survival of BRCA1Δ11/Δ11 p53+/− mice makes it a surrogate model to study the link between BRCA1 and aging. Consistent with the contribution of DDR cascade in BRCA1 deficiency, enhanced ATM and CHK2 activity was observed in BRCA1Δ11/Δ11 embryos. Consistently, absence of ATM, CHK2 or 53BP1 can mean escape of embryonic lethality in BRCA1 knockout mice and suppression of accelerated aging albeit at the expense of entering a tumor-prone state [120,123] (Figure 3). These observations highlight the critical role of genetic integrity, whose compromise may disturb organismal homeostasis and result in senescence and cancer.
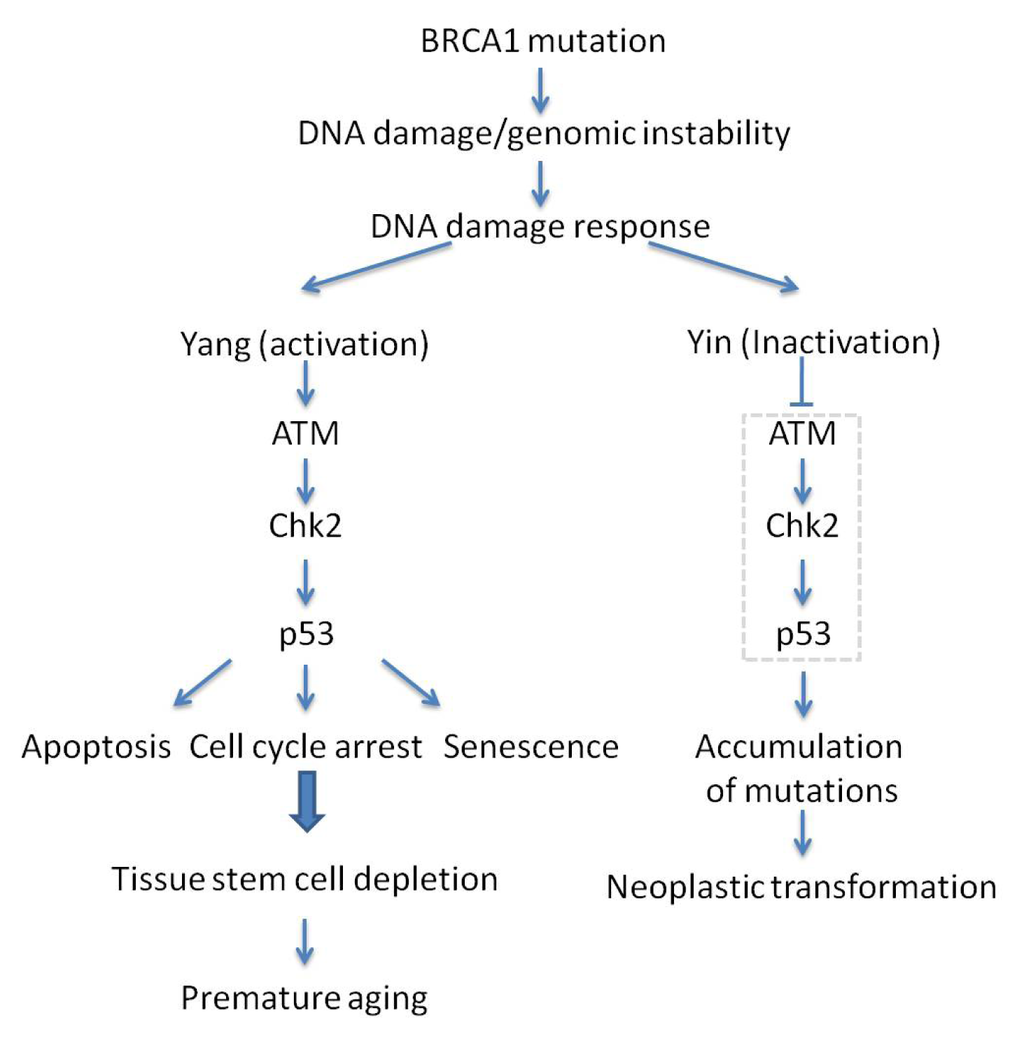
Figure 3.
Yin and Yang of the ATM-CHK2-p53 signaling pathway upon BRCA1 mutation-associated premature aging and tumorigenesis. ATM-CHK2-p53 signaling pathway senses DNA damage/genomic instability and acts as a gatekeeper to eliminate mutations, but, as a side effect, it may also lead to premature aging.
3.4. p16 and Bmi1
It is evident that p16, another important component within the DDR network, is largely involved in the senescence program, acting not only as an effector but also a biomarker of senescence [124–129]. The expression of p16 is markedly elevated with age in both rodents and human tissues [128,129]. Nevertheless, mice deficient in p16 displayed an increased incidence of tumorigenesis and loss of inactivation of p16 accounts for more than 30% of human tumors [130–133]. Hence, p16 is ubiquitously linked to both tumorigenesis and senescence. Bmi1, a polycomb group protein, is a transcriptional repressor of p16. Overexpression of Bmi1 could extend the replicative life span of primary cells but promote the formation of lymphomas [42,134–137]. Conversely, mouse embryonic fibroblasts deficient in Bmi1 possess high p16 activity and undergo premature senescence, which can be relieved partially in the absence of p16 [42]. However, the median survival of Bmi1−/− mice cannot be extended by p16 deficiency, but instead it could be improved by complete loss of CHK2 [33]. It is postulated that this may be due to the inhibition of ROS-induced DDR.
Collectively, p53/p21 and p16/Rb pathways are not only important for DDR signaling, but also critical in maintaining cellular and genomic homeostasis. Several lines of evidence indicate p53/p21 and p16/Rb pathways are collaborative. For instance, MEF derived from p21 and p16 double knockout mice displayed no evidence of cellular senescence to Ras-induced senescence and have a higher incidence of cancer compared to either of the single KO mice [138,139].
4. Will the Yin-Yang of DDR Be Beneficial for Clinical Treatment of Cancer?
The key mechanism of the most prevalent cancer therapy, radiation therapy and chemotherapy, is to damage DNA and consequently trigger DDR, tumor growth arrest, apoptosis and senescence [140–142]. The extensive DNA damage induced by these current therapies inevitably puts patients to severe side effect risk such as hair loss and bone marrow suppression [143]. Given the frequent loss of critical DDR proteins in cancer, new possibilities for tumor intervention have been postulated to re-establish the barrier or even induce tumor to senescence through exogenously introducing or molecular targeting of proteins involved in the DDR signaling. Recent studies found that chemotherapy or gene therapy, by modulating the activity of p16, p53, pRb or p21, could navigate tumor cells to senescent cells and have a substantial therapeutic effect on tumor inhibition [61,144–146]. More recently, a number of senescence-inducing small molecules entered clinical trials [147–151]. Certainly, the safety of pro-senescence therapy needs to be carefully evaluated before translating it into a clinically relevant context. Senescence is frequently accompanied with oxidative stress, altered tissue microenvironment and release of inflammatory cytokines [147,152,153]. All of these could potentially promote cancer and aging phenotypes [40,154]. Greater understanding of the molecular mechanisms involved in senescence and tumors will provide valuable new insights into how to bypass undesired side effects in senescence-inducing treatment. In the near future, it is warranted to consider the combination of pro-senescence strategies with already established treatments.
5. Conclusions
Organisms are natural perfectionists and dedicated hard workers. In case of any threat, a myriad of mechanisms engage in an intricate interplay to keep damage to a minimum. Networks of molecules have evolved to work coordinately to maintain the homeostasis of the body. However, imbalanced DNA damage response, upon genotoxic stress, can endanger cellular homeostasis, leading to the transition from a healthy to a disease state, including senescence and cancer. As we learned from ancient Chinese philosophy, finding the balance between “Yin and Yang” will ensure both health and longevity.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China to Liu Cao (81130042 and 31171323) and from the University Innovation Team support plan of Liaoning to Liu Cao (LT2011011).
Conflict of Interest
The authors declare no conflict of interest.
References
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res 2012, 751, 158–246. [Google Scholar]
- Lopez-Girona, A.; Tanaka, K.; Chen, X.B.; Baber, B.A.; McGowan, C.H.; Russell, P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 11289–11294. [Google Scholar]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol 2009, 21, 245–255. [Google Scholar]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 2000, 14, 2745–2756. [Google Scholar]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol 2001, 21, 4129–4139. [Google Scholar]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar]
- Suzuki, K.; Kodama, S.; Watanabe, M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J. Biol. Chem 1999, 274, 25571–25575. [Google Scholar]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar]
- Kim, J.S.; Krasieva, T.B.; Kurumizaka, H.; Chen, D.J.; Taylor, A.M.; Yokomori, K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J. Cell Biol 2005, 170, 341–347. [Google Scholar]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003, 22, 5612–5621. [Google Scholar]
- Dart, D.A.; Adams, K.E.; Akerman, I.; Lakin, N.D. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem 2004, 279, 16433–16440. [Google Scholar]
- Lupardus, P.J.; Byun, T.; Yee, M.C.; Hekmat-Nejad, M.; Cimprich, K.A. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev 2002, 16, 2327–2332. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res 2010, 108, 73–112. [Google Scholar]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar]
- Noon, A.T.; Goodarzi, A.A. 53BP1-mediated DNA double strand break repair: Insert bad pun here. DNA Repair 2011, 10, 1071–1076. [Google Scholar]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev 2011, 25, 409–433. [Google Scholar]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; Ziv, Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar]
- Khanna, K.K.; Keating, K.E.; Kozlov, S.; Scott, S.; Gatei, M.; Hobson, K.; Taya, Y.; Gabrielli, B.; Chan, D.; Lees-Miller, S.P.; Lavin, M.F. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat. Genet 1998, 20, 398–400. [Google Scholar]
- Lakin, N.D.; Hann, B.C.; Jackson, S.P. The ataxia-telangiectasia related protein ATR mediates DNA-dependent phosphorylation of p53. Oncogene 1999, 18, 3989–3995. [Google Scholar]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar]
- Meulmeester, E.; Maurice, M.M.; Boutell, C.; Teunisse, A.F.; Ovaa, H.; Abraham, T.E.; Dirks, R.W.; Jochemsen, A.G. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 2005, 18, 565–576. [Google Scholar]
- Meulmeester, E.; Pereg, Y.; Shiloh, Y.; Jochemsen, A.G. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle 2005, 4, 1166–1170. [Google Scholar]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J 2004, 23, 1547–1556. [Google Scholar]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ 2003, 10, 431–442. [Google Scholar]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar]
- Dulic, V.; Kaufmann, W.K.; Wilson, S.J.; Tlsty, T.D.; Lees, E.; Harper, J.W.; Elledge, S.J.; Reed, S.I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994, 76, 1013–1023. [Google Scholar]
- Reed, S.I.; Bailly, E.; Dulic, V.; Hengst, L.; Resnitzky, D.; Slingerland, J. G1 control in mammalian cells. J. Cell Sci. Suppl 1994, 18, 69–73. [Google Scholar]
- Liu, J.; Cao, L.; Chen, J.; Song, S.; Lee, I.H.; Quijano, C.; Liu, H.; Keyvanfar, K.; Chen, H.; Cao, L.Y.; et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009, 459, 387–392. [Google Scholar]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res 1965, 37, 614–636. [Google Scholar]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar]
- Bartek, J.; Lukas, J.; Bartkova, J. DNA damage response as an anti-cancer barrier: Damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle 2007, 6, 2344–2347. [Google Scholar]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001, 11, S27–S31. [Google Scholar]
- Shay, J.W.; Roninson, I.B. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004, 23, 2919–2933. [Google Scholar]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar]
- Lundberg, A.S.; Hahn, W.C.; Gupta, P.; Weinberg, R.A. Genes involved in senescence and immortalization. Curr. Opin. Cell Biol 2000, 12, 705–709. [Google Scholar]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar]
- Carnero, A.; Hudson, J.D.; Price, C.M.; Beach, D.H. p16INK4A and p19ARF act in overlapping pathways in cellular immortalization. Nat. Cell Biol 2000, 2, 148–155. [Google Scholar]
- Sharpless, N.E. Ink4a/Arf links senescence and aging. Exp. Gerontol 2004, 39, 1751–1759. [Google Scholar]
- Brown, J.P.; Wei, W.; Sedivy, J.M. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997, 277, 831–834. [Google Scholar]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol 2009, 1, a001883. [Google Scholar]
- Bos, J.L. The ras gene family and human carcinogenesis. Mutat. Res 1988, 195, 255–271. [Google Scholar]
- Braig, M.; Schmitt, C.A. Oncogene-induced senescence: Putting the brakes on tumor development. Cancer Res 2006, 66, 2881–2884. [Google Scholar]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar]
- DiTullio, R.A., Jr; Mochan, T.A.; Venere, M.; Bartkova, J.; Sehested, M.; Bartek, J.; Halazonetis, T.D. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 2002, 4, 998–1002. [Google Scholar]
- Bartkova, J.; Bakkenist, C.J.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Sehested, M.; Lukas, J.; Kastan, M.B.; Bartek, J. ATM activation in normal human tissues and testicular cancer. Cell Cycle 2005, 4, 838–845. [Google Scholar]
- Nuciforo, P.G.; Luise, C.; Capra, M.; Pelosi, G.; d’Adda di Fagagna, F. Complex engagement of DNA damage response pathways in human cancer and in lung tumor progression. Carcinogenesis 2007, 28, 2082–2088. [Google Scholar]
- Fan, C.; Quan, R.; Feng, X.; Gillis, A.; He, L.; Matsumoto, E.D.; Salama, S.; Cutz, J.C.; Kapoor, A.; Tang, D. ATM activation is accompanied with earlier stages of prostate tumorigenesis. Biochim. Biophys. Acta 2006, 1763, 1090–1097. [Google Scholar]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar]
- Lazzerini Denchi, E.; Attwooll, C.; Pasini, D.; Helin, K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol. Cell Biol 2005, 25, 2660–2672. [Google Scholar]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 2007, 21, 43–48. [Google Scholar]
- Ohtani, N.; Takahashi, A.; Mann, D.J.; Hara, E. Cellular senescence: A double-edged sword in the fight against cancer. Exp. Dermatol 2012, 21, 1–4. [Google Scholar]
- Giaimo, S.; d’Adda di Fagagna, F. Is cellular senescence an example of antagonistic pleiotropy? Aging Cell 2012, 11, 378–383. [Google Scholar]
- Larsson, L.G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol 2011, 21, 367–376. [Google Scholar]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar]
- Dumble, M.; Moore, L.; Chambers, S.M.; Geiger, H.; van Zant, G.; Goodell, M.A.; Donehower, L.A. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood 2007, 109, 1736–1742. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol 1994, 4, 1–7. [Google Scholar]
- Liu, G.; Chen, X. Regulation of the p53 transcriptional activity. J. Cell Biochem 2006, 97, 448–458. [Google Scholar]
- Soussi, T.; Wiman, K.G. Shaping genetic alterations in human cancer: The p53 mutation paradigm. Cancer Cell 2007, 12, 303–312. [Google Scholar]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res 2007, 35, 7475–7484. [Google Scholar]
- Garcia-Cao, I.; Garcia-Cao, M.; Martin-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002, 21, 6225–6235. [Google Scholar]
- Serrano, M.; Blasco, M.A. Cancer and ageing: Convergent and divergent mechanisms. Nat. Rev. Mol. Cell Biol 2007, 8, 715–722. [Google Scholar]
- Benson, E.K.; Zhao, B.; Sassoon, D.A.; Lee, S.W.; Aaronson, S.A. Effects of p21 deletion in mouse models of premature aging. Cell Cycle 2009, 8, 2002–2004. [Google Scholar]
- Zhao, B.; Benson, E.K.; Qiao, R.; Wang, X.; Kim, S.; Manfredi, J.J.; Lee, S.W.; Aaronson, S.A. Cellular senescence and organismal ageing in the absence of p21(CIP1/WAF1) in ku80(−/−) mice. EMBO Rep 2009, 10, 71–78. [Google Scholar]
- Davis, T.; Singhrao, S.K.; Wyllie, F.S.; Haughton, M.F.; Smith, P.J.; Wiltshire, M.; Wynford-Thomas, D.; Jones, C.J.; Faragher, R.G.; Kipling, D. Telomere-based proliferative lifespan barriers in Werner-syndrome fibroblasts involve both p53-dependent and p53-independent mechanisms. J. Cell Sci 2003, 116, 1349–1357. [Google Scholar]
- Stoyanova, T.; Roy, N.; Bhattacharjee, S.; Kopanja, D.; Valli, T.; Bagchi, S.; Raychaudhuri, P. p21 cooperates with DDB2 protein in suppression of ultraviolet ray-induced skin malignancies. J. Biol. Chem 2012, 287, 3019–3028. [Google Scholar]
- Choudhury, A.R.; Ju, Z.; Djojosubroto, M.W.; Schienke, A.; Lechel, A.; Schaetzlein, S.; Jiang, H.; Stepczynska, A.; Wang, C.; Buer, J.; et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007, 39, 99–105. [Google Scholar]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–638. [Google Scholar]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar]
- Martin-Caballero, J.; Flores, J.M.; Garcia-Palencia, P.; Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res 2001, 61, 6234–6238. [Google Scholar]
- Stivala, L.A.; Cazzalini, O.; Prosperi, E. The cyclin-dependent kinase inhibitor p21CDKN1A as a target of anti-cancer drugs. Curr. Cancer Drug Targets 2012, 12, 85–96. [Google Scholar]
- Roy, N.; Stoyanova, T.; Dominguez-Brauer, C.; Park, H.J.; Bagchi, S.; Raychaudhuri, P. DDB2, an essential mediator of premature senescence. Mol. Cell Biol 2010, 30, 2681–2692. [Google Scholar]
- Itoh, T.; Cado, D.; Kamide, R.; Linn, S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc. Natl. Acad. Sci. USA 2004, 101, 2052–2057. [Google Scholar]
- Yoon, T.; Chakrabortty, A.; Franks, R.; Valli, T.; Kiyokawa, H.; Raychaudhuri, P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene 2005, 24, 469–478. [Google Scholar]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. DDB2 decides cell fate following DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 10690–10695. [Google Scholar]
- Stoyanova, T.; Yoon, T.; Kopanja, D.; Mokyr, M.B.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol. Cell Biol 2008, 28, 177–187. [Google Scholar]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res 2010, 704, 12–20. [Google Scholar]
- Fotedar, R.; Bendjennat, M.; Fotedar, A. Role of p21WAF1 in the cellular response to UV. Cell Cycle 2004, 3, 134–137. [Google Scholar]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett 2002, 179, 1–14. [Google Scholar]
- Gartel, A.L. Is p21 an oncogene? Mol. Cancer Ther 2006, 5, 1385–1386. [Google Scholar]
- Nickeleit, I.; Zender, S.; Kossatz, U.; Malek, N.P. p27kip1: A target for tumor therapies? Cell Div 2007, 2, 13. [Google Scholar]
- Slingerland, J.; Pagano, M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell Physiol 2000, 183, 10–17. [Google Scholar]
- Loda, M.; Cukor, B.; Tam, S.W.; Lavin, P.; Fiorentino, M.; Draetta, G.F.; Jessup, J.M.; Pagano, M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med 1997, 3, 231–234. [Google Scholar]
- Porter, P.L.; Malone, K.E.; Heagerty, P.J.; Alexander, G.M.; Gatti, L.A.; Firpo, E.J.; Daling, J.R.; Roberts, J.M. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat. Med 1997, 3, 222–225. [Google Scholar]
- Yang, R.M.; Naitoh, J.; Murphy, M.; Wang, H.J.; Phillipson, J.; deKernion, J.B.; Loda, M.; Reiter, R.E. Low p27 expression predicts poor disease-free survival in patients with prostate cancer. J. Urol 1998, 159, 941–945. [Google Scholar]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; Kaushansky, K.; Roberts, J.M. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996, 85, 733–744. [Google Scholar]
- Muraoka, R.S.; Lenferink, A.E.; Law, B.; Hamilton, E.; Brantley, D.M.; Roebuck, L.R.; Arteaga, C.L. ErbB2/Neu-induced, cyclin D1-dependent transformation is accelerated in p27-haploinsufficient mammary epithelial cells but impaired in p27-null cells. Mol. Cell Biol 2002, 22, 2204–2219. [Google Scholar]
- Besson, A.; Gurian-West, M.; Chen, X.; Kelly-Spratt, K.S.; Kemp, C.J.; Roberts, J.M. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev 2006, 20, 47–64. [Google Scholar]
- Cordon-Cardo, C.; Koff, A.; Drobnjak, M.; Capodieci, P.; Osman, I.; Millard, S.S.; Gaudin, P.B.; Fazzari, M.; Zhang, Z.F.; Massague, J.; Scher, H.I. Distinct altered patterns of p27KIP1 gene expression in benign prostatic hyperplasia and prostatic carcinoma. J. Natl. Cancer Inst 1998, 90, 1284–1291. [Google Scholar]
- Fero, M.L.; Randel, E.; Gurley, K.E.; Roberts, J.M.; Kemp, C.J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998, 396, 177–180. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Alberg, A.J.; Helzlsouer, K.J. Epidemiology, prevention, and early detection of breast cancer. Curr. Opin. Oncol 1997, 9, 505–511. [Google Scholar]
- Brody, L.C.; Biesecker, B.B. Breast cancer susceptibility genes. BRCA1 and BRCA2. Medicine 1998, 77, 208–226. [Google Scholar]
- Paterson, J.W. BRCA1: A review of structure and putative functions. Dis. Markers 1998, 13, 261–274. [Google Scholar]
- Rahman, N.; Stratton, M.R. The genetics of breast cancer susceptibility. Annu. Rev. Genet 1998, 32, 95–121. [Google Scholar]
- Scully, R.; Livingston, D.M. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 2000, 408, 429–432. [Google Scholar]
- Zhang, J.; Powell, S.N. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol. Cancer Res 2005, 3, 531–539. [Google Scholar]
- Deng, C.X.; Scott, F. Role of the tumor suppressor gene BRCA1 in genetic stability and mammary gland tumor formation. Oncogene 2000, 19, 1059–1064. [Google Scholar]
- Zheng, L.; Li, S.; Boyer, T.G.; Lee, W.H. Lessons learned from BRCA1 and BRCA2. Oncogene 2000, 19, 6159–6175. [Google Scholar]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the BRCA1 full-length isoform. Genes Dev 2003, 17, 201–213. [Google Scholar]
- Cao, L.; Kim, S.; Xiao, C.; Wang, R.H.; Coumoul, X.; Wang, X.; Li, W.M.; Xu, X.L.; de Soto, J.A.; Takai, H.; et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon BRCA1 deficiency. EMBO J. 2006, 25, 2167–2177. [Google Scholar]
- Hakem, R.; de la Pompa, J.L.; Elia, A.; Potter, J.; Mak, T.W. Partial rescue of BRCA1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat. Genet 1997, 16, 298–302. [Google Scholar]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors BRCA1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet 2001, 28, 266–271. [Google Scholar]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; Deng, C.X.; Finkel, T. A selective requirement for 53BP1 in the biological response to genomic instability induced by BRCA1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar]
- Wang, W.; Wu, J.; Zhang, Z.; Tong, T. Characterization of regulatory elements on the promoter region of p16(INK4a) that contribute to overexpression of p16 in senescent fibroblasts. J. Biol. Chem 2001, 276, 48655–48661. [Google Scholar]
- Palmero, I.; McConnell, B.; Parry, D.; Brookes, S.; Hara, E.; Bates, S.; Jat, P.; Peters, G. Accumulation of p16INK4a in mouse fibroblasts as a function of replicative senescence and not of retinoblastoma gene status. Oncogene 1997, 15, 495–503. [Google Scholar]
- Tsutsui, T.; Kumakura, S.; Yamamoto, A.; Kanai, H.; Tamura, Y.; Kato, T.; Anpo, M.; Tahara, H.; Barrett, J.C. Association of p16(INK4a) and pRb inactivation with immortalization of human cells. Carcinogenesis 2002, 23, 2111–2117. [Google Scholar]
- Melk, A.; Kittikowit, W.; Sandhu, I.; Halloran, K.M.; Grimm, P.; Schmidt, B.M.; Halloran, P.F. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int 2003, 63, 2134–2143. [Google Scholar]
- Nielsen, G.P.; Stemmer-Rachamimov, A.O.; Shaw, J.; Roy, J.E.; Koh, J.; Louis, D.N. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest 1999, 79, 1137–1143. [Google Scholar]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol 2006, 7, 667–677. [Google Scholar]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M.; et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar]
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar]
- Van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar]
- Itahana, K.; Zou, Y.; Itahana, Y.; Martinez, J.L.; Beausejour, C.; Jacobs, J.J.; van Lohuizen, M.; Band, V.; Campisi, J.; Dimri, G.P. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell Biol 2003, 23, 389–401. [Google Scholar]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression. in vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar]
- Carbone, C.J.; Grana, X.; Reddy, E.P.; Haines, D.S. p21 loss cooperates with INK4 inactivation facilitating immortalization and Bcl-2-mediated anchorage-independent growth of oncogene-transduced primary mouse fibroblasts. Cancer Res 2007, 67, 4130–4137. [Google Scholar]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res 2003, 63, 2705–2715. [Google Scholar]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res 2005, 65, 2795–2803. [Google Scholar]
- Sidi, R.; Pasello, G.; Opitz, I.; Soltermann, A.; Tutic, M.; Rehrauer, H.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Induction of senescence markers after neo-adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: An exploratory analysis. Eur. J. Cancer 2011, 47, 326–332. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Chang, B.D.; Swift, M.E.; Shen, M.; Fang, J.; Broude, E.V.; Roninson, I.B. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc. Natl. Acad. Sci. USA 2002, 99, 389–394. [Google Scholar]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar]
- Schmitt, C.A. Cellular senescence and cancer treatment. Biochim. Biophys. Acta 2007, 1775, 5–20. [Google Scholar]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008, 6, 2853–2868. [Google Scholar]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol 2011, 192, 547–556. [Google Scholar]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar]
- Ancrile, B.; Lim, K.H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev 2007, 21, 1714–1719. [Google Scholar]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar]
- Dilley, T.K.; Bowden, G.T.; Chen, Q.M. Novel mechanisms of sublethal oxidant toxicity: Induction of premature senescence in human fibroblasts confers tumor promoter activity. Exp. Cell Res 2003, 290, 38–48. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).