

DerivaPredict: A User-Friendly Tool for Predicting and Evaluating Active Derivatives of Natural Products



Abstract
1. Introduction
2. Results
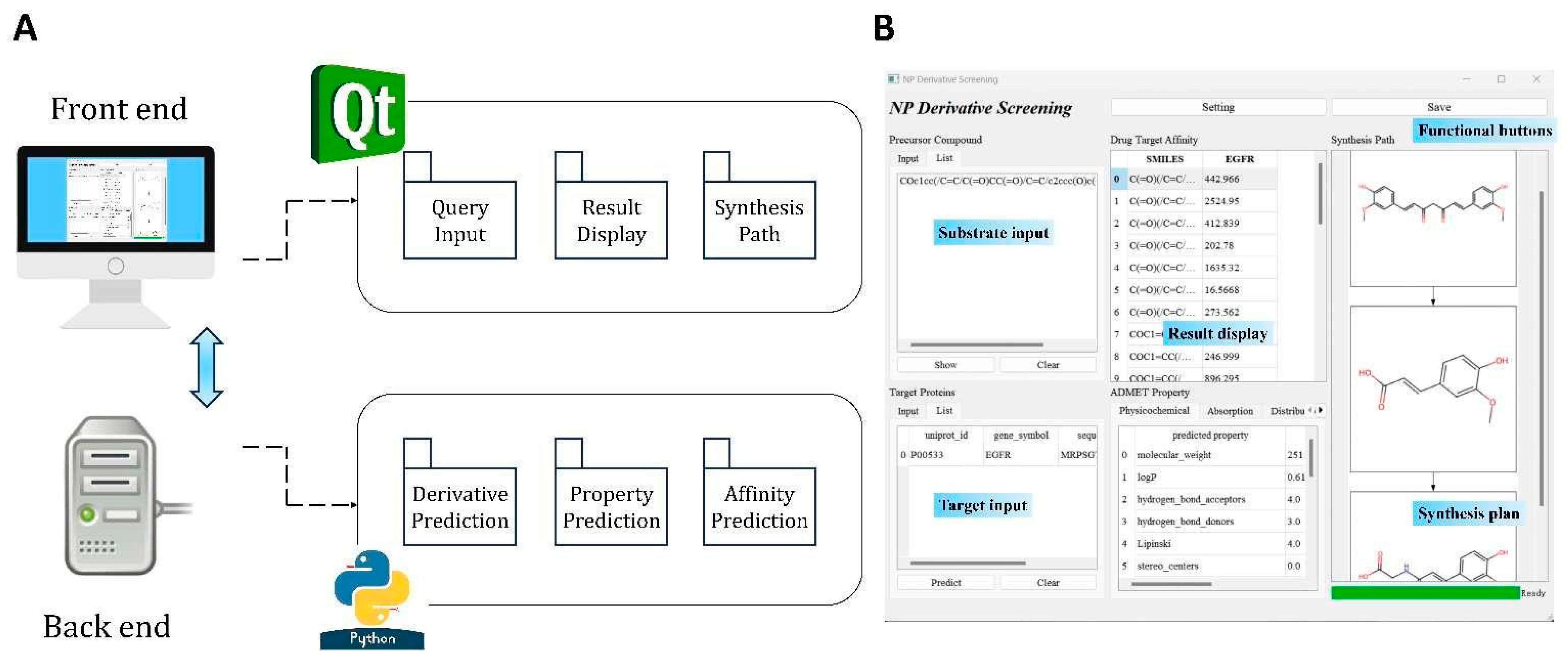
2.1. Software Integration
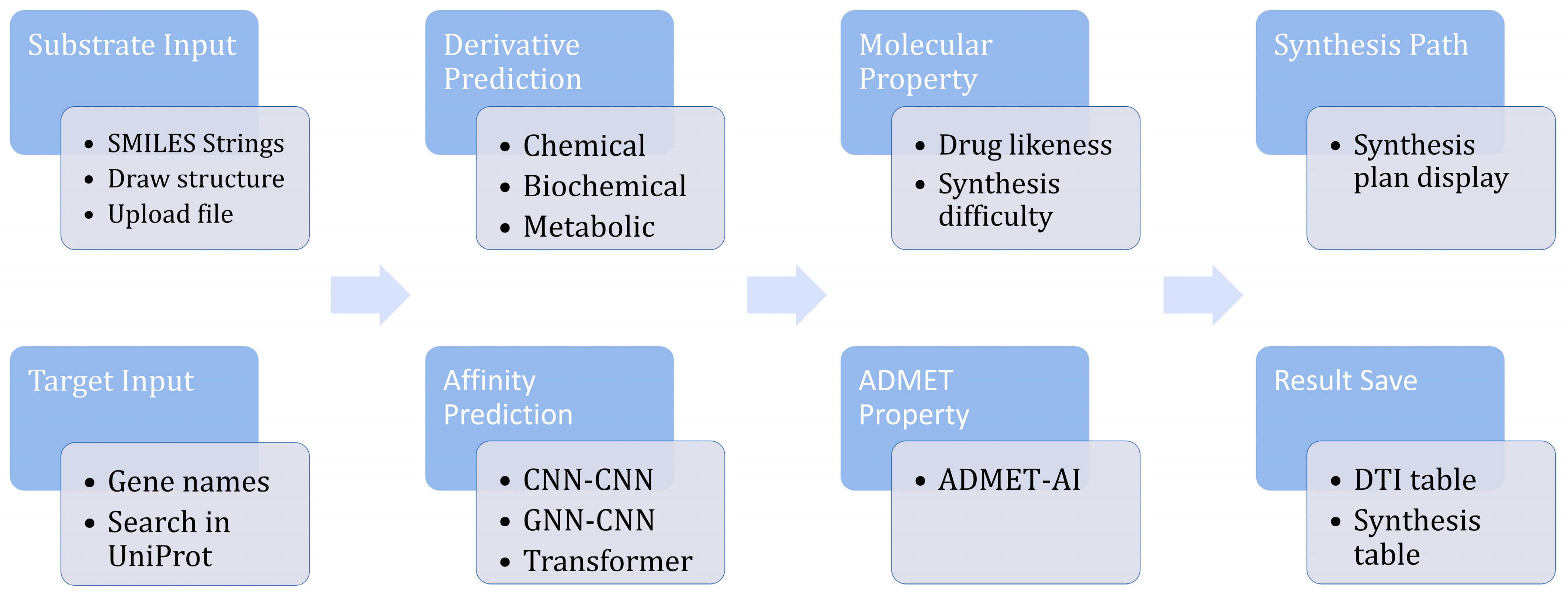
2.2. Software Functionalities
2.3. Illustrative Examples
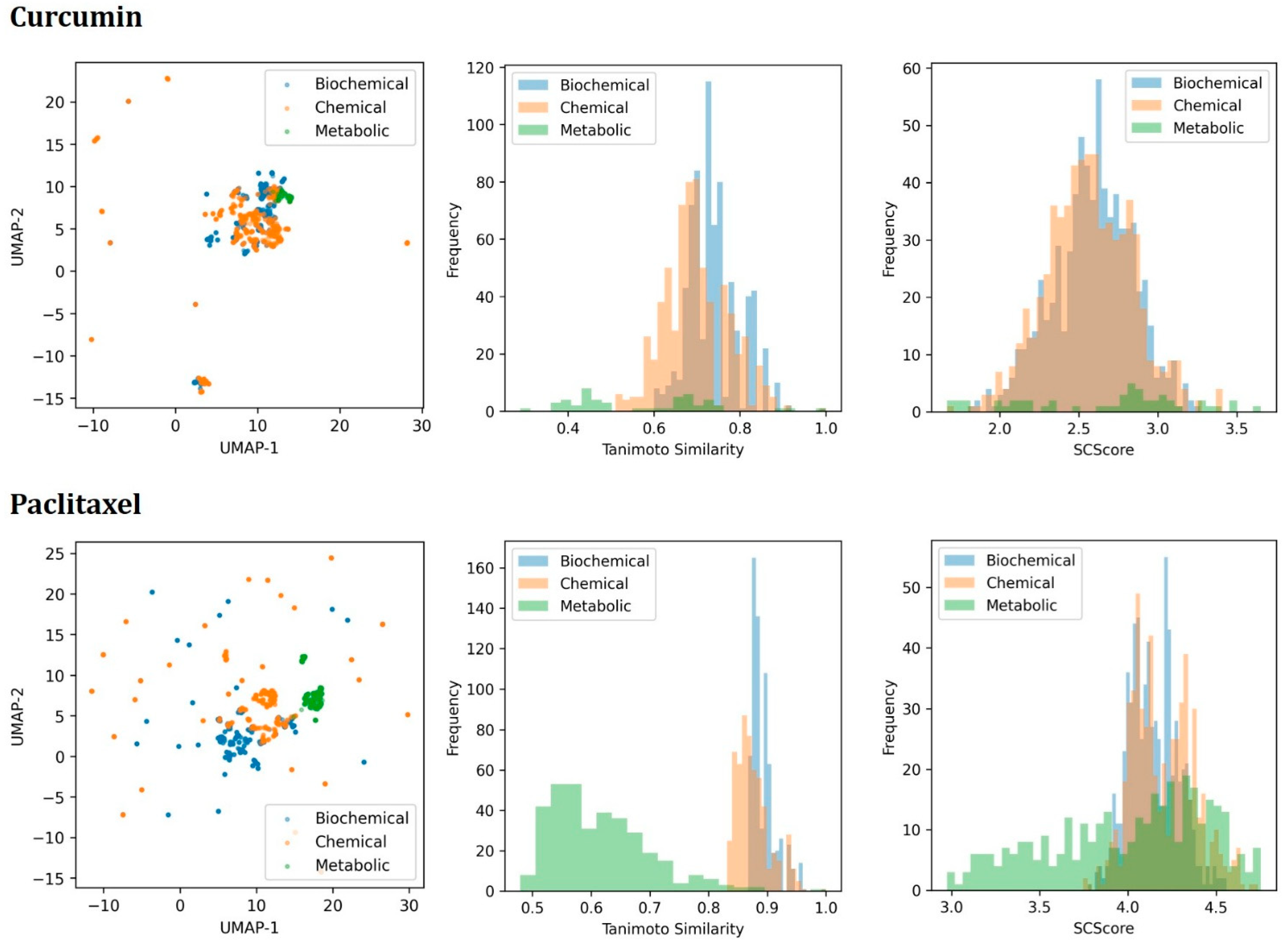
2.3.1. Structural Diversity Evaluation
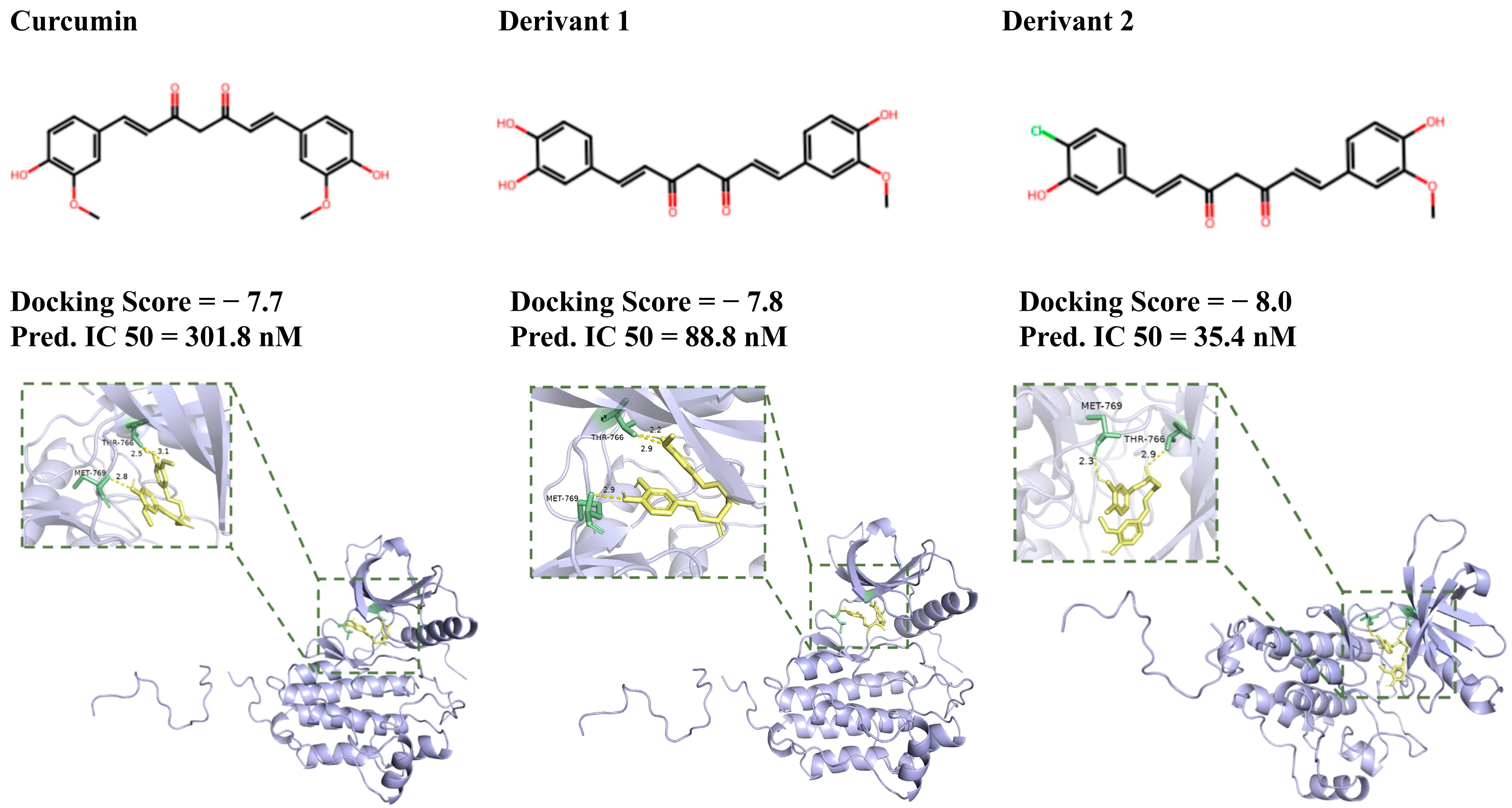
2.3.2. Pharmacological Active Prediction
3. Materials and Methods
3.1. Extraction of Reaction Templates
3.2. Generation of Potential Derivatives
3.3. Prediction of Molecular Properties
3.4. Prediction of Binding Affinity with Specific Targets
3.5. User-Defined Parameters and Initial Settings
3.6. Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Brown, D.G.; Young, R.J.; Keserü, G.M. Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug Discov. 2018, 17, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Hadadi, N.; Hatzimanikatis, V. Design of Computational Retrobiosynthesis Tools for the Design of de Novo Synthetic Pathways. Curr. Opin. Chem. Biol. 2015, 28, 99–104. [Google Scholar] [CrossRef]
- Gallo, K.; Kemmler, E.; Goede, A.; Becker, F.; Dunkel, M.; Preissner, R.; Banerjee, P. SuperNatural 3.0—A Database of Natural Products and Natural Product-Based Derivatives. Nucleic Acids Res. 2023, 51, D654–D659. [Google Scholar] [CrossRef] [PubMed]
- Poynton, E.F.; van Santen, J.A.; Pin, M.; Contreras, M.M.; McMann, E.; Parra, J.; Showalter, B.; Zaroubi, L.; Duncan, K.R.; Linington, R.G. The Natural Products Atlas 3.0: Extending the Database of Microbially Derived Natural Products. Nucleic Acids Res. 2025, 53, D691–D699. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT Online: Collection of Open Natural Products Database. J. Cheminf. 2021, 13, 2. [Google Scholar] [CrossRef]
- Fleming, N. How Artificial Intelligence Is Changing Drug Discovery. Nature 2018, 557, S55–S57. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking Drug Design in the Artificial Intelligence Era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Zeng, X.; Wang, F.; Luo, Y.; Kang, S.; Tang, J.; Lightstone, F.C.; Fang, E.F.; Cornell, W.; Nussinov, R.; Cheng, F. Deep Generative Molecular Design Reshapes Drug Discovery. Cell Rep. Med. 2022, 3, 100794. [Google Scholar] [CrossRef]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Pang, C.; Qiao, J.; Zeng, X.; Zou, Q.; Wei, L. Deep Generative Models in De Novo Drug Molecule Generation. J. Chem. Inf. Model. 2024, 64, 2174–2194. [Google Scholar] [CrossRef] [PubMed]
- Pogány, P.; Arad, N.; Genway, S.; Pickett, S.D. De Novo Molecule Design by Translating from Reduced Graphs to SMILES. J. Chem. Inf. Model. 2019, 59, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Stravs, M.A.; Dührkop, K.; Böcker, S.; Zamboni, N. MSNovelist: De Novo Structure Generation from Mass Spectra. Nat. Methods 2022, 19, 865–870. [Google Scholar] [CrossRef]
- Li, C.; Wang, C.; Sun, M.; Zeng, Y.; Yuan, Y.; Gou, Q.; Wang, G.; Guo, Y.; Pu, X. Correlated RNN Framework to Quickly Generate Molecules with Desired Properties for Energetic Materials in the Low Data Regime. J. Chem. Inf. Model. 2022, 62, 4873–4887. [Google Scholar] [CrossRef] [PubMed]
- Moret, M.; Friedrich, L.; Grisoni, F.; Merk, D.; Schneider, G. Generative Molecular Design in Low Data Regimes. Nat. Mach. Intell. 2020, 2, 171–180. [Google Scholar] [CrossRef]
- Colby, S.M.; Nuñez, J.R.; Hodas, N.O.; Corley, C.D.; Renslow, R.R. Deep Learning to Generate in Silico Chemical Property Libraries and Candidate Molecules for Small Molecule Identification in Complex Samples. Anal. Chem. 2020, 92, 1720–1729. [Google Scholar] [CrossRef]
- Joo, S.; Kim, M.S.; Yang, J.; Park, J. Generative Model for Proposing Drug Candidates Satisfying Anticancer Properties Using a Conditional Variational Autoencoder. ACS Omega 2020, 5, 18642–18650. [Google Scholar] [CrossRef]
- Kotsias, P.-C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct Steering of de Novo Molecular Generation with Descriptor Conditional Recurrent Neural Networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; Priyakumar, U.D. MolGPT: Molecular Generation Using a Transformer-Decoder Model. J. Chem. Inf. Model. 2022, 62, 2064–2076. [Google Scholar] [CrossRef]
- Wang, S.; Song, T.; Zhang, S.; Jiang, M.; Wei, Z.; Li, Z. Molecular Substructure Tree Generative Model for de Novo Drug Design. Brief. Bioinform. 2022, 23, bbab592. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Jones, D.T. Improving Protein Function Prediction with Synthetic Feature Samples Created by Generative Adversarial Networks. Nat. Mach. Intell. 2019, 730143. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, J.; Jun, J.J.; Xie, X.-Q. Deep Convolutional Generative Adversarial Network (dcGAN) Models for Screening and Design of Small Molecules Targeting Cannabinoid Receptors. Mol. Pharm. 2019, 16, 4451–4460. [Google Scholar] [CrossRef]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.-C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de Novo Molecular Generation Method Using Latent Vector Based Generative Adversarial Network. J. Cheminf. 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Sousa, T.; Correia, J.; Pereira, V.; Rocha, M. Generative Deep Learning for Targeted Compound Design. J. Chem. Inf. Model. 2021, 61, 5343–5361. [Google Scholar] [CrossRef]
- Abbasi, M.; Santos, B.P.; Pereira, T.C.; Sofia, R.; Monteiro, N.R.C.; Simões, C.J.V.; Brito, R.M.M.; Ribeiro, B.; Oliveira, J.L.; Arrais, J.P. Designing Optimized Drug Candidates with Generative Adversarial Network. J. Cheminform. 2022, 14, 40. [Google Scholar] [CrossRef]
- Gu, Y.; Xu, Z.; Yang, C. Empowering Graph Neural Network-Based Computational Drug Repositioning with Large Language Model-Inferred Knowledge Representation. Interdiscip. Sci. Comput. Life Sci. 2024, 1–18. [Google Scholar] [CrossRef]
- Bran, A.M.; Cox, S.; Schilter, O.; Baldassari, C.; White, A.D.; Schwaller, P. Augmenting Large Language Models with Chemistry Tools. Nat. Mach. Intell. 2024, 6, 525–535. [Google Scholar] [CrossRef]
- Wang, J.; Luo, H.; Qin, R.; Wang, M.; Wan, X.; Fang, M.; Zhang, O.; Gou, Q.; Su, Q.; Shen, C.; et al. 3DSMILES-GPT: 3D Molecular Pocket-Based Generation with Token-Only Large Language Model. Chem. Sci. 2025, 16, 637–648. [Google Scholar] [CrossRef]
- Dziekan, J.M.; Wirjanata, G.; Dai, L.; Go, K.D.; Yu, H.; Lim, Y.T.; Chen, L.; Wang, L.C.; Puspita, B.; Prabhu, N.; et al. Cellular Thermal Shift Assay for the Identification of Drug–Target Interactions in the Plasmodium Falciparum Proteome. Nat. Protoc. 2020, 15, 1881–1921. [Google Scholar] [CrossRef]
- Ji, H.; Lu, X.; Zhao, S.; Wang, Q.; Liao, B.; Bauer, L.G.; Huber, K.V.M.; Luo, R.; Tian, R.; Tan, C.S.H. Target Deconvolution with Matrix-Augmented Pooling Strategy Reveals Cell-Specific Drug-Protein Interactions. Cell Chem. Biol. 2023, 30, 1478–1487.e7. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target Identification Using Drug Affinity Responsive Target Stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef]
- Liang, Y.; Zhao, J.; Zou, H.; Zhang, J.; Zhang, T. In Vitro and in Silico Evaluation of EGFR Targeting Activities of Curcumin and Its Derivatives. Food Funct. 2021, 12, 10667–10675. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.E.M.; Yücer, R.; Dawood, M.; Hegazy, M.-E.F.; Drif, A.; Ooko, E.; Kadioglu, O.; Seo, E.-J.; Kamounah, F.S.; Titinchi, S.J.; et al. In Silico and In Vitro Screening of 50 Curcumin Compounds as EGFR and NF-κB Inhibitors. Int. J. Mol. Sci. 2022, 23, 3966. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A Deep Learning Library for Drug–Target Interaction Prediction. Bioinformatics 2021, 36, 5545–5547. [Google Scholar] [CrossRef]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. Computer-Assisted Retrosynthesis Based on Molecular Similarity. ACS Cent. Sci. 2017, 3, 1237–1245. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Bansal, P.; Morgat, A.; Axelsen, K.B.; Muthukrishnan, V.; Coudert, E.; Aimo, L.; Hyka-Nouspikel, N.; Gasteiger, E.; Kerhornou, A.; Neto, T.B.; et al. Rhea, the Reaction Knowledgebase in 2022. Nucleic Acids Res. 2022, 50, D693–D700. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. BiGG Models 2020: Multi-Strain Genome-Scale Models and Expansion across the Phylogenetic Tree. Nucleic Acids Res. 2020, 48, D402–D406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zeng, T.; Li, C.; Chen, B.; Coley, C.W.; Yang, Y.; Wu, R. Deep Learning Driven Biosynthetic Pathways Navigation for Natural Products with BioNavi-NP. Nat. Commun. 2022, 13, 3342. [Google Scholar] [CrossRef]
- Chen, B.; Li, H.; Huang, R.; Tang, Y.; Li, F. Deep Learning Prediction of Electrospray Ionization Tandem Mass Spectra of Chemically Derived Molecules. Nat. Commun. 2024, 15, 8396. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tian, S.; Allen, D.; Oler, E.; Peters, H.; Lui, V.W.; Gautam, V.; Djoumbou-Feunang, Y.; Greiner, R.; Metz, T.O. BioTransformer 3.0—A Web Server for Accurately Predicting Metabolic Transformation Products. Nucleic Acids Res. 2022, 50, W115–W123. [Google Scholar] [CrossRef] [PubMed]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58, 252–261. [Google Scholar] [CrossRef]
- Swanson, K.; Liu, G.; Catacutan, D.B.; Arnold, A.; Zou, J.; Stokes, J.M. Generative AI for Designing and Validating Easily Synthesizable and Structurally Novel Antibiotics. Nat. Mach. Intell. 2024, 6, 338–353. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep Drug–Target Binding Affinity Prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of Drug-Target Interactions via Deep Learning with Convolution on Protein Sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef]
- Shaikh, F.; Tai, H.K.; Desai, N.; Siu, S.W.I. LigTMap: Ligand and Structure-Based Target Identification and Activity Prediction for Small Molecular Compounds. J. Cheminf. 2021, 13, 1–12. [Google Scholar] [CrossRef]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable Deep Learning of Compound-Protein Affinity through Unified Recurrent and Convolutional Neural Networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef]
- Li, S.; Wan, F.; Shu, H.; Jiang, T.; Zhao, D.; Zeng, J. MONN: A Multi-Objective Neural Network for Predicting Compound-Protein Interactions and Affinities. Cell Systems 2020, 10, 308–322.e11. [Google Scholar] [CrossRef]
- Watanabe, N.; Ohnuki, Y.; Sakakibara, Y. Deep Learning Integration of Molecular and Interactome Data for Protein—Compound Interaction Prediction. J. Cheminf. 2021, 13, 44. [Google Scholar] [CrossRef]
- Ding, Q.; Zu, S.; Hou, S.; Zhang, Y.; Li, S. VISAR: An Interactive Tool for Dissecting Chemical Features Learned by Deep Neural Network QSAR Models. Bioinformatics 2020, 36, 3610–3612. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Deng, H.; Lu, H.; Zhang, Z. Predicting a Molecular Fingerprint from an Electron Ionization Mass Spectrum with Deep Neural Networks. Anal. Chem. 2020, 92, 8649–8653. [Google Scholar] [CrossRef] [PubMed]
- Mei, P.-C.; An, N.; Xiao, H.-M.; Chen, Y.-Y.; Zhu, Q.-F.; Feng, Y.-Q. QSAR-Guided Strategy for Accurate Annotation of FAHFA Regioisomers. Talanta 2025, 285, 127421. [Google Scholar] [CrossRef] [PubMed]
- Srour, A.M.; Ahmed, N.S.; Abd El-Karim, S.S.; Anwar, M.M.; El-Hallouty, S.M. Design, Synthesis, Biological Evaluation, QSAR Analysis and Molecular Modelling of New Thiazol-Benzimidazoles as EGFR Inhibitors. Bioorg. Med. Chem. 2020, 28, 115657. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Curcumin | Derivate 1 | Derivate 2 | |
|---|---|---|---|
| MW | 368.385 | 372.804 | 354.358 |
| SCScore | 1.804 | 2.109 | 2.212 |
| logP | 3.370 | 4.015 | 3.067 |
| nHBA | 6 | 5 | 6 |
| nHBD | 2 | 2 | 3 |
| QED | 0.548 | 0.566 | 0.401 |
| TPSA | 93.060 | 83.830 | 104.060 |
| BBB | 0.344 | 0.255 | 0.286 |
| BS | 0.463 | 0.578 | 0.414 |
| CYP1A2 inhibition | 0.439 | 0.665 | 0.420 |
| CYP2C19 inhibition | 0.582 | 0.703 | 0.506 |
| CYP3A4 inhibition | 0.751 | 0.771 | 0.697 |
| PAMPA | 0.739 | 0.734 | 0.663 |
| Caco2 | −5.171 | −5.084 | −5.283 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Zhang, M.; Chang, S.; Chu, G.; Ji, H. DerivaPredict: A User-Friendly Tool for Predicting and Evaluating Active Derivatives of Natural Products. Molecules 2025, 30, 1683. https://doi.org/10.3390/molecules30081683
Song Y, Zhang M, Chang S, Chu G, Ji H. DerivaPredict: A User-Friendly Tool for Predicting and Evaluating Active Derivatives of Natural Products. Molecules. 2025; 30(8):1683. https://doi.org/10.3390/molecules30081683
Chicago/Turabian StyleSong, Yu, Meng Zhang, Sihao Chang, Ganghui Chu, and Hongchao Ji. 2025. "DerivaPredict: A User-Friendly Tool for Predicting and Evaluating Active Derivatives of Natural Products" Molecules 30, no. 8: 1683. https://doi.org/10.3390/molecules30081683
APA StyleSong, Y., Zhang, M., Chang, S., Chu, G., & Ji, H. (2025). DerivaPredict: A User-Friendly Tool for Predicting and Evaluating Active Derivatives of Natural Products. Molecules, 30(8), 1683. https://doi.org/10.3390/molecules30081683