Abstract
Single nucleotide polymorphisms (SNPs) are currently the marker of choice in a variety of genetic studies. Using the high resolution melting (HRM) genotyping approach, 101 gene-based SNP markers were developed for Apostichopus japonicus, a sea cucumber species with economic significance for the aquaculture industry in East Asian countries. HRM analysis revealed that all the loci showed polymorphisms when evaluated using 40 A. japonicus individuals collected from a natural population. The minor allele frequency ranged from 0.035 to 0.489. The observed and expected heterozygosities ranged from 0.050 to 0.833 and 0.073 to 0.907, respectively. Thirteen loci were found to depart significantly from Hardy–Weinberg equilibrium (HWE) after Bonferroni corrections. Significant linkage disequilibrium (LD) was detected in one pair of markers. These SNP markers are expected to be useful for future quantitative trait loci (QTL) analysis, and to facilitate marker-assisted selection (MAS) in A. japonicus.
1. Introduction
The sea cucumber Apostichopus japonicus (Selenka 1867), naturally distributes along the coasts of China, Japan, Korea and Russia [1]. Due to their nutritional and medicinal value, they have long been exploited as an important fishery resource in East Asian countries. Over the past decade, the aquaculture of A. japonicus has become widespread along the coasts of China, due to increasing market demand and over-exploitation of wild sea cucumbers [2]. However, the rapid expansion and intensification of sea cucumber aquaculture has resulted in some severe problems, such as wide-spread disease and stock deterioration, possibly caused by inappropriate broodstock management and inbreeding depression [2]. In order to properly manage broodstock resources and efficiently enhance aquaculture production, control of inbreeding and selection of broodstock with the desired traits, such as rapid growth and disease resistance, are currently necessary for sustainable development of the A. japonicus aquaculture. Recently, marker-assisted selection (MAS) has become a valuable tool for selecting individuals with traits of interest [3]. To perform MAS, a large number of genetic markers are usually needed to determine the quantitative trait loci (QTLs) associated with economically important traits.
Single nucleotide polymorphisms (SNPs) have been shown to be the most abundant type of genetic variations in eukaryotic genomes [4], and are currently the marker of choice in a variety of genetic studies, such as high-density genetic linkage mapping and QTL analysis. However, only a limited number of SNP markers have been reported for A. japonicus [5–7]. Moreover, molecular markers developed from the expressed sequence tag (EST) databases offer several advantages over anonymous genomic markers, as (i) they can detect variation in the expressed portion of the genome, so that gene tagging could give “perfect” marker-trait associations; (ii) they could alleviate the problem of null alleles which is usually associated with markers developed from the non-transcribed regions; and (iii) they are expected to have greater transferability between species, since transcribed regions are more conserved among closely related species/genera.
Previously, our group has released a large amount of EST data by 454 sequencing of the A. japonicus transcriptome [7]. By mining our EST dataset, more than 54,000 putative SNPs have been identified, 200 of which were selected in this study for marker development. SNP validation was performed using 48 A. japonicus individuals collected from four natural populations. Genetic parameters of the validated SNP markers were evaluated using 40 A. japonicus individuals from a single natural population. These SNP markers will be useful for future QTL analysis in order to facilitate MAS in A. japonicus.
2. Results and Discussion
Transcriptomic sequences represent an important resource for rapid and cost-effective development of gene-based SNPs. For the high resolution melting (HRM)-based SNP marker development, we designed PCR primers for 200 candidate SNPs (Table 1), which were previously identified from the A. japonicus transcriptome generated by 454-FLX sequencing [7]. After PCR amplification, 159 (79.5%) amplified strong bands with expected sizes. The others were discarded without further consideration, as they produced bands larger than expected (possibly caused by introns) or resulted in poor amplification (weak or non-specific amplification). During the initial HRM screen, 63.5% (101) of the 159 successfully amplified loci showed polymorphisms in 48 individuals collected from 4 natural populations, 21.4% (34) generated non-polymorphic curves, and 15.1% (24) displayed unreliable melting curves. In this study, we showed that minor allele frequency (MAF) can serve as an important selection criterion to distinguish true SNPs from sequencing errors when performing SNP mining from 454 sequencing data (Figure 1). For example, most of the validated SNPs usually have a MAF of more than 35%, whereas most non-validated SNPs usually have a MAF of less than 25%. Although our study demonstrated that SNP markers can be efficiently developed from transcriptomic resources, it should be noted that the SNPs obtained may largely represent common genetic variations due to the low coverage of the original transcriptome sequencing, and may suffer from ascertainment bias resulting from simple sample source used in the original transcriptome sequencing.

Table 1.
Results of validation and genotyping of candidate single nucleotide polymorphisms (SNPs).
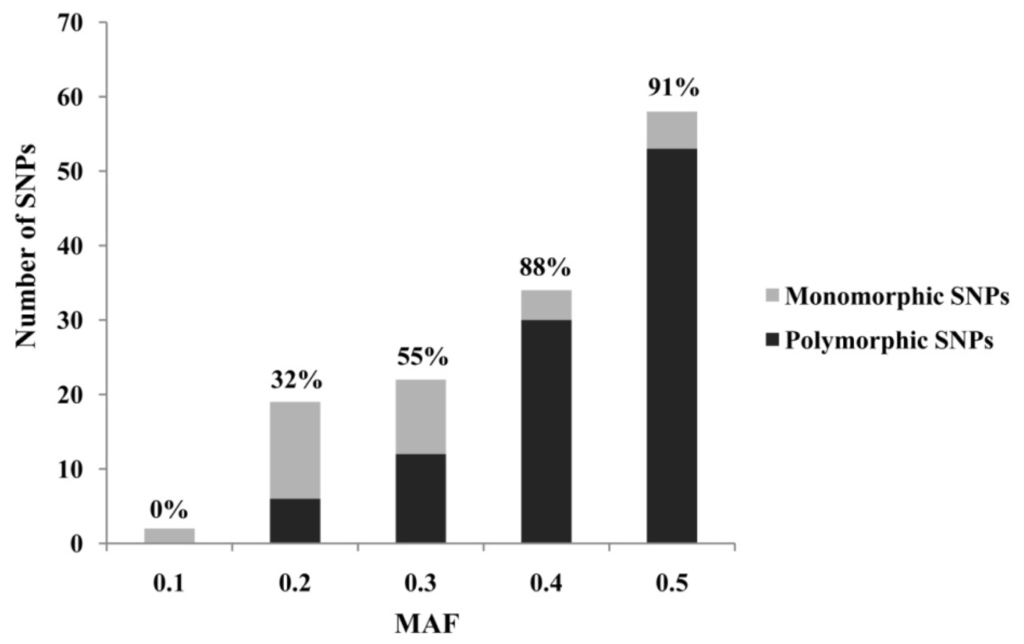
Figure 1.
Distribution of SNP minor allele frequency (MAF) for Apostichopus japonicus. The number above each bar was the polymorphic rate in respective MAF categories.
Genetic parameters of the validated SNP markers were further evaluated using 40 A. japonicus individuals from a single natural population. As expected, all 101 SNP loci were polymorphic. The minor allele frequency ranged from 0.035 to 0.489 (Table 2). The Ho ranged from 0.050 to 0.833, while the He varied from 0.073 to 0.907. Thirteen loci departed significantly (p < 0.01) from Hardy–Weinberg equilibrium (HWE) after Bonferroni correction, suggesting that these loci may be under ongoing natural selection. Significant linkage disequilibrium (LD) was detected in one pair of SNP markers (ApjSNP092_CT and ApjSNP098_CT).
Table 2.
Characterization of 101 SNPs for the sea cucumber Apostichopus japonicus.
As the gene-derived SNPs reside in or are immediately next to protein-coding sequences, they stand a better chance for identifying functional genes that are responsible for complex traits as well as simply inherited traits [8,9]. In our study, 70 SNP markers (Table 2) were developed from the EST sequences showing significant similarity to an entry in the NCBI nr database [10]. Among the annotation information, genes potentially involved in growth or immunity (e.g., epidermal growth factor receptor, Zinc finger protein 62 homolog and heat shock protein 90 kDa beta) were identified. It would be interesting to see whether any of these growth- or immune-related SNPs are highlighted in future QTL mapping of economically important traits, such as high growth rate and disease resistance.
3. Experimental Section
3.1. Sampling and DNA Extraction
A total of 48 A. japonicus individuals used for SNP marker validation were collected from four natural populations (Dalian, Yantai, Qingdao and Wendeng) in China. Genetic parameters of the validated SNP markers were further evaluated using 40 A. japonicus individuals from the Rongcheng (Shandong, China) population. Genomic DNA was extracted from the muscles of sea cucumbers by following the protocol developed by Zhan et al. [11]. The quantity and integrity of genomic DNA was determined using an Ultrospec™ 2100 pro UV/Visible Spectrophotometer (Amersham Biosciences, Uppsala, Sweden) and gel electrophoresis, respectively.
3.2. SNP Discovery and Genotyping
Our group has recently released a large amount of transcriptomic data by 454 sequencing of eight cDNA libraries constructed using more than 200 sea cucumber individuals. Potential SNPs were detected from the assembled contigs using the program GS Reference Mapper (version 2.6, Roche 454 Life Sciences: Branford, CT, USA, 2011) with default parameters (cDNA mode). More than 54,000 putative SNPs were identified from the dataset, 200 of which were selected in this study for marker development with the selection criteria of at least 3× occurrence of the minority allele and at least 6× contigs coverage (number of reads forming the contig). SNP genotyping was performed using a recently developed cost-effective HRM method [12]. For each locus, three non-modified oligonucleotides were used, corresponding to two PCR primers and one probe, primers were designed using Primer3 [13] with the following rules: (1) primer length should be at least 20 bases; (2) product size should not exceed 120 bp in order to decrease the probability of intron interference; (3) the primer Tm should be between 59 °C and 61 °C; (4) the primer GC% should be 40%–60%; and (5) the amplicon contains only one SNP site. Probes were designed using OligoCalc [14] with the following criteria: (1) SNP site locates in the middle of the probe; (2) the length of probe is between 20 and 35 bases; (3) Tm is about 60 °C; (4) the 3′ end of each probe is blocked by two mismatch bases; and (5) no overlap between primes and probe. Each SNP locus was first amplified by an asymmetrical PCR with HRM fluorescent dye in the PCR master mix and then interrogated by an unlabeled probe. The 48 individuals of A. japonicus collected from four natural populations were used for SNP marker validation. PCR amplifications were carried out in a 10 μL reaction mixture containing 20 ng of genomic DNA, 1× PCR buffer, 0.2 mM dNTPs, 1.5 mM MgCl2, 0.5 U Taq DNA polymerase (Takara, Dalian, China), 0.1 μM forward primer, 0.5 μM reverse primer and 1× LCGreen Plus (Idaho technology inc., Salt Lake City, Utah, USA). The amplification was programmed as: an initial denaturation at 95 °C for 5 min, followed by 55 cycles of 95 °C for 40 s, 60 °C for 40 s and 72 °C for 40 s, finishing with a final elongation at 72 °C for 5 min. The PCR products were checked by gel electrophoresis, and those with correct PCR product sizes were then subjected to probe testing. An aliquot of the appropriate probe was added in each reaction to a final concentration of 5 μM. The PCR product and probe mixture were denatured at 95 °C for 15 min and then slowly cooled to 4 °C. HRM genotyping was immediately performed on a Light Scanner instrument (HR96 model, Idaho technology inc., Salt Lake City, Utah, USA) with continuous melting curve acquisition (10 acquisitions per °C) during a 0.1 °C/s ramp from 40 to 95 °C.
3.3. Data Analysis
Data were retrieved and analyzed using the Light Scanner software followed by manual curation of the obtained genotype calls. POPGENE [15] was used to analyze allele frequency, expected (He) and observed (Ho) heterozygosities, and tests for deviation from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD).
4. Conclusions
In summary, 101 gene-based SNPs were successfully developed from the transcriptome sequences of A. japonicus. These developed markers are expected to be useful for future QTL analysis, and to facilitate MAS in A. japonicus.
Acknowledgments
Financial support for this work was provided by the National Key Technology R&D Program of China (2011BAD13B05 and 2011BAD13B06), and the National High Technology Research and Development Program of China (2012AA10A412).
References
- Sloan, N.A. References. In Echinodermata; Keegan, B.F., O’Connor, B.D.S., Eds.; A.A. Balkema: Rotterdam, The Netherlands, 1985; pp. 109–124. [Google Scholar]
- Chen, J.X. Present Status and Prospects of Sea Cucumber Industry in China. In Advances in Sea Cucumber Aquaculture and Management; Lovatelli, A., Conand, C., Purcell, S., Uthicke, S., Hamel, J.F., Mercier, A., Eds.; FAO: Rome, Italy, 2004; pp. 25–38. [Google Scholar]
- Collard, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar]
- Cho, R.J.; Mindrinos, M.; Richards, D.R.; Sapolsky, R.J.; Anderson, M.; Drenkard, E.; Dewdney, J.; Reuber, T.L.; Stammers, M.; Federspiel, N.; et al. Genome-wide mapping with biallelic markers in Arabidopsis thaliana. Nat. Genet 1999, 23, 203–207. [Google Scholar]
- Sun, W.J.; Li, Q.; Kong, L.F. Characterization of thirteen single nucleotide polymorphism markers in the sea cucumber (Apostichopus japonicus). Conserv. Genet. Resour 2010, 2, 141–144. [Google Scholar]
- Yang, A.F.; Sun, D.F.; Liu, S.K.; Dong, Y.; Chen, Z.; Zhou, Z.C. Characterization of fifteen SNP markers by mining EST in sea cucumber, Apostichopus japonicus. J. Genet. 2012, 91, e49–e53. [Google Scholar]
- Du, H.X.; Bao, Z.M.; Hou, R.; Wang, S.; Su, H.L.; Yan, J.J.; Tian, M.L.; Li, Y.; Wei, W.; Hu, X.L.; et al. Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka, 1867). PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Tsumura, Y.; Kado, T.; Takahashi, T.; Tani, N.; Ujino-Ihara, T.; Iwata, H. Genome scan to detect genetic structure and adaptive genes of natural populations of Cryptomeria japonica. Genetics 2007, 176, 2393–2403. [Google Scholar]
- Namroud, M.C.; Beaulieu, J.; Juge, N.; Laroche, J.; Bousquet, J. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol. Ecol 2008, 17, 3599–3613. [Google Scholar]
- NCBI nr database. Available online: ftp://ftp.ncbi.nlm.nih.gov/blast/db/ accessed on 25 January 2012.
- Zhan, A.B.; Bao, Z.M.; Lu, W.; Hu, X.L.; Peng, W.; Wang, M.L.; Hu, J.J. Development and characterization of 45 novel microsatellite markers for sea cucumber (Apostichopus japonicus). Mol. Ecol. Notes 2007, 7, 1345–1348. [Google Scholar]
- Wang, S.; Zhang, L.L.; Meyer, E.; Matz, M.V. Construction of a high-resolution genetic linkage map and comparative genome analysis for the reef-building coral Acropora millepora. Genome Biol 2009, 10. [Google Scholar] [CrossRef]
- Rozen, S.; Skaletsky, H. References. In Bioinformatics Methods and Protocols: Methods in Molecular Biology; Krawetz, S., Misener, S., Eds.; Humana: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res 2007, 35, 43–46. [Google Scholar]
- Yeh, F.C.; Boyle, T.J.B. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg. J. Bot 1997, 129, 157. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).