Abstract
1,4-Dihydropyridines (DHPs) have been used in the reduction of carbon-carbon double bonds under microwave irradiation without solvent. The efficiency of the reactions is dramatically dependent on the steric effects in the DHPs and on the electronic effects in the olefins.
Introduction
Hantzsch 1,4-dihydropyridines (DHPs) continue to attract much attention because of their use as NAD(P)H models to probe the mechanism of hydrogen transfer [,,] and their applications in medicine as calcium channel modulators, vasodilators and antihypertensive agents [,,].
Results and Discussion
In this communication, we wish to report our results on the synthesis of some 1,4-DHPs and their behavior under microwave irradiation in redox reactions with olefins, a system that has been poorly studied, contrary to the mimetic reduction of carbonyl compounds.
The most commonly used method used to prepare 1,4-DHPs is the Hantzsch synthesis [] involving condensation of β-ketoesters with ammonia-aldehyde mixtures or of enaminoesters and aldehydes. In those experiments, the reagents are heated for several hours in a solvent maintained at its boiling point. Conventional heating has sometimes been replaced by microwave induced heating [,,,] but, surprisingly, this last procedure has never been used for the preparation of 4-unsubstituted derivatives. We have been able to synthesize such 4-unsubstituted DHPs under microwave irradiation and in the absence of solvent by using alkyl acetoacetates in the presence of hexamethylenetetramine as the source of the ammonia-formaldehyde mixture. Ammonium acetate has been added to the reaction media in order to maintain the stoichiometric balance between ammonia and methanal. In that way, diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (1a), for example, has been isolated in 63% yield after irradiation for 100 seconds in a Synthewave 402 focused microwave reactor.
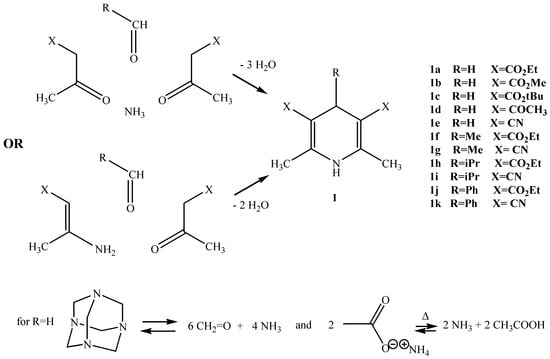
Scheme 1.
We then explored the synthetic potential of such DHPs for the reduction of olefins substituted by electron-withdrawing groups (carbonyl and nitro). Indeed, a few years ago, Ohno et al [,] effected those reactions in boiling benzene and explained the role of silica gel as catalyst. However heating times as long as 18 hours were required to obtain satisfactory yields. Our aim was to test the possibility of working without solvent and to decrease the heating period by performing the experiments under microwave irradiation, as we recently described for the Meerwein-Ponndorf- Verley reduction of carbonyl compounds []. We also compared the performance of two kinds of microwave ovens: a multimode oven and a monomode one which allows for automatic temperature control. In our experiments the monomode microwave oven has been set at 150 °C. For ethyl crotonate (Table 1, entry 5) and cyclohex-2-en-1-one (Table 1, entry 6), the temperature has been lowered to 120°C to avoid loss of product (b.p.= 136 °C and 168 °C respectively). In most experiments a slight excess of DHP has been used to ensure completion of the reduction.
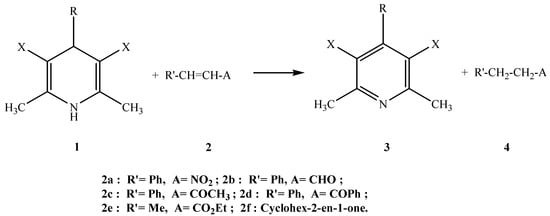
Scheme 2.

Table 1.
Reduction by 1a of carbon-carbon double bonds
| Entry | Substrate | Multimode 250 W, 8 min 1.5 eq DHP | Multimode 800 W, 8 min 1.5 eq DHP | Monomode 150 W, 4 min 1.5 eq DHP | Monomode 150 W, 6 min 1eq DHP | |||||
| %2* | %4 | %2* | %4 | %2* | %4 | %2* | %4 | %3 | ||
| 1 | 2a | 15 | 82 | 0 | 79 | 0 | 78 | 0 | 78 | 86 |
| 2 | 2b | 5 | 44 | 25 | 50 | 25 | 50 | - | - | - |
| 3 | 2c | 0 | 75 | 4 | 96 | 27 | 60 | 27 | 78 | 81 |
| 4 | 2d | 0 | 99 | 19 | 52 | 19 | 81 | 17 | 83 | 70 |
| 5 | 2e | 0 | 0 | 0 | 0 | 0 | 0 | - | - | - |
| 6 | 2f | 0 | 50 | 52 | 48 | 52 | 48 | - | - | - |
* substrate recovered
As it can be seen by examination of the data collected in Table 1, comparable results can be obtained with both kinds of ovens. In particular, we observed no reaction with the α−β-unsaturated ester, probably due to the poor electron-withdrawing effect of the ester group. In the other cases, ourresults are quite promising as, when the hydrogen transfer occurred, yields ranged from 44% to 99%. In addition, reaction times could be considerably reduced as they did not exceed a few minutes. Let us also emphasize that even when equimolar amounts of reagents were used, pyridines 3 and alkanes 4 have been both isolated in good yields. Thus our procedure of reduction of olefins in this mimetic system seems to be limited to conjugated esters only.
We also examined the substituent effects in the heterocycle for 1,4-DHP/nitrostyrene systems (Table 1, entry 1; Table 2). For that purpose, nitrostyrene 2a was reduced by several 1,4-DHPs in the multimode microwave oven for 8 minutes at 250 W. Our results are summarized in Table 2. The electron-withdrawing effect of 3,5-substituents is difficult to estimate but it clearly appears that the hydrogen transfer is strongly disfavoured by the presence of a substituent on the carbon atom at the 4 position of the DHP and larger the substituent is, the lower the yield of the reaction. The presence of the bulky tert-butyl ester groups in positions 3 and 5 of the DHP also disfavors the reduction process.
Table 2.
Reduction of nitrostyrene by different DHPs.
| 1,4-DHP (1,5 eq) | Yield | |
| % 2a (substrate recovered) | % 4a | |
| 1a | 15 | 82 |
| 1b | 0 | 77 |
| 1c | 2 | 31 |
| 1d | 0 | 91 |
| 1e | 20 | 65 |
| 1f | 65 | 19 |
| 1g | 2 | 55 |
| 1h | 100 | 0 |
| 1i | 65 | 8 |
| 1j | 100 | 0 |
| 1k | 100 | 0 |
To complete this work, we examined the possibility of carrying out the reduction using a DHP anchored on a soluble polymer as a potential recyclable reducing reagent. For that purpose, we reacted poly(styrene-co-allyl alcohol) with ethyl acetoacetate under microwave irradiation and solvent-free conditions as earlier described []. The so-obtained polymeric ester was then treated with a mixture of ethyl acetoacetate, hexamethylenetetramine, and ammonium acetate, again in a microwave oven. The polymer-anchored reducing heterocycle (whose structure has been ascertained by MAS NMR: δ C=O 168.4 and 169.7 ppm) was finally exposed under microwave in the presence of nitrostyrene to afford the reduction product. Optimization of this last step is now in progress.
Conclusions
A convenient synthesis of 4-unsubstituted DHPs under microwave irradiation and solvent-free conditions has been developed. In addition, these compounds can be readily used to reduce carbon- carbon double bonds substituted by strong electron-withdrawing groups under similar environmental friendly conditions. Preliminary experiments also demonstrate that applications of this reaction with the polymer-supported DHP 1a can reasonably be foreseen.
Experimental
General
Microwave irradiations were performed in a Synthewave 402 Prolabo instrument (monomode) or in a Normatron Normandie-Labo oven (multimode). Silica gel 60 Merck (63-200μm) was used in the reduction of olefins. NMR spectra were recorded on a Bruker AC300 spectrometer. Reagents and products have been described previously: 1a,h,j and 3a [], 1b,d,e,k [], 1c [], 1f,g,i []; 2 and 4 are available from commercial sources. Melting points and NMR spectra of our compounds are in agreement with literature data.
Diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (1a).
A mixture of ethyl acetoacetate (20 mmol), hexamethylenetetramine (8 mmol) and ammonium acetate (10mmol) is irradiated for 100s (45W, Synthewave S402). After cooling, the solid is filtered, washed with ethanol and dried under vacuum; m.p. 188-190°C; 1H-NMR (CDCl3) δ: 1.24 (t, 6H, CO2CH2CH3), 2.17 (s, 6H, CH3C(NH)=), 3.25 (s, 2H, H-4), 4.12 (q, 4H, CO2CH2CH3), 5.22 (s, 1H, H-1); 13C-NMR (CDCl3) δ: 14.5, 19.2, 24.8, 59.7, 99.5, 144.8,168.1.
Typical procedure for the reduction of olefins by DHP
Silica gel (4g), nitrostyrene (0.6g, 4 mmol) and DHP 1a (1.5g, 6 mmol) are diluted in methylene chloride (20 mL). The solvent is quickly evaporated in vacuo. The resulting solid is irradiated for 4 min (150W, Synthewave S402) under a nitrogen atmosphere, cooled and filtered. The filtrate is stirred for 20 min with 6N HCl (20 mL). The organic layer is dried on MgSO4 and concentrated in vacuum to afford 2-phenylnitroethane (yield 78%); 1H-MR (CDCl3) δ: 3.31 (t, 2H, H-2 ), 4.60 (t, 2H, H-1), 7.20-7.36 (m, 5H, aromatic hydrogens); 13C-MR (CDCl3) δ: 33.4, 76.3, 127.4, 128.6, 128.9, 135.7.
References
- Zhu, X-Q.; Liu, Y-C.; Cheng, J-P. J. Org. Chem. 1999, 64, 8980–8981.
- Cheng, J-P.; Lu, Y.; Zhu, X-Q.; Sun, Y.; Bi, F.; He, J. J. Org. Chem. 2000, 65, 3853–3857. [PubMed]
- Zhu, X-Q.; Liu, Y-C.; Zhao, B-J.; Cheng, J-P. J. Org. Chem. 2001, 66, 370–375. [PubMed]
- Schleifer, K-J. J. Med. Chem. 1999, 42, 2204–2211. [PubMed]
- Visentin, S.; Amiel, P.; Frittero, R.; Boschi, D.; Roussel, C.; Giusta, L.; Carbone, E.; Gasco, A. J. Med. Chem. 1999, 42, 1422–1427. [PubMed]
- Jiang, J-L.; Li, A-H.; Jang, S-Y.; Chang, L.; Melman, N.; Moro, S.; Ji, X-D.; Lobkowsky, E.; Clardy, J.; Jacobson, K. J. Med. Chem. 1999, 42, 3055–3065. [PubMed]
- Hantzch, A. Justus Liebigs Ann. Chem. 1882, 215, 1–82.
- Suarez, M.; Loupy, A.; Perez, E.; Moran, L.; Gerona, G.; Morales, A.; Autie, M. Heterocyclic Commun. 1996, 2, 275–280.
- Zhang, Y-W.; Shen, Z-X.; Pan, B.; Lu, X-H.; Chen, M-H. Synth. Commun. 1995, 25, 857–862.
- Alajarin, R.; Jordan, P.; Vaquero, J.; Alvarez-Builla, J. Synthesis 1995, 389–391.
- Khadilkar, B.; Chitnavis, A. Indian J. Chem. 1995, 34B, 652–653.
- Nakamura, K.; Fujii, M.; Ohno, A.; Oka, S. Tetrahedron Lett. 1984, 25, 3983–3986.
- Fujii, M.; Nakamura, K.; Yasui, S.; Oka, S.; Ohno, A. Bull. Chem. Soc. Jpn. 1988, 61, 495–500.
- Barbry, D.; Torchy, S. Tetrahedron Lett. 1997, 38, 2959–2960.
- Vanden Eynde, J-J.; Rutot, D. Tetrahedron 1999, 55, 2687–2694.
- Vanden Eynde, J-J.; Delfosse, F.; Mayence, A.; Van Haverbeke, Y. Tetrahedron 1995, 51, 6511–6516.
- Kurfuerst, A.; Trska, P.; Golfer, I. Collect. Czech. Chem. Commun. 1984, 49, 2393–2399.
- Dommisse, R.; Lepoivre, J.; Alderweireldt, F. Bull. Soc. Chim. Belges 1977, 86, 633–635.
- Loev, B.; Snader, K. J. Org. Chem. 1965, 30, 1914–1916.
- Samples Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.