Abstract
A palladium-catalyzed aromatic dual C-H acylations followed with intramolecular cyclizations have been developed by the assistance of bidentate N-(quinolin-8-yl)benzamide. This tandem process involves the formation of three new chemical bonds, providing access to novel quinoline-substituted hydroxyl isoindolones skeleton under simple reaction conditions. The deuterium-labeled competition reaction has revealed that C-H bond cleavage is the turnover limiting step.
1. Introduction
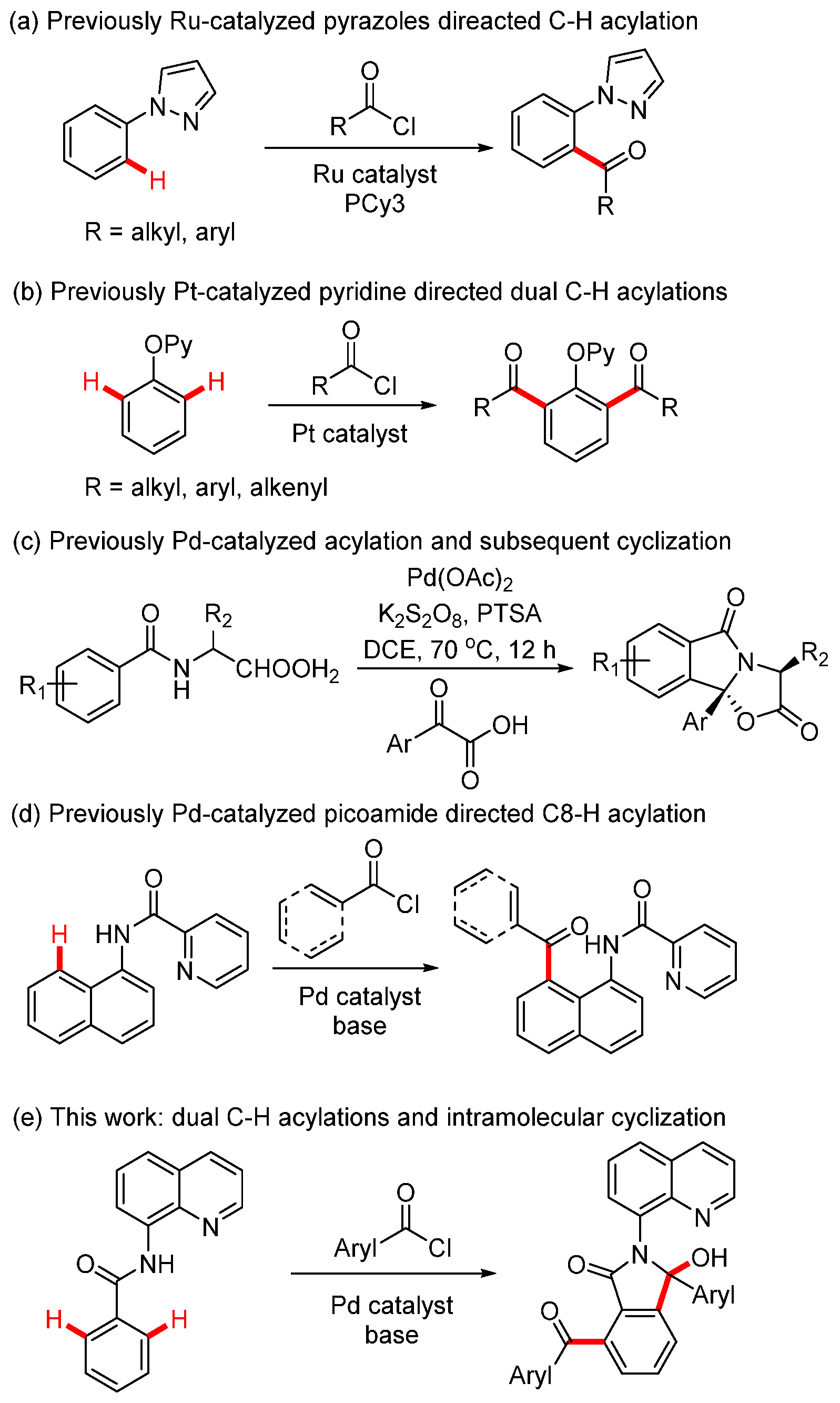
Transition metal-catalyzed C-H acylation reactions have been intensively investigated as the ketone products serve as valuable moieties in natural products, functional materials, and drug discovery [1]. Several reagents have been developed in catalytic C-H acylations, including aldehyde [2], α-oxocarboxylic acids [3], alcohol [4], aryl methanes [5], cyclopropenones [6], anhydrides [7], ketenes [8], and acyl fluorides [9]. Acyl chlorides are less commonly used in C-H acylations since they are typically employed in Friedel–Crafts acylation under Lewis acid catalysis [10]. However, utilizing acyl chlorides in C-H acylations presents a promising strategy, as only a base is required for the cleavage of hydrogen chloride and no external oxidant is needed for substrate or metal catalyst turnover. To the best of our knowledge, very few reactions have described the C-H acylations with acyl chlorides assisted by directing group strategy. In 2013, the Frost group reported the Ru-catalyzed aromatic C-H acylation of arylpyrazoles, demonstrating good functional group tolerance of both aryl and alkyl acyl chlorides (Scheme 1a) [11]. In 2017, the Huo group reported Pt-catalyzed dual C-H acylation of 2-(aryloxy)pyridines, yielding diacylated products (Scheme 1b) [12]. In 2019, Wang and coworkers reported Pd-catalyzed ortho-acylation of N-benzoyl α-amino acid derivatives and subsequent intramolecular cyclizations for the synthesis of oxazoloisoindolinones (Scheme 1c) [13]. Later, Wu’s group described palladium-catalyzed C8-H acylation of 1-Naphthylamines with acyl chlorides (Scheme 1d) [14].
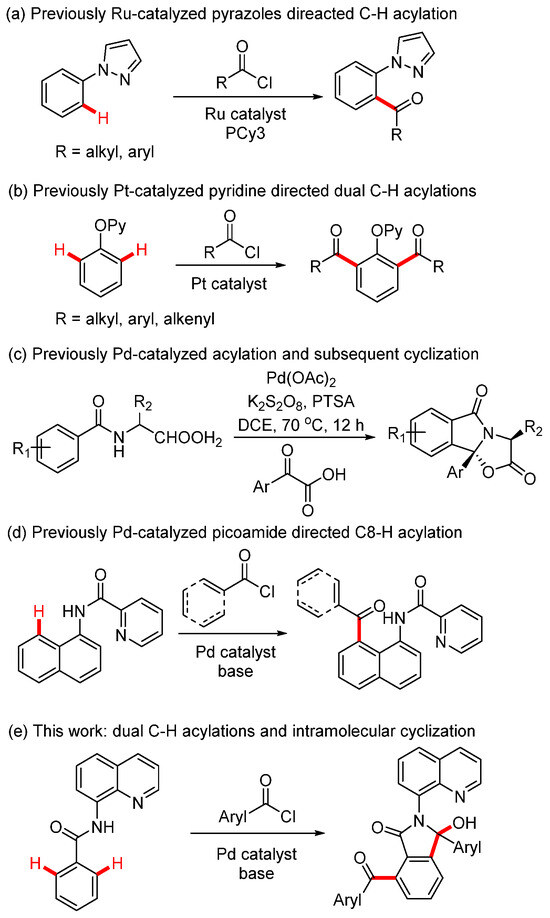
Scheme 1.
Metal-catalyzed C-H acylations from acyl chlorides.
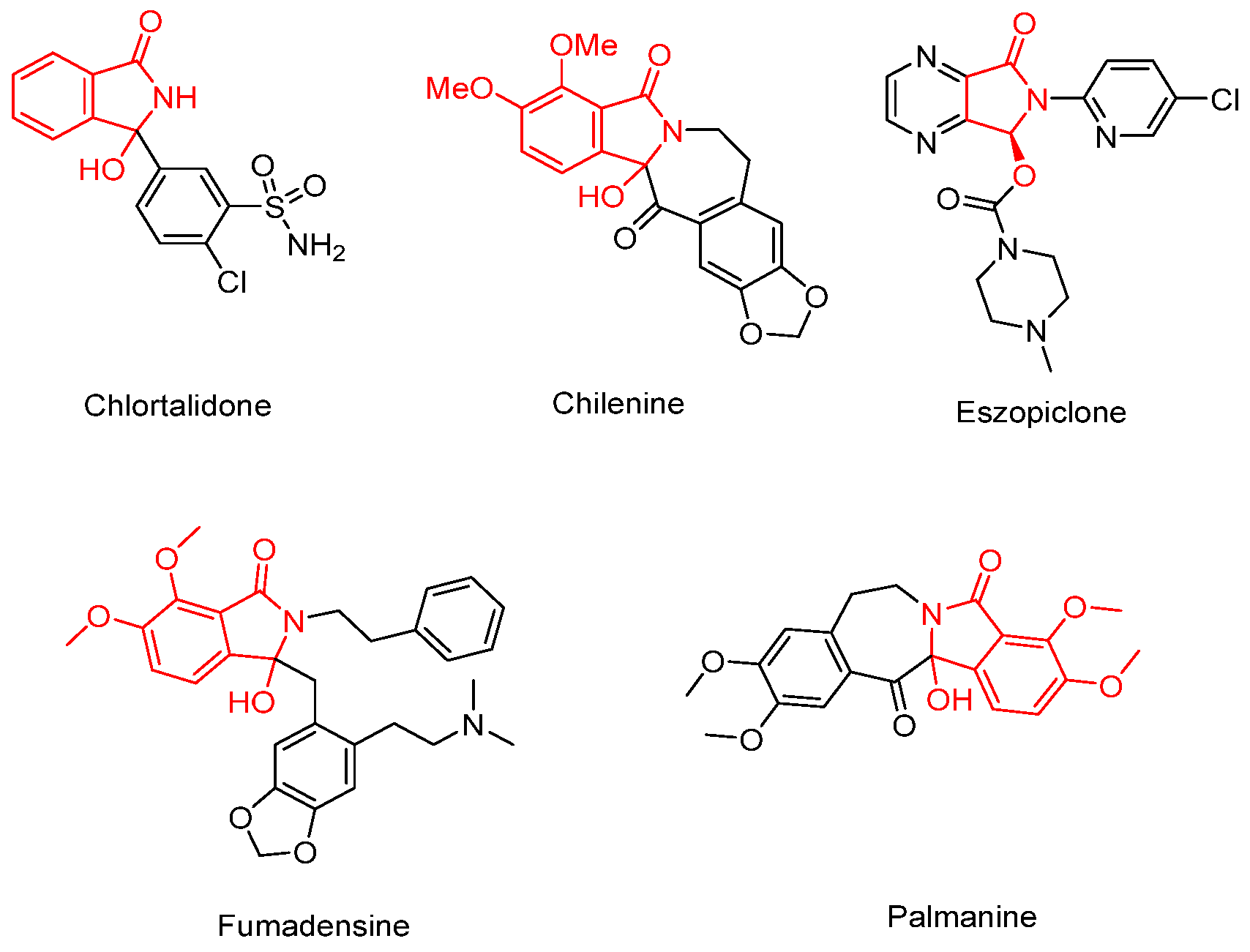
Hydroxyl isoindolones represent a useful class of heterocycles that exist in many natural products and drugs, such as chlortalidone, chilenine, eszopicloneand, palmanine, and fumadensine (Figure 1) [15,16,17,18,19]. Therefore, the diversity of synthetic methods for the preparation of the hydroxyl isoindolone core structure has attracted the attention of organic chemists, who continue to make efforts in this area. Herein, we wish to present our findings on Pd-catalyzed dual acylation with acyl chlorides and intramolecular cyclization, facilitated by N-(quinolin-8-yl)benzamide-directed ortho C-H activation, resulting in novel quinoline-substituted hydroxyl isoindolones (Scheme 1e).
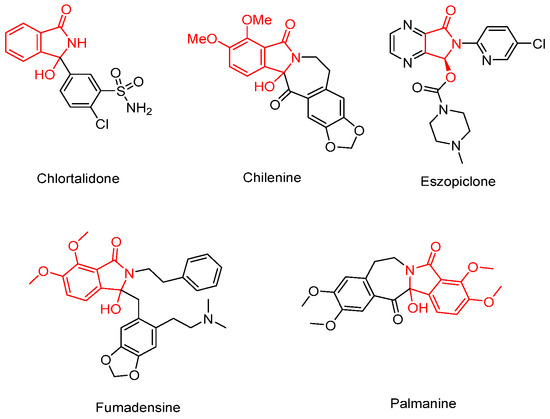
Figure 1.
Bioactive compounds with a hydroxyl isoindolone skeleton.
2. Results and Discussion
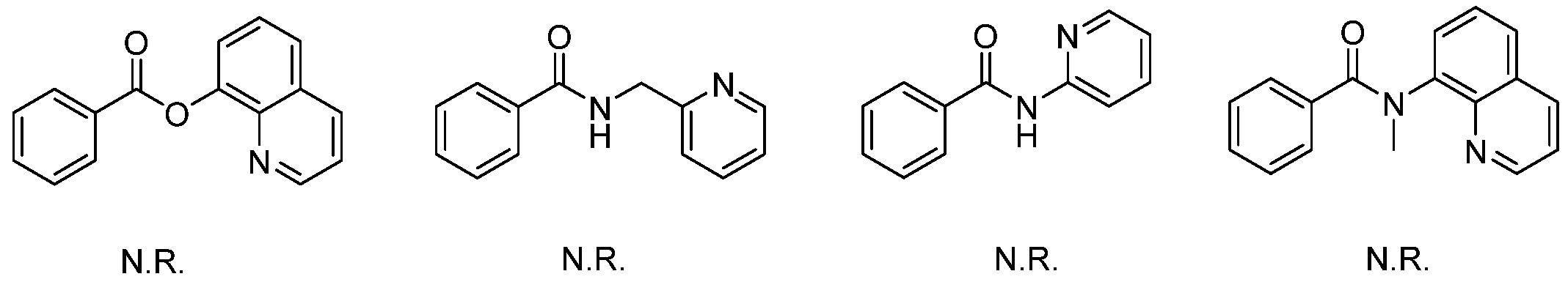
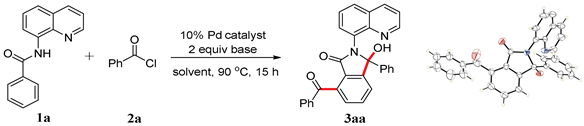
We initiated the optimization process using N-(quinolin-8-yl)benzamide and benzoyl chloride as model substrates. The reaction yielded an unexpected product 3aa in 39% yield under Pd(OAc)2 catalysis, utilizing KOAc as the base and toluene as the solvent (Table 1, entry 1). The structure of 3aa was determined through NMR and HRMS analysis and was fully confirmed by X-ray diffraction (CDCC 2371075). The product was generated via dual acylations and intramolecular cyclization [20,21]. The subsequent optimization involved screening various metal catalysts, bases, solvents, and concentrations to enhance the yield. Different bases were tested in this reaction, and the results indicated that NaOAc was the optimal choice (entry 4). In addition to Pd(OAc)2, other palladium(II) catalysts, such as PdCl2(MeCN)2, PdCl2, Pd2(dba)3, and Pd(TFA)2, were also compatible in this tandem reaction, although they produced the corresponding product in lower yields (entries 9–12). The solvent screening included both polar and non-polar solvents (entries 13–20), and the highest yield was obtained using xylene as the solvent (entry 18). Subsequently, the reaction conditions, including concentration, temperature, and duration, were optimized (entries 21–25), resulting in a maximum yield of 83% with 2 mL of xylene at 90 °C for 15 h (entry 22). With lower Pd catalyst loading, the yield of the product decreased to 67% (5 mol% Pd(OAc)2) and 60% (2.5 mol% Pd(OAc)2). The product was isolated in 68% yield when the reaction was performed under air (entry 28). When benzoyl bromide was employed as the acylation reagent, the yield of the product decreased to 42% (entry 29). No reaction occurred in the absence of the palladium catalyst or base (entries 30–31). It is noteworthy that aside from the starting materials and product 3aa, no other products or intermediates were detected by thin-layer chromatography (TLC) or isolated from chromatography during the optimization process. In addition to N-(quinolin-8-yl)benzamide as the directing group, other bidentate substrates were also tested in this reaction system; however, the results indicated that they were not promising for this transformation (Scheme 2).

Table 1.
Optimization of the reaction conditions a.
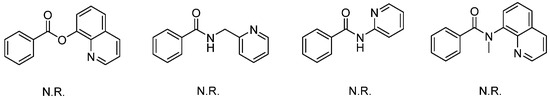
Scheme 2.
Screening of other bidentate-directing substrates.
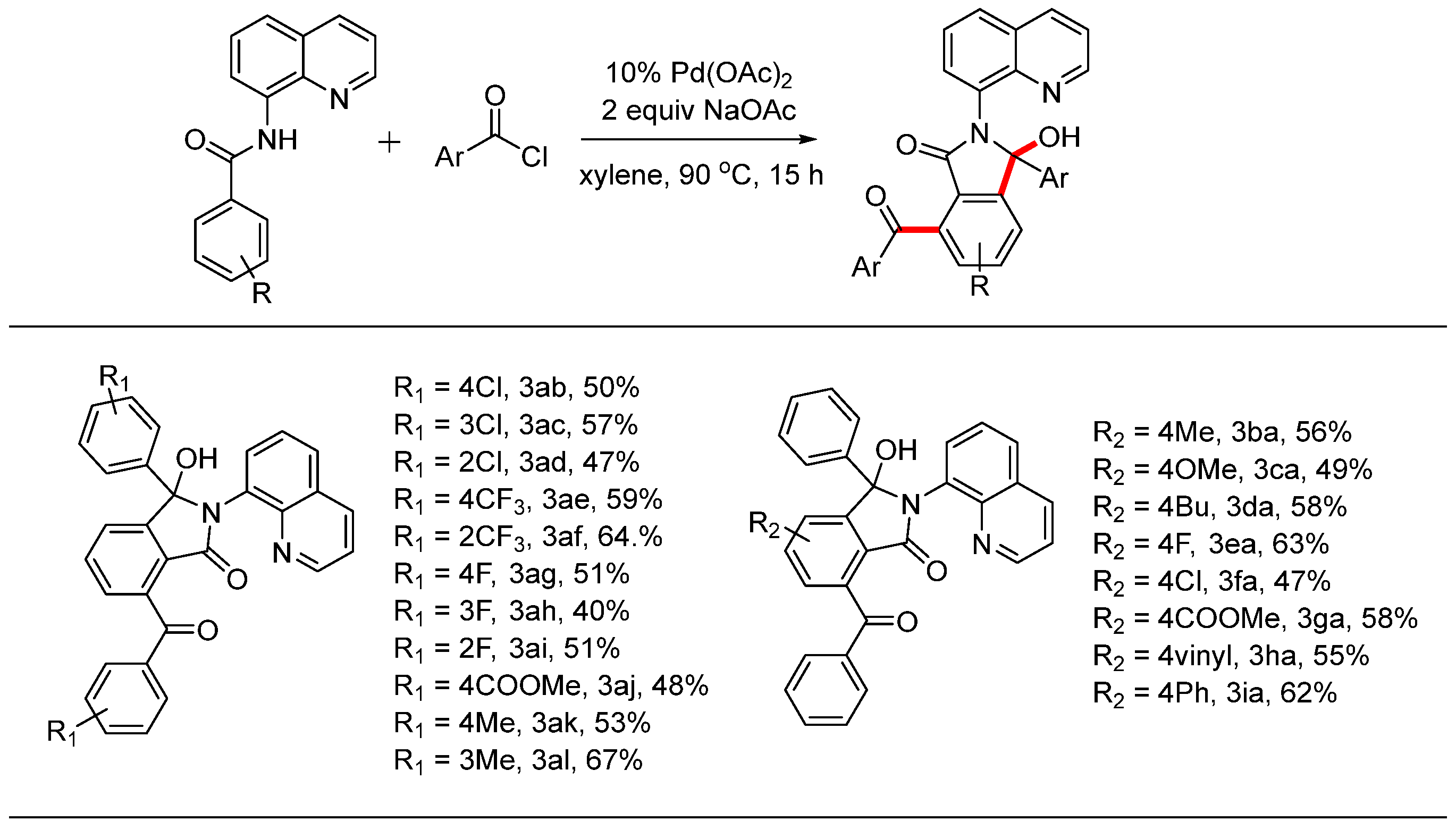
With the optimal reaction conditions established, we investigated the substrate scope using various substituted N-(quinolin-8-yl)benzamide derivatives, and the results are summarized in Scheme 3. The reaction conditions proved compatible with aromatic acyl chloride derivatives containing functional groups such as -F, -Cl, -CF3, -COOMe, and -Me, yielding the desired products in moderate yields. When comparing substituents in different positions on the phenyl ring, the steric effect did not significantly impact the yield of the product. For instance, the yield of product 3af (64%) with ortho-substitution was higher than that of the para-substituted product 3ae (59%). Similar results were observed for products with chloride groups (3ab, 3ac, and 3ad), fluorine groups (3ag, 3ah, and 3ai), and methyl groups (3ak and 3al). Next, we also examined the scope of N-(quinolin-8-yl)benzamide derivatives. The introduction of various groups, including both electron-donating (-Me, -OMe, and -nBu) and electron-withdrawing (-F, -Cl, and -COOMe) substituents, was compatible with the reaction conditions, resulting in the desired products in moderate yields. Additionally, vinyl and phenyl groups at the para position successfully underwent this reaction transformation, yielding the corresponding products 3ha and 3ia in 55% and 62%, respectively.
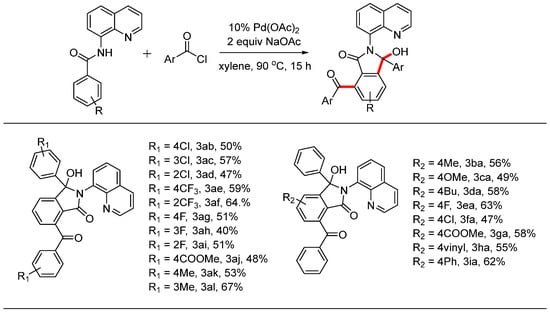
Scheme 3.
Pd-catalyzed dual C-H aroylations and cyclization reactions.
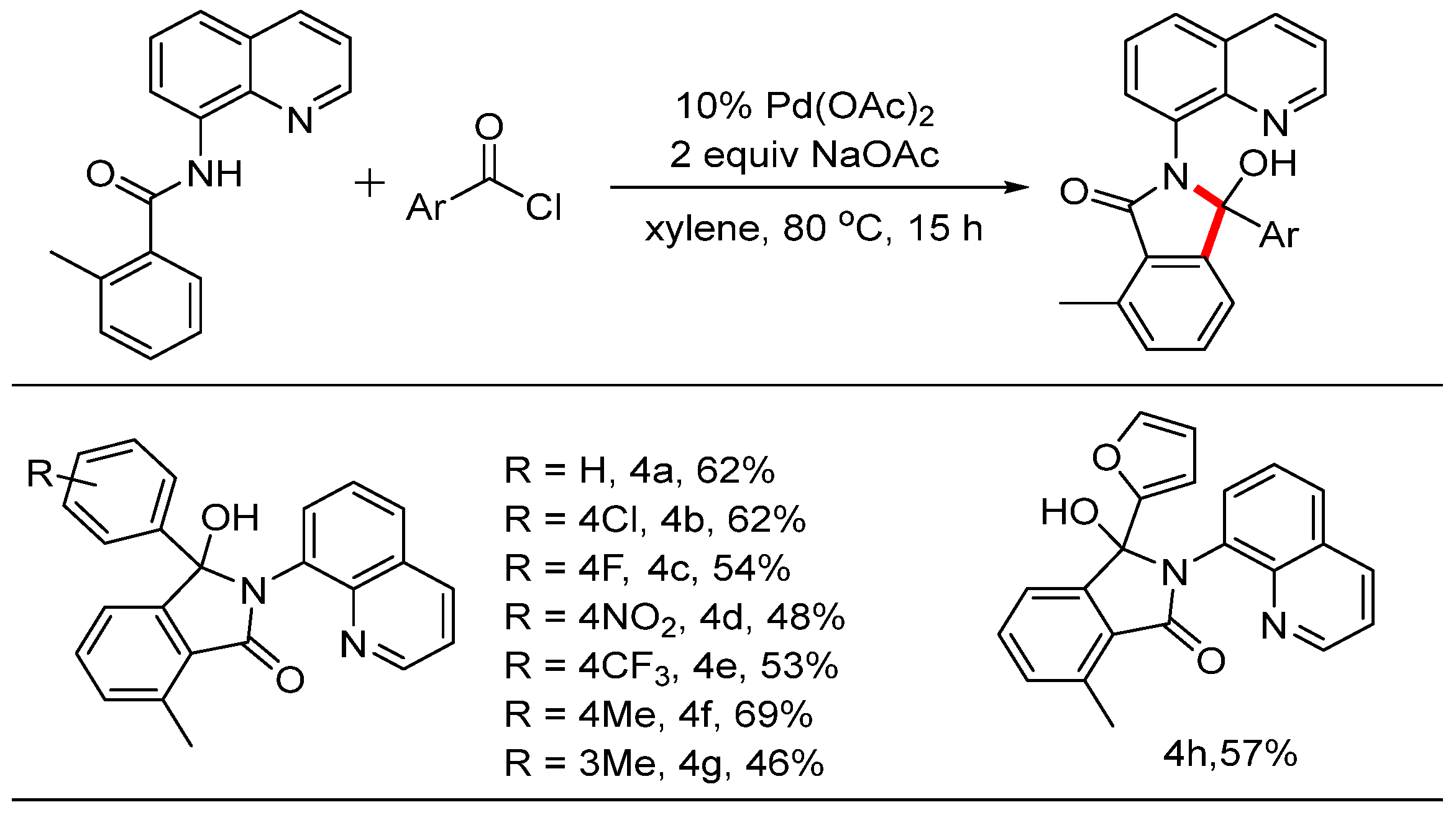
When the ortho position of the phenyl ring was occupied by a methyl group, the reaction yielded only mono-acylation and cyclization products. The scope of this reaction was also investigated, and the results are summarized in Scheme 4. When benzoyl chloride was subjected to these reaction conditions, product 4a was isolated with a yield of 62%. The structure was determined by NMR and HRMS and was fully confirmed by X-ray diffraction analysis (CDCC 2371076). Electron-withdrawing groups such as -Cl, -F, -NO2, and -CF3 at the para position were tolerated in this system, resulting in products with moderate yields. To observe the steric effect, the methyl group at the para position produced product 4f with a yield of 69%, while the meta-substituted substrate yielded product 4g, also of 69%. Notably, when 2-furoyl chloride was used in this reaction, the desired product 4h was isolated with a yield of 57%. To test whether the reaction could be scaled up to generate preparatively useful quantities of material (Scheme 5), we attempted a gram-scale reaction using 1.24 g (5 mmol) of starting materials. Gratifyingly, this was converted into the product 1.40 g with a yield of 60%.
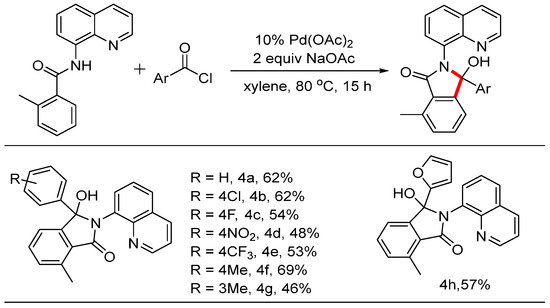
Scheme 4.
Pd-catalyzed mono C-H aroylation and cyclization reactions.
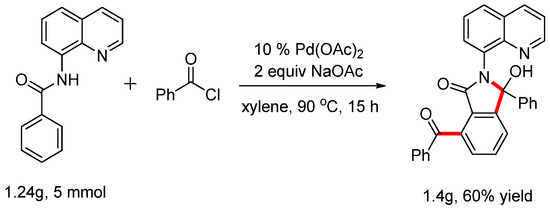
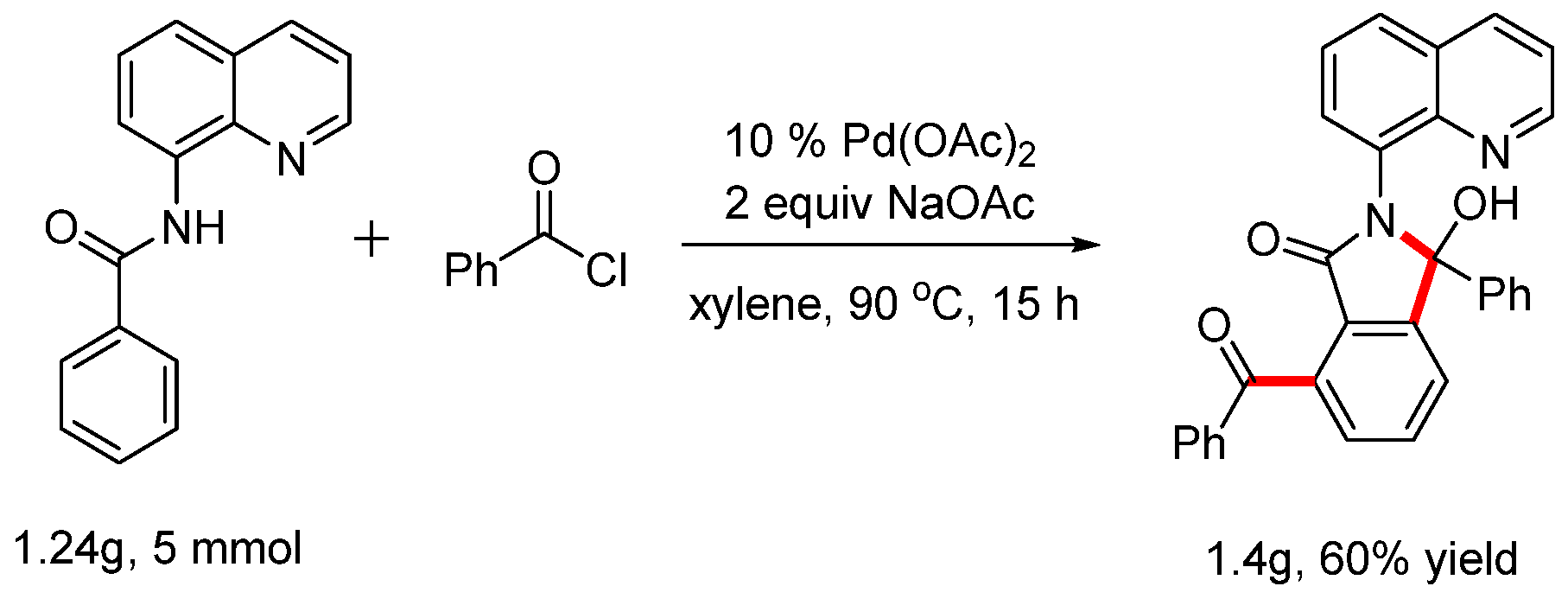
Scheme 5.
Gram-scale reaction.
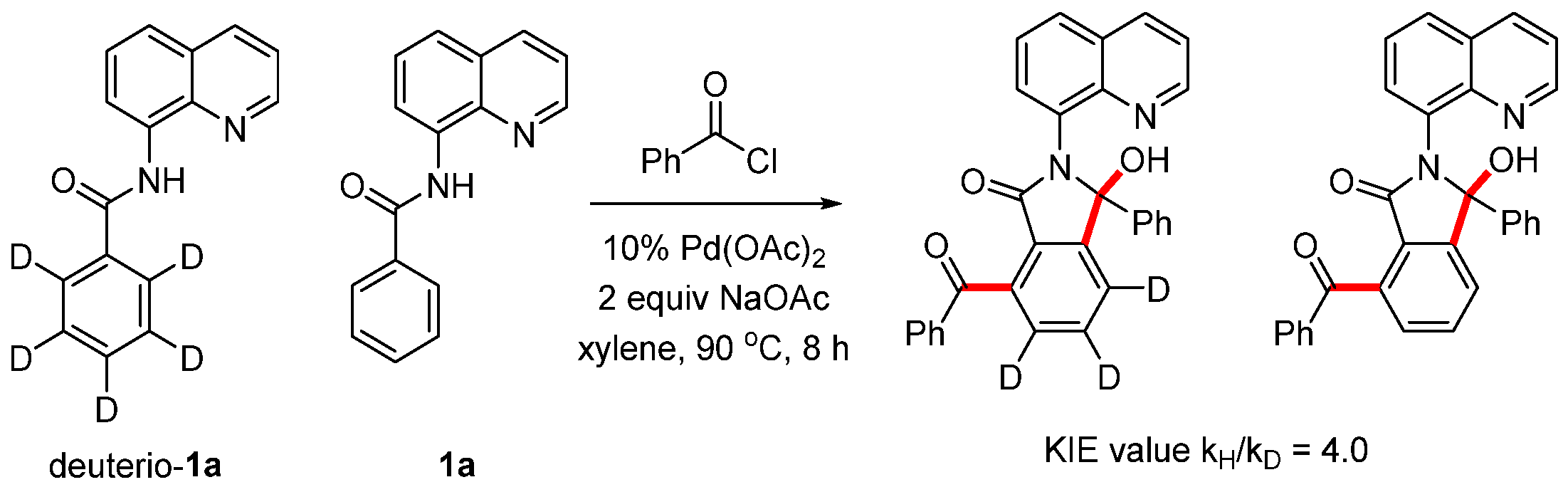
To investigate the catalytic mechanism, we conducted a competition experiment using equimolar amounts of deuterio-1a and N-(quinolin-8-yl)benzamide 1a with benzoyl chloride under our standard conditions for 8 h. This resulted in an intermolecular kinetic isotope effect (kH/kD) of 4.0. These results indicate that C-H activation is the rate-determining step of this reaction (Scheme 6).
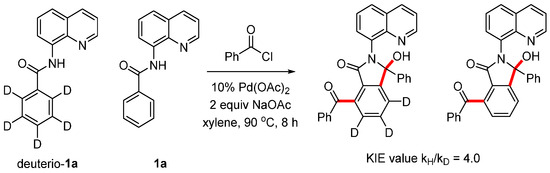
Scheme 6.
Kinetic isotope effect experiment.
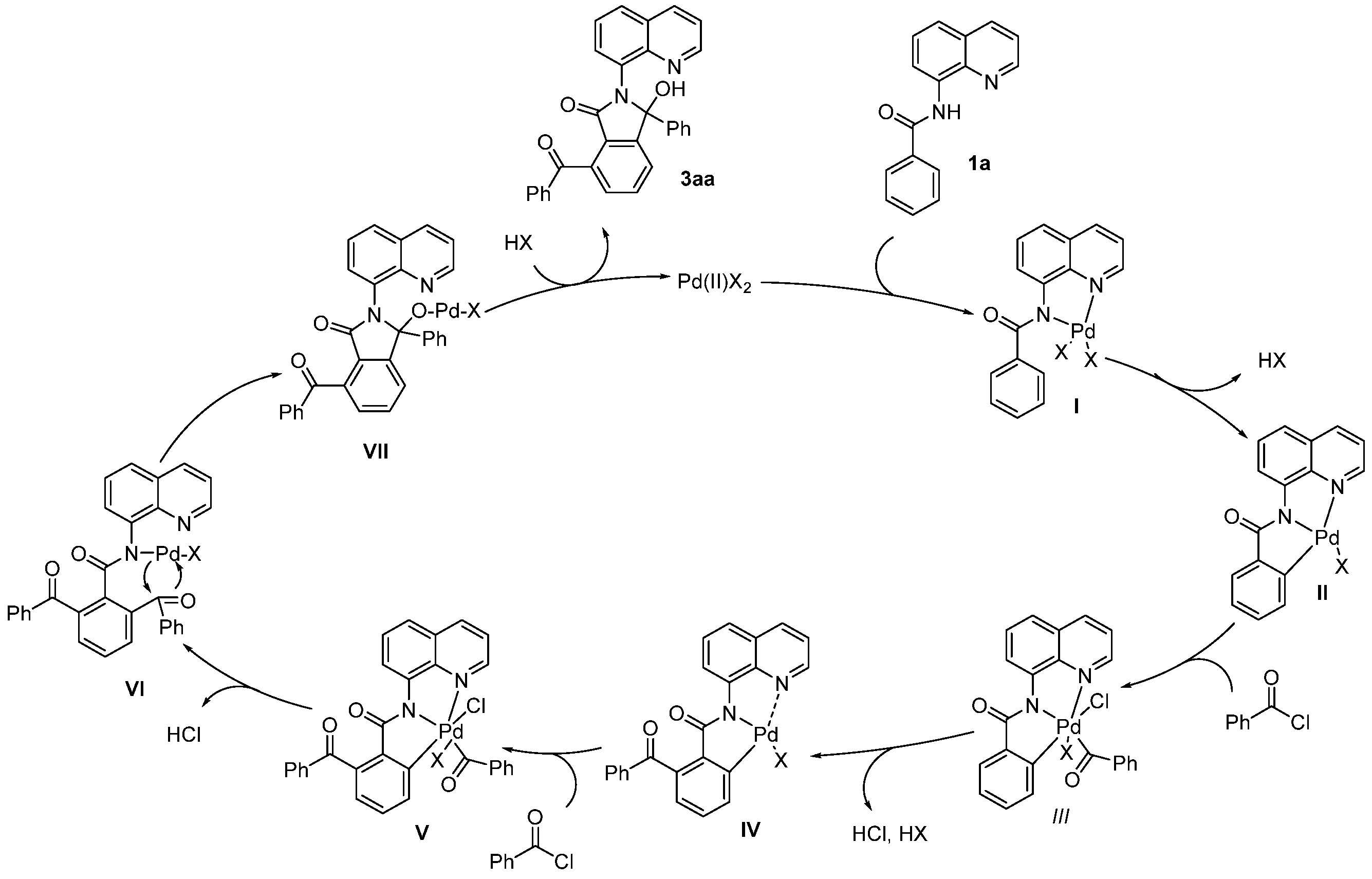
Based on the experimental results and previous literature [13,14], a plausible mechanism is proposed in Scheme 7. First, palladium intermediate I is generated by the coordination of two nitrogen atoms from the amide and quinoline of substrate 1a to the Pd catalyst, followed by a concerted metalation–deprotonation process that produces intermediate II. Next, the oxidative addition of acyl chlorides to II leads to the formation of intermediate III, which undergoes reductive elimination to yield the mono-acylated Pd intermediate IV. A similar oxidative addition and reductive elimination occur to generate Pd intermediates V and VI, in which the Pd atom coordinates to the nitrogen atom of benzamide. The intramolecular cyclization of VI produces intermediate VII, which is followed by protonation to yield the final product 3aa, along with the simultaneous release of a Pd(II) species to complete the catalytic cycle.
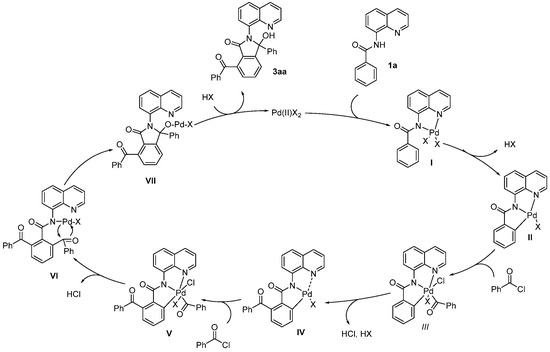
Scheme 7.
Proposed mechanism.
3. Materials and Methods
3.1. General Information
1H NMR and 13C NMR were recorded in CDCl3 or DMSO-d6 at room temperature on the Bruker AVANCE NEO 600 (151 MHz for 13C NMR), Billerica, MA, USA. The chemical shifts scale is based on internal TMS. The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; qui, quintet; sxt, sextet. The coupling constants, J, are reported in Hertz (Hz). Mass spectroscopy data were collected on an HRMS-ESI instrument. Unless otherwise noted, all reagents were obtained from commercial suppliers and used without further purification. All solvents were purified and dried according to standard methods prior to use. Products were purified by flash column chromatography on 100–200 mesh silica gel, SiO2.
3.2. Typical Procedure for the Preparation of Benzamides
All benzamides 1 were synthesized from the corresponding benzoic acids or benzoyl chlorides and 8-aminoquinoline. The deuterated amides were synthesized according to a literature method, and the spectral properties are consistent with the literature values [22,23,24,25,26]. The following amides were synthesized according to the literature procedures [27].
3.3. General Procedure for the Synthesis of Compound 3
A Schlenk tube was equipped with a magnetic stir bar and charged with substituted N-(quinolin-8-yl)benzamide 1 (0.1 mmol), 2 (0.25 mmol), NaOAc (0.2 mmol, 16 mg), Pd(OAc)2 (0.01 mmol, 2.3 mg), and xylene (2 mL). Then, the flask was sealed under N2 and stirred at 90 °C for 16 h. After the reaction was quenched by the addition of water, the mixture was extracted with dichloromethane and the combined organic layer was dried over sodium sulfate. The concentration in vacuo followed by silica gel column purification with petroleum ether/ethyl acetate eluent gave the desired product 3.
7-Benzoyl-3-hydroxy-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (3aa, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 83% yield (38 mg), mp 115–116 °C. 1H NMR (600 MHz, CDCl3) δ 10.72 (s, 1H), 8.82 (dd, J = 4.4, 1.7 Hz, 1H), 8.19 (dd, J = 8.3, 1.7 Hz, 1H), 7.93–7.89 (m, 2H), 7.70 (t, J = 7.6 Hz, 1H), 7.64 (dd, J = 8.2, 1.4 Hz, 1H), 7.60 (dd, J = 7.6, 1.4 Hz, 1H), 7.55–7.48 (m, 3H), 7.47–7.38 (m, 6H), 7.18–7.11 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 196.6, 167.2, 150.7, 148.4, 143.6, 139.4, 138.1, 137.9, 137.7, 137.7, 137.1, 134.3, 133.6, 133.2, 132.0, 129.8, 129.4, 128.8, 128.2, 127.9, 127.5, 127.3, 127.2, 127.1, 126.9, 126.8, 124.0, 123.7, 121.2, 93.3. HRMS(ESI) m/z [M + H]+ Calcd for C30H20N2O3: 457.5010, found: 457.5086.
7-(4-Chlorobenzoyl)-3-(4-chlorophenyl)-3-hydroxy-2-(quinolin-8-yl)isoindolin-1-one (3ab, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 50% yield (26.25 mg), mp 125–128 °C. 1H NMR (600 MHz, CDCl3) δ 10.87 (s, 1H), 8.82 (dd, J = 4.4, 1.7 Hz, 1H), 8.22 (d, J = 8.2 Hz, 1H), 7.86–7.81 (m, 2H), 7.73 (d, J = 7.6 Hz, 1H), 7.71–7.67 (m, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.55 (dd, J = 7.5, 0.9 Hz, 1H), 7.50–7.44 (m, 3H), 7.39–7.37 (m, 2H), 7.35–7.32 (m, 2H), 7.16–7.11 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 195.0, 167.0, 150.2, 148.5, 139.9, 138.2, 137.1, 135.5, 134.1, 133.6, 130.9, 129.5, 128.8, 128.4, 128.1, 128.0, 127.6, 124.3, 121.4, 92.9. HRMS(ESI) m/z [M + H]+ Calcd for C30H18Cl2N2O3: 525.0694, found: 525.0770.
7-(3-Chlorobenzoyl)-3-(3-chlorophenyl)-3-hydroxy-2-(quinolin-8-yl)isoindolin-1-one (3ac, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 57% yield (30 mg), mp 120–123 °C. 1H NMR (600 MHz, CDCl3) δ 10.61 (s, 1H), 8.80 (d, J = 4.4 Hz, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.90 (s, 1H), 7.76–7.70 (m, 2H), 7.65 (d, J = 8.2 Hz, 1H), 7.59–7.42 (m, 7H), 7.33 (d, J = 8.0 Hz, 1H), 7.25–7.21 (m, 1H), 7.11 (d, J = 5.0 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 195.2, 167.3, 150.6, 149.1, 143.9, 142.2, 139.1, 138.5, 137.2, 135.1, 134.6, 134.0, 133.7, 133.4, 132.3, 130.1, 129.9, 129.8, 129.6, 128.8, 128.4, 128.2, 128.1, 127.4, 127.3, 127.2, 125.2, 124.8, 121.8, 93.1. HRMS(ESI) m/z [M + H]+ Calcd for C30H18Cl2N2O3: 525.0694, found: 525.0775.
7-(2-Chlorobenzoyl)-3-(2-chlorophenyl)-3-hydroxy-2-(quinolin-8-yl)isoindolin-1-one (3ad, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 47% yield (25 mg), mp 114–117 °C. 1H NMR (600 MHz, CDCl3) δ 8.83 (dd, J = 4.4, 1.7 Hz, 1H), 8.18 (dd, J = 8.3, 1.7 Hz, 1H), 8.08 (dd, J = 6.9, 2.7 Hz, 1H), 7.84 (dd, J = 7.6, 1.4 Hz, 1H), 7.69 (d, J = 6.7 Hz, 2H), 7.64 (ddd, J = 11.0, 7.9, 1.5 Hz, 2H), 7.48–7.44 (m, 2H), 7.41 (dt, J = 6.4, 4.6 Hz, 2H), 7.39–7.34 (m, 1H), 7.29–7.27 (m, 1H), 7.15–7.12 (m, 1H), 7.08 (tt, J = 7.4, 5.3 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 195.1, 167.3, 149.3, 148.2, 143.3, 138.2, 138.1, 137.5, 136.2, 133.4, 133.1, 132.9, 132.5, 132.0, 130.9, 130.9, 130.7, 130.2, 130.0, 129.3, 128.9, 127.2, 127.2, 126.4, 126.2, 124.0, 121.2, 91.5. HRMS(ESI) m/z [M + H]+ Calcd for C30H18Cl2N2O3: 525.0694, found: 525.0774.
3-Hydroxy-2-(quinolin-8-yl)-7-(4-(trifluoromethyl)benzoyl)-3-(4-(trifluoromethyl)phenyl)isoindolin-1-one (3ae, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 59% yield (34.9 mg), mp 95–98 °C. 1H NMR (600 MHz, CDCl3) δ 10.98 (s, 1H), 8.84 (d, J = 4.4 Hz, 1H), 8.23 (d, J = 8.2 Hz, 1H), 8.01 (d, J = 7.9 Hz, 2H), 7.76 (t, J = 7.6 Hz, 1H), 7.68 (d, J = 8.1 Hz, 3H), 7.57 (dd, J = 21.6, 7.9 Hz, 4H), 7.52–7.44 (m, 5H) 13C NMR (151 MHz, CDCl3) δ 195.3, 167.0, 150.0, 148.6, 143.6, 139.7, 138.3, 136.8, 133.8, 131.8, 129.7 (d, J = 256.7 Hz), 128.1, 127.8, 127.1, 126.9, 125.7 (q, J = 3.7 Hz), 125.3 (q, J = 3.8 Hz), 124.6, 121.5, 92.9. HRMS(ESI) m/z [M + H]+ Calcd for C32H18F6N2O3: 593.1222, found: 593.1296.
3-Hydroxy-2-(quinolin-8-yl)-7-(2-(trifluoromethyl)benzoyl)-3-(2-(trifluoromethyl)phenyl)isoindolin-1-one (3af, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 64% yield (37.9 mg), mp 108–110 °C. 1H NMR (600 MHz, CDCl3) δ 10.15 (s, 1H), 8.69 (dd, J = 4.2, 1.6 Hz, 1H), 8.50 (dd, J = 7.4, 1.6 Hz, 1H), 8.09 (dd, J = 8.3, 1.7 Hz, 1H), 7.68–7.63 (m, 4H), 7.59 (d, J = 7.5 Hz, 2H), 7.55–7.48 (m, 5H), 7.46–7.42 (m, 2H), 7.38 (dd, J = 8.3, 4.2 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 195.2, 165.6, 148.3, 138.6, 137.9, 137.8, 134.6, 134.5, 131.8, 131.2, 130.3, 129.3, 128.9 (q, J = 200.8 Hz), 128.1, 127.5, 127.0 (q, J = 3.7 Hz), 124.6, 122.8(q, J = 3.8 Hz), 122.2, 121.8. HRMS(ESI) m/z [M + H]+ Calcd for C32H18F6N2O3: 593.1222, found: 593.1296.
7-(4-Fluorobenzoyl)-3-(4-fluorophenyl)-3-hydroxy-2-(quinolin-8-yl)isoindolin-1-one (3ag, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 51% yield (25 mg), mp 123–126 °C. 1H NMR (600 MHz, CDCl3) δ 10.10 (s, 1H), 8.81 (dd, J = 4.4, 1.8 Hz, 1H), 8.19 (dd, J = 8.3, 1.8 Hz, 1H), 7.95–7.90 (m, 2H), 7.72 (t, J = 7.6 Hz, 1H), 7.66 (dd, J = 8.3, 1.4 Hz, 1H), 7.59–7.35 (m, 7H), 7.10–7.04 (m, 2H), 6.85 (t, J = 8.7 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 194.6, 166.9, 161.5 (d, J = 255.6 Hz), 150.3, 148.5, 143.4, 138.1, 137.2, 135.4 (d, J = 3.0 Hz), 133.6 (d, J = 2.9 Hz), 133.4, 133.3, 132.2 (d, J = 9.4 Hz), 131.8, 129.4, 128.5 (d, J = 8.2 Hz), 127.8, 127.5, 126.9 (d, J = 14.1 Hz), 124.2, 121.3, 115.6, 115.5 (d, J = 21.9 Hz), 114.9, 92.9. HRMS(ESI) m/z [M + H]+ Calcd for C30H18F2N2O3: 493.1285, found: 493.1358.
7-(3-Fluorobenzoyl)-3-(3-fluorophenyl)-3-hydroxy-2-(quinolin-8-yl)isoindolin-1-one (3ah, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 40% yield (19.7 mg), mp 108–110 °C. 1H NMR (600 MHz, CDCl3) δ 10.77 (s, 1H), 8.90–8.78 (m, 1H), 8.22 (s, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.56 (t, J = 8.4 Hz, 2H), 7.52 (d, J = 7.7 Hz, 1H), 7.47 (dt, J = 15.9, 7.2 Hz, 2H), 7.38 (q, J = 7.3 Hz, 1H), 7.24–7.19 (m, 2H), 7.15 (q, J = 7.5 Hz, 1H), 7.09 (d, J = 7.9 Hz, 1H), 6.84 (t, J = 8.5 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 195.0, 166.9, 163.4 (d, J = 247.0Hz), 161.8, 150.1, 139.1, 136.9, 133.6, 130.1 (d, J = 6.5 Hz), 129.8 (d, J = 7.8 Hz), 129.5, 128.0, 124.4, 122.3, 121.4, 120.5 (d, J = 21.7 Hz), 115.9 (d, J = 22.4 Hz), 115.2 (d, J = 23.2 Hz), 114.0, 113.8, 92.8. HRMS(ESI) m/z [M + H]+ Calcd for C30H18F2N2O3: 493.1285, found: 493.1360.
7-(2-Fluorobenzoyl)-3-(2-fluorophenyl)-3-hydroxy-2-(quinolin-8-yl)isoindolin-1-one (3ai, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 51% yield (25 mg), mp 125–127 °C. 1H NMR (600 MHz, CDCl3) δ 8.81 (ddd, J = 12.1, 4.4, 1.7 Hz, 1H), 8.18 (ddd, J = 28.3, 8.4, 1.8 Hz, 1H), 7.90 (td, J = 7.6, 1.9 Hz, 1H), 7.86–7.84 (m, 1H), 7.73–7.64 (m, 2H), 7.63–7.56 (m, 2H), 7.56–7.50 (m, 1H), 7.47–7.39 (m, 3H), 7.23–7.17 (m, 1H), 7.11 (ddt, J = 7.3, 5.0, 2.4 Hz, 1H), 7.05 (dd, J = 10.9, 8.2 Hz, 1H), 6.95–6.91 (m, 1H), 6.78 (ddd, J = 11.6, 8.8, 5.4 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 193.2, 167.5, 158.4 ((d, J = 249.8 Hz)), 149.3, 148.5, 139.1, 135.0 (d, J = 8.9 Hz), 134.9, 133.3, 133.2, 131.5, 130.7 (d, J = 8.5 Hz), 129.4 (d, J = 2.4 Hz), 127.8, 127.4, 127.3, 124.1 (d, J = 3.6 Hz), 123.8, 122.0, 121.4, 116.8, 116.6 (d, J = 22.3 Hz), 115.8 (d, J = 21.7 Hz), 90.8. HRMS(ESI) m/z [M + H]+ Calcd for C30H18F2N2O3: 493.1285, found: 493.1364.
Methyl4-(1-hydroxy-1-(4-(methoxycarbonyl)phenyl)-3-oxo-2-(quinolin-8-yl)isoindoline-4-carbonyl)benzoate (3aj, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 48% yield (27.5 mg), mp 111–113 °C. 1H NMR (600 MHz, CDCl3) δ 10.80 (s, 1H), 8.81 (dd, J = 4.5, 1.8 Hz, 1H), 8.19–8.13 (m, 1H), 8.09–8.05 (m, 2H), 7.94 (d, J = 8.4 Hz, 2H), 7.86–7.78 (m, 2H), 7.73 (t, J = 7.6 Hz, 1H), 7.65–7.61 (m, 1H), 7.58 (d, J = 7.5 Hz, 1H), 7.54 (dd, J = 7.6, 1.4 Hz, 1H), 7.52–7.45 (m, 3H), 7.44 (dd, J = 8.3, 4.4 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 3.87 (s, 3H), 3.81 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 196.0, 167.3, 166.8, 166.5, 150.4, 149.0, 144.9, 143.7, 140.8, 138.5, 137.3, 134.3, 134.0, 133.5, 132.1, 130.3, 130.0, 129.9, 129.8, 129.7, 128.6, 128.0, 127.4, 127.3, 127.1, 124.9, 121.8, 93.4, 77.6, 77.4, 77.2, 52.7, 52.4. HRMS(ESI) m/z [M + H]+ Calcd for C34H24N2O7: 573.1584, found: 573.1572.
3-Hydroxy-7-(4-methylbenzoyl)-2-(quinolin-8-yl)-3-(p-tolyl)isoindolin-1-one (3ak, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 53% yield (25.6 mg), mp 115–118 °C. 1H NMR (600 MHz, CDCl3) δ 10.29 (s, J = 229.3 Hz, 1H), 8.81 (dt, J = 4.4, 2.6 Hz, 1H), 8.17 (d, J = 8.2 Hz, 1H), 7.81 (d, J = 7.8 Hz, 2H), 7.68 (t, J = 7.5 Hz, 1H), 7.61 (dd, J = 15.7, 7.9 Hz, 2H), 7.50 (d, J = 7.4 Hz, 1H), 7.47 (d, J = 7.6 Hz, 1H), 7.43 (q, J = 6.9 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 7.9 Hz, 2H), 2.34 (s, 3H), 2.19 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 196.0, 167.2, 150.7, 144.2, 137.7, 136.6, 134.7, 133.2, 129.8, 129.4, 129.1, 128.9, 128.7, 127.6, 127.2, 126.5, 123.9, 121.2, 93.3, 21.6, 20.9. HRMS(ESI) m/z [M + H]+ Calcd for C32H24N2O3: 485.1787, found: 485.1864.
3-Hydroxy-7-(3-methylbenzoyl)-2-(quinolin-8-yl)-3-(m-tolyl)isoindolin-1-one (3al, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 67% yield (32.4 mg), mp 122–125 °C. 1H NMR (600 MHz, CDCl3) δ 10.66 (s, 1H), 8.86–8.76 (m, 1H), 8.15 (d, J = 8.3 Hz, 1H), 7.80 (s, 1H), 7.70 (dd, J = 17.2, 8.4 Hz, 2H), 7.63 (d, J = 7.9 Hz, 2H), 7.53 (dd, J = 11.6, 7.6 Hz, 2H), 7.43 (q, J = 6.7 Hz, 2H), 7.37–7.25 (m, 4H), 7.08 (t, J = 7.6 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 2.37 (s, 3H), 2.22 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 196.6, 167.2, 150.7, 148.4, 143.6, 139.4, 138.1, 137.9, 137.7, 137.6, 137.1, 134.3, 133.6, 133.2, 132.0, 129.8, 129.4, 128.8, 128.2, 127.9, 127.5, 127.3, 127.2, 127.1, 126.9, 124.0, 123.7, 121.2, 93.3, 21.3, 21.2. HRMS(ESI) m/z [M + H]+ Calcd for C32H24N2O3: 485.1787, found: 485.1864.
7-Benzoyl-3-hydroxy-5-methyl-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (3ba, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 56% yield (26.4 mg), mp 125–127 °C. 1H NMR (600 MHz, CDCl3) δ 10.71 (s, 1H), 8.78 (d, J = 4.5 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 7.7 Hz, 2H), 7.59 (dd, J = 13.5, 7.8 Hz, 2H), 7.50 (t, J = 7.7 Hz, 1H), 7.45–7.37 (m, 6H), 7.31 (d, J = 27.5 Hz, 2H), 7.19–7.11 (m, 3H), 2.45 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 196.7, 167.4, 151.1, 148.6, 144.7, 143.7, 139.9, 138.2, 137.6, 137.3, 133.9, 133.5, 132.0, 129.8, 129.5, 128.7, 128.5, 128.2, 128.1, 127.3, 127.0, 126.8, 124.7, 124.7, 121.4, 93.3, 22.0. HRMS(ESI) m/z [M + H]+ Calcd for C31H22N2O3: 471.1630, found: 471.1706.
7-Benzoyl-3-hydroxy-5-methoxy-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (3ca, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 49% yield (23.8 mg), mp 118–120 °C.1H NMR (600 MHz, CDCl3) δ 8.81 (dd, J = 4.4, 1.7 Hz, 1H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 8.10–8.06 (m, 1H), 7.95–7.91 (m, 2H), 7.60 (dd, J = 8.0, 1.4 Hz, 1H), 7.56 (d, J = 7.5 Hz, 1H), 7.53–7.49 (m, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.43–7.39 (m, 5H), 7.19–7.15 (m, 2H), 7.15–7.12 (m, 1H), 7.03 (d, J = 2.2 Hz, 1H), 6.94 (d, J = 2.2 Hz, 1H), 3.84 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 196.1, 167.1, 164.1, 153.2, 148.5, 139.9, 139.2, 137.0, 133.7, 133.6, 132.1, 130.2, 129.9, 129.5, 128.6, 128.5, 128.2, 128.2, 127.3, 127.1, 126.7, 121.4, 119.7, 114.9, 108.6, 93.1, 56.1. HRMS(ESI) m/z [M + H]+ Calcd for C31H22N2O4: 487.1580, found: 487.1657.
7-Benzoyl-5-(tert-butyl)-3-hydroxy-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (3da, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 58% yield (29.7 mg), mp 117–120 °C. 1H NMR (600 MHz, CDCl3) δ 10.77 (s, 1H), 8.80 (dd, J = 4.5, 1.8 Hz, 1H), 8.15 (dd, J = 8.2, 1.9 Hz, 1H), 7.95–7.91 (m, 2H), 7.62–7.58 (m, 1H), 7.57 (d, J = 1.6 Hz, 1H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H), 7.53–7.49 (m, 2H), 7.44–7.37 (m, 6H), 7.15 (dt, J = 15.0, 6.9 Hz, 3H), 1.34 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 197.08, 167.31, 158.04, 150.82, 148.58, 143.80, 139.96, 138.17, 137.36, 137.21, 133.86, 133.53, 132.09, 129.87, 129.51, 128.55, 128.16, 128.11, 127.37, 127.07, 126.75, 125.51, 124.80, 121.41, 121.04, 93.57, 35.86, 31.34. HRMS(ESI) m/z [M + H]+ Calcd for C34H28N2O3: 513.2100, found: 513.2176.
7-Benzoyl-5-fluoro-3-hydroxy-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (3ea, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 63% yield (29.9 mg), mp 97–100 °C. 1H NMR (600 MHz, CDCl3) δ 11.27–10.51 (s, 1H), 8.81 (dd, J = 4.4, 1.7 Hz, 1H), 8.17 (dd, J = 8.4, 1.8 Hz, 1H), 7.91 (d, J = 7.6 Hz, 2H), 7.62 (dd, J = 15.5, 7.9 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.46–7.40 (m, 6H), 7.25–7.22 (m, 1H), 7.21–7.13 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 194.8, 166.4, 165.68 (d, J = 214.0 Hz), 148.7, 143.6, 139.2, 138.3, 136.7, 133.9, 133.5, 132.1, 129.8, 129.6, 128.7, 128.5, 128.4, 127.6, 127.1, 126.7, 121.5, 115.9 (d, J = 24.9 Hz), 111.8 (d, J = 23.8 Hz), 93.0. HRMS(ESI) m/z [M + H]+ Calcd for C30H19FN2O3: 475.1380, found: 475.1459.
7-Benzoyl-5-chloro-3-hydroxy-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (3fa, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 47% yield (23 mg), mp 96–100 °C. 1H NMR (600 MHz, CDCl3) δ 10.77 (s, 1H), 8.81 (dd, J = 4.4, 1.6 Hz, 1H), 8.17 (d, J = 8.3 Hz, 1H), 7.91 (d, J = 7.6 Hz, 2H), 7.64 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 7.5 Hz, 1H), 7.57–7.49 (m, 2H), 7.47–7.39 (m, 7H), 7.19 (t, J = 7.1 Hz, 2H), 7.16 (dd, J = 8.3, 6.0 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 194.8, 166.4, 152.4, 148.7, 143.6, 139.9, 139.2, 139.1, 138.3, 136.8, 133.9, 133.4, 132.1, 129.9, 129.6, 128.7, 128.5, 128.4, 128.2, 127.7, 127.1, 126.7, 125.5, 124.7, 121.5, 93.1. HRMS(ESI) m/z [M + H]+ Calcd for C30H19ClN2O3: 491.1084, found: 491.1161.
7-Benzoyl-3-hydroxy-1-oxo-3-phenyl-2-(quinolin-8-yl)isoindoline-5-carboxylate (3ga, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 58% yield (29.8 mg), mp 113–115 °C. 1H NMR (600 MHz, CDCl3) δ 8.83 (dd, J = 4.3, 2.0 Hz, 1H), 8.20 (d, J = 8.2 Hz, 2H), 8.13 (d, J = 1.6 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.67 (dd, J = 8.2, 1.8 Hz, 1H), 7.61 (dd, J = 7.6, 1.5 Hz, 1H), 7.54 (d, J = 7.3 Hz, 1H), 7.49–7.46 (m, 1H), 7.43 (ddd, J = 14.5, 8.4, 5.7 Hz, 5H), 7.17 (dt, J = 14.9, 6.9 Hz, 3H), 3.92 (d, J = 1.9 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 195.3, 166.3, 165.4, 150.9, 148.6, 143.4, 138.9, 138.1, 137.7, 136.7, 134.7, 133.7, 133.3, 131.9, 130.5, 129.7, 129.4, 129.1, 128.5, 128.3, 128.2, 127.6, 127.0, 126.6, 125.3, 121.4, 93.3, 52.6. HRMS(ESI) m/z [M + H]+ Calcd for C32H22N2O5: 515.1529, found: 515.1609.
7-Benzoyl-3-hydroxy-3-phenyl-2-(quinolin-8-yl)-5-vinylisoindolin-1-one (3ha, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 55% yield (26.5 mg), mp 120–123 °C. 1H NMR (600 MHz, CDCl3) δ 10.61 (s, J = 99.1 Hz, 1H), 8.81 (d, J = 4.6 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 7.7 Hz, 2H), 7.60 (dd, J = 12.8, 7.8 Hz, 2H), 7.55 (s, 1H), 7.53–7.48 (m, 2H), 7.45–7.37 (m, 6H), 7.21–7.11 (m, 3H), 6.76 (dd, J = 17.6, 10.8 Hz, 1H), 5.87 (d, J = 17.6 Hz, 1H), 5.41 (d, J = 10.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 196.2, 166.8, 151.2, 148.4, 142.9, 139.5, 137.8, 137.0, 135.3, 133.4, 129.7, 129.3, 128.4, 128.1, 128.0, 126.9, 126.6, 126.2, 125.8, 121.5, 121.2, 117.7, 93.2. HRMS(ESI) m/z [M + H]+ Calcd for C32H22N2O3: 483.5390, found: 483.1703.
7-Benzoyl-3-hydroxy-3,5-diphenyl-2-(quinolin-8-yl)isoindolin-1-one (3ia, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 62% yield (32.9 mg), mp 118–121 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.83 (dd, J = 4.2, 1.8 Hz, 1H), 8.32 (d, J = 8.3 Hz, 1H), 7.89 (ddd, J = 13.3, 5.6, 3.2 Hz, 4H), 7.75–7.72 (m, 2H), 7.69–7.65 (m, 2H), 7.57 (ddd, J = 29.5, 14.9, 7.5 Hz, 5H), 7.50–7.45 (m, 3H), 7.43–7.40 (m, 1H), 7.17–7.09 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 195.4, 165.0, 151.3, 149.9, 145.0, 143.7, 139.3, 138.2, 137.3, 136.7, 133.7, 133.4, 129.5, 129.3, 128.8, 128.7, 128.6, 128.2, 128.1, 127.8, 127.3, 127.1, 126.7, 126.6, 126.1, 126.0, 122.0, 121.6, 92.7. HRMS(ESI) m/z [M + H]+ Calcd for C36H24N2O3: 533.1787, found: 533.1861.
3.4. General Procedure for the Synthesis of Compound 4
A Schlenk tube was equipped with a magnetic stir bar and charged with 2-methyl-N-(quinolin-8-yl)benzamide 1i (0.1 mmol), 2 (0.25 mmol), NaOAc (0.2 mmol, 16 mg), Pd(OAc)2 (0.01 mmol, 2.3 mg), and xylene (2 mL). Then, the flask was sealed under N2 and stirred at 80 °C for 16 h. After the reaction was quenched by the addition of water, the mixture was extracted with dichloromethane, and the combined organic layer was dried over sodium sulfate. The concentration in vacuo followed by silica gel column purification with petroleum ether/ethyl acetate eluent gave the desired product 4.
3-Hydroxy-7-methyl-3-phenyl-2-(quinolin-8-yl)isoindolin-1-one (4a, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 62% yield (22.7 mg), mp 130–132 °C. 1H NMR (600 MHz, CDCl3) δ 9.85 (s, 1H), 8.80 (dd, J = 4.3, 1.7 Hz, 1H), 8.18 (dd, J = 8.3, 1.8 Hz, 1H), 7.69 (dd, J = 8.2, 1.4 Hz, 1H), 7.59 (dd, J = 7.5, 1.4 Hz, 1H), 7.50–7.44 (m, 2H), 7.43–7.40 (m, 3H), 7.29 (d, J = 7.6 Hz, 1H), 7.20–7.10 (m, 4H), 2.81 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.2, 151.4, 149.3, 140.6, 138.3, 138.2, 133.2, 132.6, 131.4, 129.8, 128.3, 128.1, 127.9, 127.2, 126.9, 126.8, 121.7, 120.6, 92.7, 17.9. HRMS(ESI) m/z [M + Na]+ Calcd for C24H18N2O2: 389.1368, found: 389.1262.
3-(4-Chlorophenyl)-3-hydroxy-7-methyl-2-(quinolin-8-yl)isoindolin-1-one (4b, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 62% yield (24.8 mg), mp 107–109 °C. 1H NMR (600 MHz, CDCl3) δ 10.08–9.88 (m, 1H), 8.79 (d, J = 4.3 Hz, 1H), 8.20 (d, J = 8.3 Hz, 1H), 7.72 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 7.4 Hz, 1H), 7.52–7.41 (m, 3H), 7.34 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 7.6 Hz, 1H), 7.14 (dd, J = 15.7, 7.9 Hz, 3H), 2.80 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.0, 151.0, 149.3, 139.4, 138.4, 138.4, 134.1, 133.3, 132.5, 131.7, 129.9, 128.5, 128.5, 128.1, 127.4, 126.7, 121.8, 120.5, 92.3, 17.9. HRMS(ESI) m/z [M + Na]+ Calcd for C24H17ClN2O2: 423.0979, found: 423.0876.
3-(4-Fluorophenyl)-3-hydroxy-7-methyl-2-(quinolin-8-yl)isoindolin-1-one (4c, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 54% yield (20.7 mg), mp 114–116 °C. 1H NMR (600 MHz, CDCl3) δ 9.96 (d, J = 90.1 Hz, 1H), 8.84–8.79 (m, 1H), 8.23 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.56 (dd, J = 7.5, 1.5 Hz, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.40–7.33 (m, 2H), 7.30 (d, J = 7.6 Hz, 1H), 7.17 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 8.7 Hz, 2H), 2.80 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.6, 163.0 (d, J = 246.6 Hz), 150.7, 148.9, 138.0, 136.1 (d, J = 3.0 Hz), 132.9, 132.1, 131.2, 129.5, 128.5 (d, J = 8.3 Hz), 127.6, 126.9, 126.3, 121.4, 120.1, 114.9, 114.7, 92.0, 17.5. HRMS(ESI) m/z [M + H]+ Calcd for C24H17FN2O2: 385.1274, found: 385.1346.
3-Hydroxy-7-methyl-3-(4-nitrophenyl)-2-(quinolin-8-yl)isoindolin-1-one (4d, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 48% yield (19.7 mg), mp 118–120 °C. 1H NMR (600 MHz, CDCl3) δ 10.28 (s, 1H), 8.82 (tt, J = 4.5, 1.3 Hz, 1H), 8.25 (ddt, J = 8.5, 4.1, 1.8 Hz, 1H), 8.04–7.99 (m, 2H), 7.75 (ddt, J = 8.1, 3.4, 1.6 Hz, 1H), 7.60 (t, J = 8.5 Hz, 3H), 7.55–7.45 (m, 3H), 7.35–7.30 (m, 1H), 7.13 (d, J = 7.5 Hz, 1H), 2.80 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.8, 150.3, 149.4, 148.3, 147.8, 144.3, 138.7, 138.5, 133.7, 133.5, 132.4, 132.0, 129.9, 128.3, 128.1, 127.4, 126.6, 123.7, 121.9, 120.5, 91.9, 17.9. HRMS(ESI) m/z [M + H]+ Calcd for C24H17N3O4: 412.1219, found: 412.1286.
3-Hydroxy-7-methyl-2-(quinolin-8-yl)-3-(4-(trifluoromethyl)phenyl)isoindolin-1-one (4e, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 53% yield (23.0 mg), mp 123–126 °C. 1H NMR (600 MHz, CDCl3) δ 10.11 (s, 1H), 8.82 (d, J = 4.3 Hz, 1H), 8.23 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 7.4 Hz, 1H), 7.54 (t, J = 9.0 Hz, 3H), 7.46 (dt, J = 21.6, 7.9 Hz, 4H), 7.31 (d, J = 7.6 Hz, 1H), 7.14 (d, J = 7.5 Hz, 1H), 2.81 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.0, 150.8, 149.4, 145.0, 138.6, 138.4, 133.9 (d, J = 3.4 Hz), 132.6, 131.8, 129.9 (d, J = 205.4 Hz), 128.2, 127.5 (d, J = 6.5 Hz), 126.6, 125.4, 125.4, 125.4, 125.4, 121.9, 120.6, 92.2, 17.9. HRMS(ESI) m/z [M + H]+ Calcd for C25H17F3N2O2: 457.1242, found: 457.1136.
3-Hydroxy-7-methyl-2-(quinolin-8-yl)-3-(p-tolyl)isoindolin-1-one (4f, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 69% yield (26.2 mg), mp 116–119 °C. 1H NMR (600 MHz, CDCl3) δ 9.81 (s, 1H), 8.80 (dd, J = 4.4, 1.7 Hz, 1H), 8.20–8.16 (m, 1H), 7.70 (dd, J = 8.2, 1.4 Hz, 1H), 7.62–7.58 (m, 1H), 7.51–7.41 (m, 3H), 7.28 (t, J = 7.9 Hz, 3H), 7.18 (d, J = 7.5 Hz, 1H), 6.97 (d, J = 8.0 Hz, 2H), 2.81 (s, 3H), 2.21 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.75, 151.14, 148.84, 137.78, 137.40, 137.19, 132.75, 130.94, 129.39, 128.60, 127.42, 126.84, 126.43, 126.30, 121.24, 120.16, 92.37, 20.91, 17.48. HRMS(ESI) m/z [M + Na]+ Calcd for C25H20N2O2: 403.1525, found: 403.1424.
3-Hydroxy-7-methyl-2-(quinolin-8-yl)-3-(m-tolyl)isoindolin-1-one (4g, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 46% yield (17.5 mg), mp 115–117 °C. 1H NMR (600 MHz, CDCl3) δ 9.69 (s, 1H), 8.81 (d, J = 4.2 Hz, 1H), 8.20 (d, J = 8.3 Hz, 1H), 7.72 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 7.4 Hz, 1H), 7.52–7.43 (m, 3H), 7.28 (d, J = 7.6 Hz, 1H), 7.24 (s, 1H), 7.19 (t, J = 6.7 Hz, 2H), 7.06 (t, J = 7.6 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 2.80 (s, 3H), 2.20 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.8, 151.2, 140.1, 137.9, 137.5, 132.8, 131.0, 129.5, 128.5, 127.8, 127.5, 127.1, 123.7, 121.3, 120.2, 92.4, 21.3, 17.5. HRMS(ESI) m/z [M + Na]+ Calcd for C25H20N2O2: 403.1525, found: 403.1424.
3-(Furan-2-yl)-3-hydroxy-7-methyl-2-(quinolin-8-yl)isoindolin-1-one (4h, new compound): Following the general procedure, the title compound was isolated by flash chromatography (eluent: petrol ether/ethyl acetate = 2/1~1/1) as a white solid in 57% yield (20.3 mg), mp 122–125 °C. 1H NMR (600 MHz, CDCl3) δ 8.87 (dd, J = 4.4, 1.8 Hz, 1H), 8.31 (dd, J = 8.3, 1.7 Hz, 1H), 7.84 (dd, J = 8.2, 1.3 Hz, 2H), 7.64 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.53 (dd, J = 8.3, 4.4 Hz, 1H), 7.42 (dd, J = 15.2, 7.5 Hz, 2H), 7.26 (d, J = 1.4 Hz, 1H), 6.50 (d, J = 3.3 Hz, 1H), 6.20 (dd, J = 3.3, 1.8 Hz, 1H), 2.88 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.2, 148.8, 147.9, 142.7, 138.0, 132.6, 131.5, 129.3, 127.5, 127.5, 126.9, 126.5, 121.3, 119.9, 109.9, 109.3, 88.9, 17.4. HRMS(ESI) m/z [M + Na]+ Calcd for C22H16N2O3: 379.1161, found: 379.1054.
4. Conclusions
In conclusion, we have developed a novel dual C-H acylations and intramolecular cyclization sequence using bidentate N-(quinolin-8-yl)benzamide and aromatic acyl chlorides under oxidant-free and ligand-free conditions. This process involves the formation of three new chemical bonds and provides straightforward access to new hydroxyl isoindolono-quinoline skeletons. Further studies and applications of this innovative synthetic methodology are currently underway in our laboratory.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29225397/s1. Kinetic isotope effect measurements. Copies of 1H and 13C NMR Spectra for products. Single-crystal information (3aa and 4a).
Author Contributions
Synthesis and characterization, H.X. and Y.Y. (Yuchen Yang); data curation, H.X. and Y.Y. (Yuzhu Yang); writing—original draft preparation, H.X. and Y.Y. (Yuzhu Yang); writing—review and editing, F.L. and Y.Y. (Yuzhu Yang); funding acquisition, Y.Y. (Yuzhu Yang). All authors have read and agreed to the published version of the manuscript.
Funding
Y.Y. is grateful for financial support from State Key Laboratory of Functions and Applications of Medicinal Plants and the West Light Foundation of the Chinese Academy of Science.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We acknowledge the analytical testing support from the Analysis and Testing Center, Natural Products Research Center of Guizhou Province.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef]
- Wang, H.; Li, T.; Hu, D.; Tong, X.; Zheng, L.; Xia, C. Acylation of Arenes with Aldehydes through Dual C–H Activations by Merging Photocatalysis and Palladium Catalysis. Org. Lett. 2021, 23, 3772–3776. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Li, Z.; Wang, G. Direct Decarboxylative Meta-Selective Acylation of Arenes via an Ortho-Ruthenation Strategy. ACS Catal. 2018, 8, 11875–11881. [Google Scholar] [CrossRef]
- Liao, Y.; Jiang, C.; Qiang, C.; Liu, P.; Sun, P. HAT-Mediated Electrochemical C(sp2)–H Acylation of Quinolines with Alcohols. Org. Lett. 2023, 25, 7327–7331. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Qian, C.; Lin, D.; Zeng, W.; Liu, X. Palladium-Catalyzed Cascade Oxidation/sp2 C–H Acylation of Azoarenes with Aryl Methanes. Org. Lett. 2013, 15, 5444–5447. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhou, X.; Xu, Y.; Li, X. Rhodium(III)-Catalyzed Acylation of C(sp3)–H Bonds with Cyclopropenones. Org. Lett. 2017, 19, 3644–3647. [Google Scholar] [CrossRef]
- Suzuki, H.; Sasamori, F.; Matsuda, T. Rhodium-Catalyzed C(sp2)–H Alkoxycarbonylation/Acylation of Indolines with Anhydrides as a Carbonyl Source. Org. Lett. 2022, 24, 1141–1145. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y.; Kong, L.; Zhou, X.; Tang, G.; Lan, Y.; Li, X. Mild Acylation of C(sp3)–H and C(sp2)–H Bonds under Redox-Neutral Rh(III) Catalysis. ACS Catal. 2016, 6, 7744–7748. [Google Scholar] [CrossRef]
- Liu, S.; He, B.; Li, H.; Zhang, X.; Shang, Y.; Su, W. Facile Synthesis of Alkylidene Phthalides by Rhodium-Catalyzed Domino C−H Acylation/Annulation of Benzamides with Aliphatic Carboxylic Acids. Chem. Eur. J. 2021, 27, 15628–15633. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Use of Solid Catalysts in Friedel−Crafts Acylation Reactions. Chem. Rev. 2006, 106, 1077–1104. [Google Scholar] [CrossRef]
- Liu, P.; Frost, C. Ruthenium-Catalyzed C–H Functionalization of Arylpyrazoles: Regioselective Acylation with Acid Chlorides. Org. Lett. 2013, 15, 5862–5865. [Google Scholar] [CrossRef] [PubMed]
- McAteer, D.; Javed, E.; Huo, L.; Huo, S. Platinum-Catalyzed Double Acylation of 2-(Aryloxy)pyridines via Direct C–H Activation. Org. Lett. 2017, 19, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Wang, X.; Wang, G. Diastereoselective Synthesis of Oxazoloisoindolinones via Cascade Pd-Catalyzed ortho-Acylation of N-Benzoyl α-Amino Acid Derivatives and Subsequent Double Intramolecular Cyclizations. J. Org. Chem. 2019, 84, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yang, F.; Wu, Y.; Wu, Y. Palladium-Catalyzed C8-H Acylation of 1-Naphthylamines with Acyl Chlorides. Org. Lett. 2019, 21, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Topliss, J.G.; Konzelman, L.M.; Sperber, N.; Roth, F.E. Antihypertensive Agents. III. 3-Hydroxy-3-phenylphthalimidines. J. Med. Chem. 1964, 7, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.G.; Danishefsky, S.J. The total synthesis of chilenine: Novel constructions of cyclic enamides. Tetrahedron Lett. 1989, 30, 2747–2752. [Google Scholar] [CrossRef]
- McKenzie, W.S.; Rosenbery, M.J. Mass. Dent. Soc. 2007, 56, 44–45. [Google Scholar]
- Zarga, M.H.A.; Sabri, S.S.; Firdous, S.; Shamma, M. Sesquiterpene acids from Inula Viscosa. Phytochemistry 1987, 26, 1233–1234. [Google Scholar] [CrossRef]
- Zhu, W.; Tong, S.; Zhu, J.; Wamg, M. Intramolecular Arylation of Tertiary Enamides through Pd(OAc)2-Catalyzed Dehydrogenative Cross-Coupling Reaction: Construction of Fused N-Heterocyclic Scaffolds and Synthesis of Isoindolobenzazepine Alkaloids. J. Org. Chem. 2019, 84, 2870–2878. [Google Scholar] [CrossRef]
- Sharma, S.; Park, E.; Park, J.; Kim, I. Tandem Rh(III)-Catalyzed Oxidative Acylation of Secondary Benzamides with Aldehydes and Intramolecular Cyclization: The Direct Synthesis of 3-Hydroxyisoindolin-1-ones. Org. Lett. 2012, 14, 906–909. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, N.; Huang, J.; Lu, S.; Zhu, Y.; Yu, X.; Zhao, K. Efficient Synthesis of Hydroxyl Isoindolones by a Pd-Mediated C-H Activation/Annulation Reaction. Chem. Eur. J. 2013, 19, 11184–11188. [Google Scholar] [CrossRef]
- Roane, J.; Daugulis, O. A General Method for Aminoquinoline-Directed, Copper-Catalyzed Sp2 C–H Bond Amination. J. Am. Chem. Soc. 2016, 138, 4601–4607. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.; Klimovica, K.; Daugulis, O. Copper-Catalyzed, Directing Group-Assisted Fluorination of Arene and Heteroarene C–H Bonds. J. Am. Chem. Soc. 2013, 135, 9342–9345. [Google Scholar] [CrossRef]
- Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. Copper-Mediated C–H/C–H Biaryl Coupling of Benzoic Acid Derivatives and 1,3-Azoles. Angew. Chem. Int. Ed. 2013, 52, 4457–4461. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.D.; Roane, J.; Daugulis, O. Directed Amination of Non-Acidic Arene C–H Bonds by a Copper–Silver Catalytic System. Angew. Chem. Int. Ed. 2013, 52, 6043–6046. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Chatani, N. Rhodium-Catalyzed Alkylation of C–H Bonds in Aromatic Amides with α,β-Unsaturated Esters. Org. Lett. 2014, 16, 5148–5151. [Google Scholar] [CrossRef]
- Suess, A.M.; Ertem, M.Z.; Cramer, C.J.; Stahl, S.S. Divergence between Organometallic and Single-Electron-Transfer Mechanisms in Copper(II)-Mediated Aerobic C–H Oxidation. J. Am. Chem. Soc. 2013, 135, 9797–9804. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).