1. Introduction
Fatty acids (FAs) are organic compounds characterized by a long carbon chain and a carboxylic acid functional group responsible for several functions in human physiology [
1,
2]. The chronically elevated concentration of FAs in plasma is correlated to human disorders [
3,
4] such as diabetes [
5], atherosclerosis [
6], and obesity [
7]. FAs’ high lipophilicity is responsible for their low water solubility; thus, their trafficking requires specialized proteins such as fatty acid binding proteins (FABPs) [
8]. Based on their localization, FABPs are classified into different families; FABP4 (aP2 or A-FABP) is the subtype mainly produced in the adipocytes [
9]. The research on FABP4 inhibitors started when it was reported that knockout animal models of FABP4 naturally developed protective effects against insulin resistance [
10] and other events, such as metabolic syndrome and atherosclerosis [
11,
12,
13]. Indeed, pharmacological approaches with small molecules of FABP4is demonstrated similar results in the phenotype of FABP4-deficient mice [
14].
This protein family also has a role in cancer progression [
15]. In fact, renal cell carcinoma, bladder, prostate, and other cancer cells [
16,
17,
18] were reported with a non-physiological expression of FABPs. The classical FABP4i, BMS309403, was reported to weaken the migration and invasion of colon cancer cells. These results highlighted the tendency of FABP4 to promote colon cancer metastasis and invasion [
19]. It is known that FABP4 leads to abnormal metastasis and aggression in ovarian cancer, contributing to poor prognosis for this [
20] and other types of cancers, such as glioblastoma [
21].
All of these recent findings suggest that FABP4 targeting may represent a valid and promising therapeutic strategy against oncological conditions.
Various effective FABP4is were developed recently, but unfortunately, none are currently in the clinical research phase [
14,
22]. Computer-aided drug design is promising in drug discovery and an effective tool for identifying molecular hits such as FABP4is [
23,
24,
25,
26,
27,
28]. In line with our recent interest in developing new antitumor compounds and identifying novel bioactive heterocycles [
29,
30,
31,
32,
33,
34], we herein report the first application of SMD to evaluate the binding properties of FABP4is to the targeted protein.
MD simulations of proteins at the atomic level are a well-established method for describing the behavior of proteins and the protein-ligand interactions involved in cell signaling for disease processes. A special kind of MD simulation is the SMD simulation. With this method, a protein or molecule can be given a directing vector as part of the SMD simulation to examine how it reacts to outside stimuli [
29]. SMD simulations, which provide atomic-level resolution of force-probe events, proved to be essential supplements to current experimental techniques [
30]. In a typical SMD simulation, one terminus of the molecule is subjected to an external vector while the opposing terminus is fixed in space. This allows researchers to examine how the molecule reacts to mechanical stress and how it can clarify the structure-function relationship of a macromolecular complex that involves either protein-ligand or protein-protein interactions [
31,
32]. SMD simulations for protein-ligand complexes may be used to determine stabilizing interactions, which help inform the development of drugs that can quickly and readily bind to the active binding site to increase or decrease the activity of the desired protein. SMD was developed to address simple questions from protein conformational changes relative to the study of relevant residues involved in particular interactions. Recently, it was also used to evaluate drug-binding affinity for drug screening. The unbinding force obtained from receptor-ligand dissociation and SMD simulations can be used to estimate a ligand’s binding affinity with reasonable accuracy [
33,
34,
35].
The use of computational simulations proved very advantageous to the pharmaceutical industry, where high precision, low cost, and reduced time and labor in drug development are all priorities. The molecular mechanics Poisson–Boltzmann solvent accessible surface area (MM/PBSA) method is one of the most extensively used computational approaches for calculating the binding affinity, i.e., the free energy of binding (Δ
Gbind), between substrate and protein. It was also proven that the MM/PBSA and SMD have a strong connection in assessing the binding affinity of small molecules to a protein [
36]. For example, recently, SMD simulations were used to evaluate the binding affinity of antiviral compounds [
37,
38], studies which also correlated simulation results and MM/PBSA data. These simulations were also used to evaluate molecules for the treatment of cancer-specific inhibitors of allosteric BcrAbl fusion protein for the treatment of chronic myeloid leukemia [
39] and histone deacetylases (HDAC) [
40].
All of these studies demonstrate that SMD is a powerful tool for predicting the mechanisms of efficient drug binding and a potent hypothesis-generating tool for screening new drug candidates. Due to our recent interest in the design of small molecules able to inhibit FABP4 by using structure- and ligand-based computational tools, we herein report the first application of SMD to evaluate the binding affinity of small molecules as inhibitors of FABP4. The aim of this study was to optimize a simple method to employ SMD for the fast evaluation of novel small FABP4is molecules.
2. Results
The ligand binding sites of FABPs, shown in X-ray crystallographic studies, have been known for decades. However, the mechanism of how the ligands access their binding sites remains challenging to identify, and computational SMD proved the existence of basically one ligand escaping its route despite identifying at least two possible portals. In all successful dissociations, the palmitate molecule was established to come out from essentially the same region [
41]. The same area was used in our study to dissociate the selected ligands.
The key component for running the SMD simulation is the initial configuration of the system. The Jarzynski equation requires configurations sampled from an equilibrium distribution with
λ =
λ0. These configurations can be obtained in two ways: through a Monte Carlo simulation or by periodically saving snapshots from a molecular dynamics simulation of the system with the reaction coordinate constrained to the desired initial value. The second option was used in this work. The two key parameters fundamental to interpreting the reaction unfolding are the acceleration estimated in pm/ps
2 and the distance measured in Å. These settings allowed us to calculate and obtain the total energy measured in kcal/mol required for the ligand to travel to a previously set distance. Structural heterogeneity was ensured by selecting different molecules as a measurement of pairwise similarity calculated by using circular fingerprints (
Figure 1). Extended-connectivity fingerprints (ECFP) are circular topological fingerprints optimized and designed for molecular characterization, similarity searching, and ligand-based molecular modeling. They are among the most common and best-performing similarity search tools in drug discovery, and they are used in many applications [
42,
43]. The similarity matrix from the ECFP evaluation was also compared with the Tanimoto similarity index, and a similar pattern was also identified in the Tanimoto matrix (
Figure S1,
Supplementary Materials), confirming the good dissimilarity between the chosen compounds.
Table 1 reports the p
Ki values obtained among the various molecule sets between the calculated energies of binding by docking scoring functions, the SMD simulations’ calculated total energies and F
max, and their experimental inhibition constants.
Figure 2 states the total energies and the max force (F
max) obtained from each SMD experiment. Simulations were carried out in triplicate with double-blind conditions using different hardware systems to ensure the homogeneity and repeatability of the experiment.
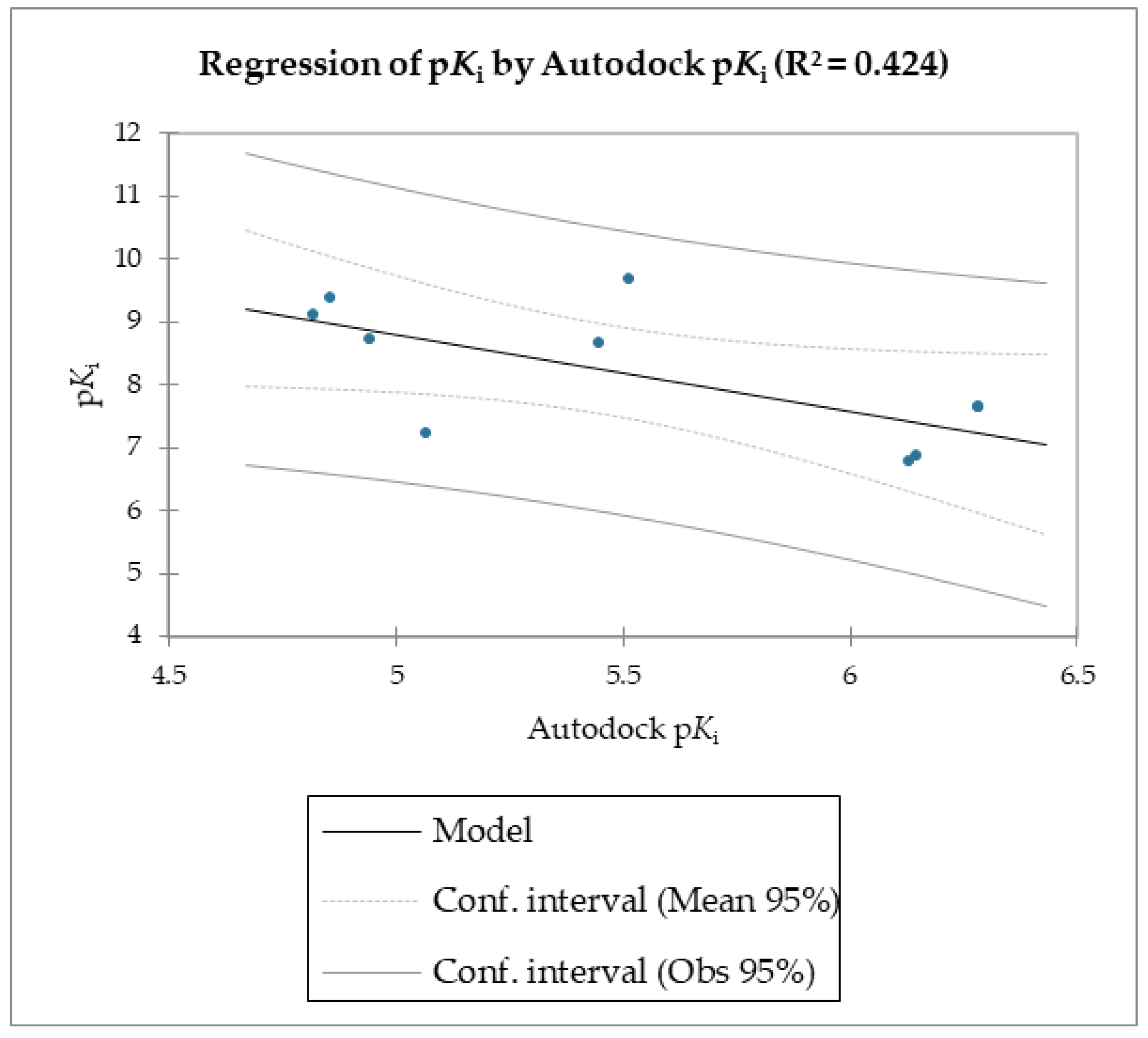
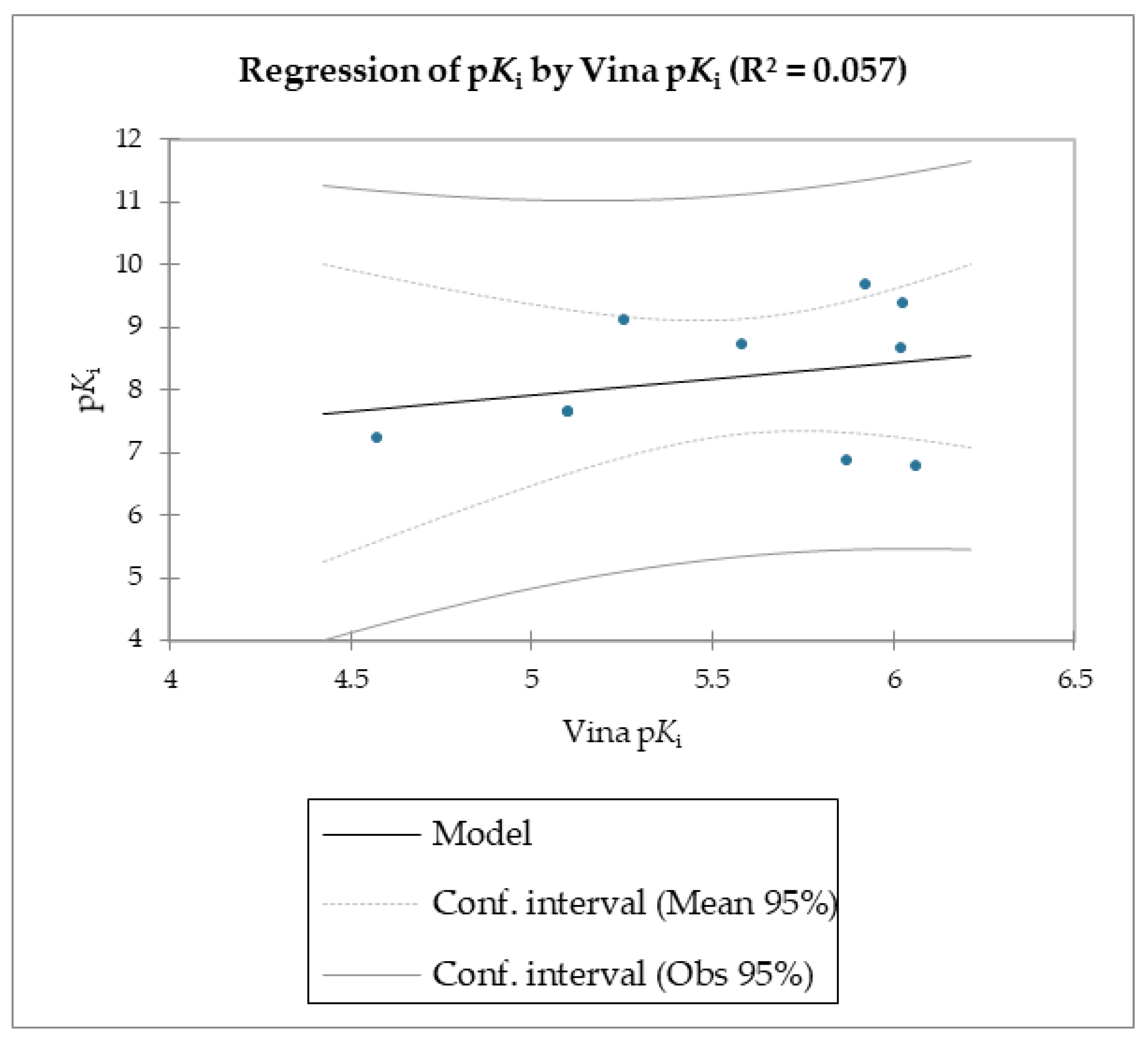
The MMPB(GB)SA and the SMD simulations were superior overall at linearly correlating the experimental data (employing relative and not absolute binding value) for the studied molecules compared to the scoring functions of Autodock and Vina, which were the least able to correlate the calculated free energy with the experimental data (
Table 2,
Table 3 and
Table 4,
Figure 3 and
Figure 4).
The reported data clearly shows the higher capability of SMD to score the affinities of small molecules against FABP4 compared to Autodock and Vina. In fact, as the value of the reference constant increased for each subset of molecules, we consistently obtained the increase of Fmax required to extract the ligand from its receptor, following a linear correlation. Conversely, the Autodock and Vina scoring functions were not as accurate as the SMD data in potency evaluation, as they showed poor correlation between the increased potency of the compounds and the calculated binding energies.
To compare the SMD simulations’ energy evaluation with a more accurate energy of binding calculation and also include the effect of solvation, the experimental values were also compared to the MM-PB(GB)SA binding energies.
SMD simulation was recently shown to be as accurate as the MM/PBSA and molecular mechanics generalized Born surface area (MM/GBSA) techniques in predicting the binding affinity of small ligands to proteins. If a pulling speed of
v = 0.005 nm/ps is used, then SMD simulation is computationally around 30-fold quicker than the MM/PBSA method, indicating its considerable potential for drug design [
37]. Previous research found a strong link between F
max values and experimental free energies from 5 up to 20 protein–ligand systems, proving that a small dataset helps evaluate such a tool [
37,
44].
The correlation between the experimental and calculated p
Ki using the MM/PB(GB)SA and the SMD F
max methods are reported in
Figure 5,
Figure 6 and
Figure 7.
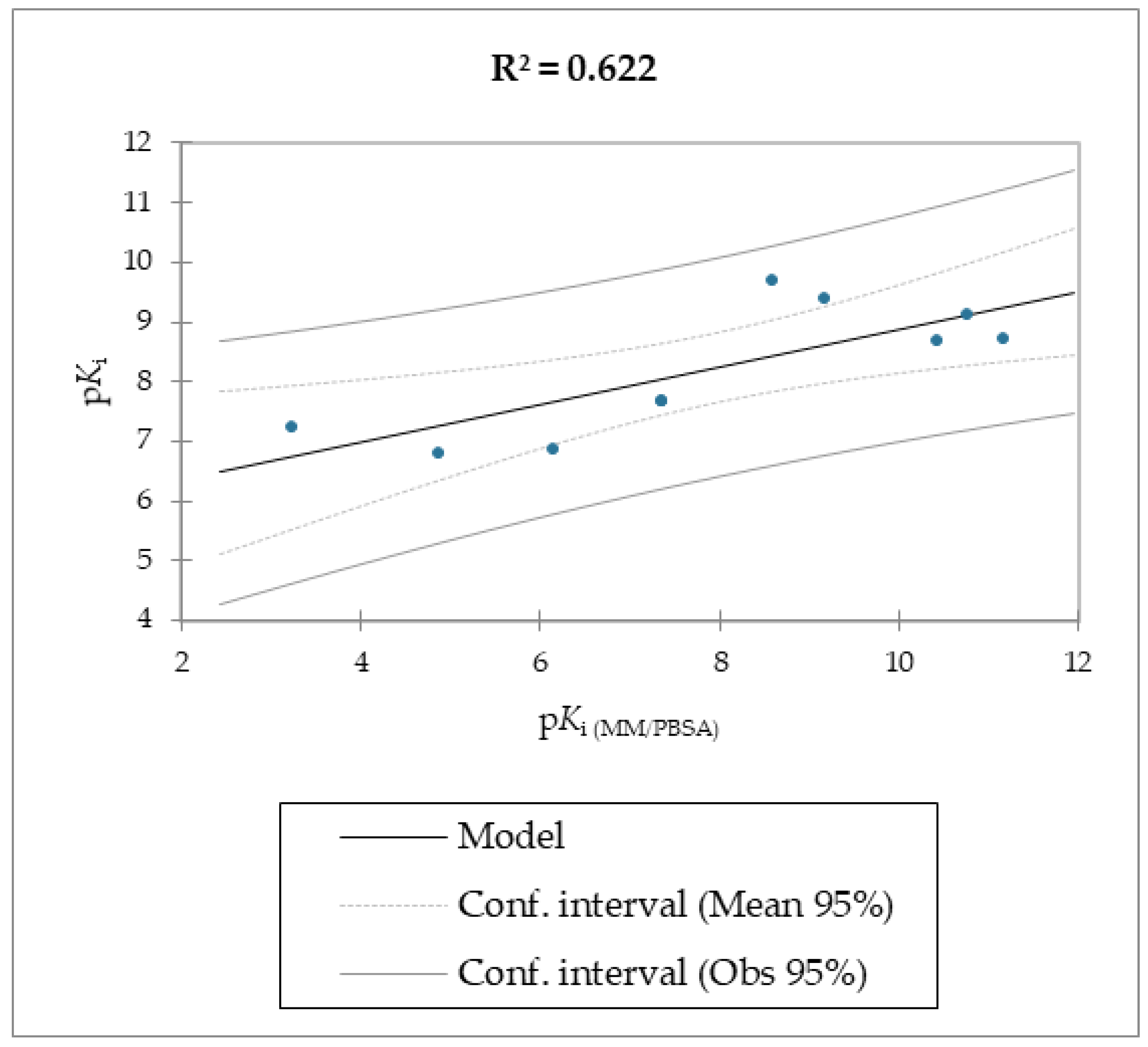
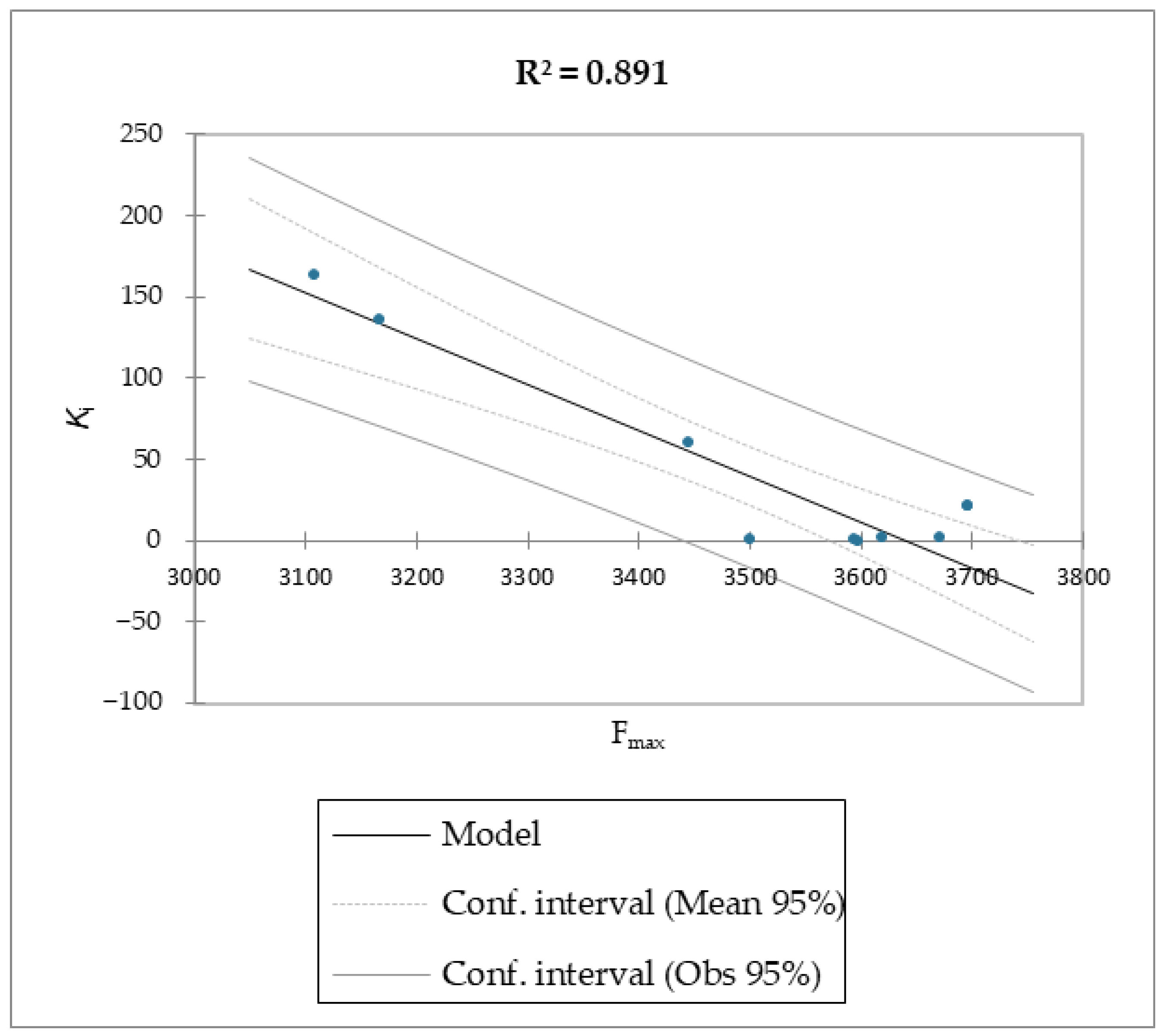
Regarding molecules
1–
9, MM/PBSA and MM/GBSA’s scoring functions underperformed the SMD-derived data. Notably, the F
max overperformed all of the other data with a linear relationship and an R
2 of 0.891, whereas the MM/PBSA and MM/GBSA models resulted in R
2 values of 0.622 and 0.472, respectively. The three linear models were calculated using linear regression. The measured
Ki was used as a dependent variable for the SMA-derived model, and the SMA F
max was used as the explanatory variable. Regarding the two models derived from the MM/PBSA and MM/GBSA models’ calculations, the experimental p
Ki was used as a dependent variable, and the p
Ki, which was calculated using the MM/PBSA and MM/GBSA models, was used as the explanatory variable. The predictive capabilities for the last two models, measured as R
2, were 0.622 and 0.472 for MM/PBSA and MM/GBSA, respectively, confirming the data model from the F
max linear model was better than the others derived from the MM/PBSA and MM/GBSA models’ calculations. The evaluation of the models was retrieved through several standard ways, as reported in
Table 5.
The three equations calculated from the models are:
Moreover, given the R
2 of the F
max model, 89% of the variability of the dependent variable is explained by the explanatory variable. Considering the p-value of the computed F statistic shown in the ANOVA table (
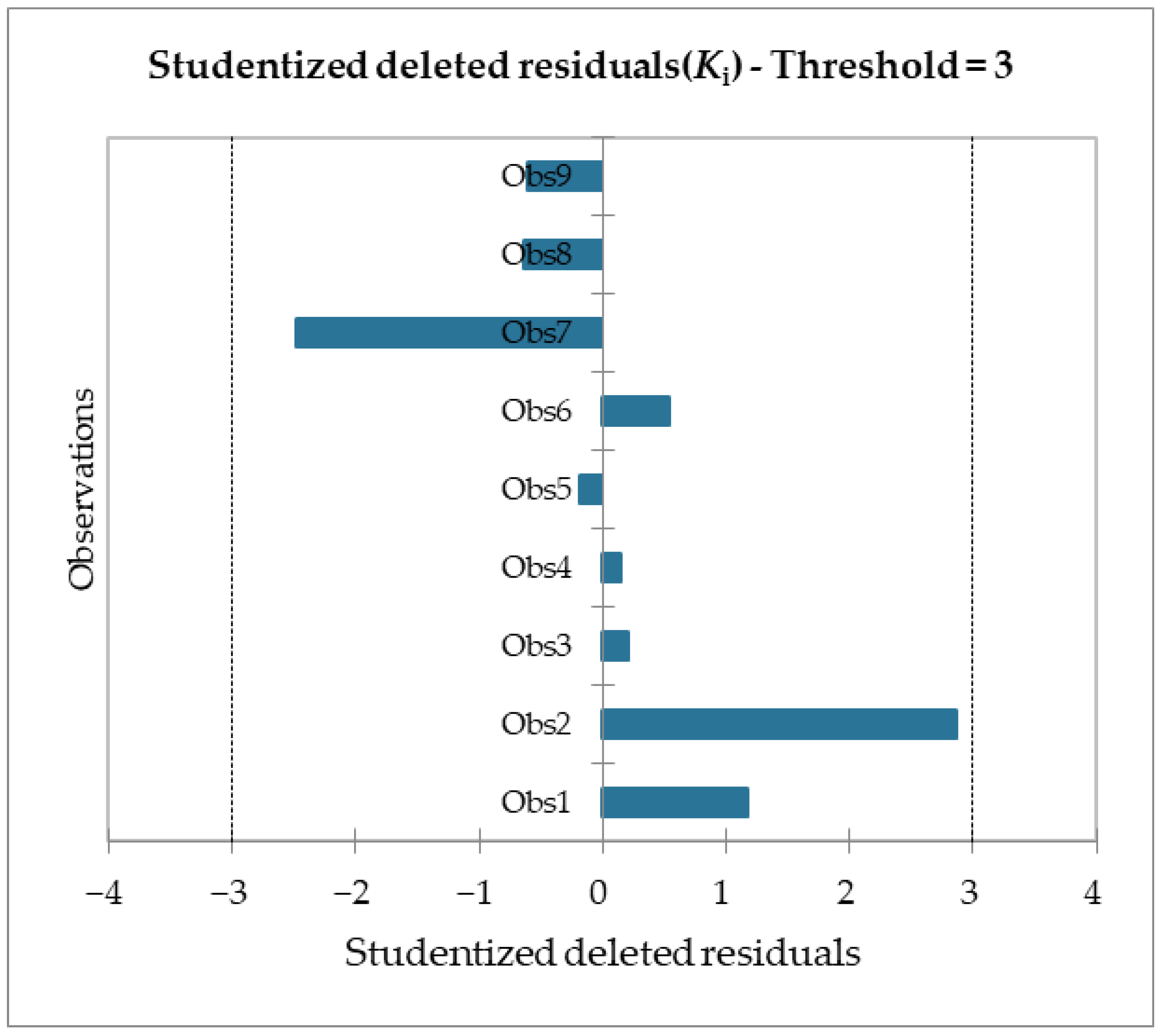
Table 6) and given a significance level of 5%, the information brought by the explanatory variables is significantly better than what a basic mean would bring. Outliers were checked with studentized deleted residuals and Cook’s distances, as reported in
Figure 8 and
Figure 9, and both confirmed the presence of no outliers among our training data.
Finally, to verify the predictive capabilities of the F
max model toward low-activity and inactive compounds, a second set of molecules was used; the structures of the molecules are reported in
Table 7. Molecule
10 (used as a negative control with an experimental p
Ki of 4.3) was outlined as the least active, with the lowest calculated SMD energy of 337.71 kcal/mol and an F
max of 2387.26 pN. Another five molecules were added to this second set as true negatives. Molecules
11–
15 (decoy compounds) were generated by employing the DUD-E webserver and using BMS309403 (a FABP4i) as the reference compound [
45]. The decoy compounds have similar physicochemical properties but different 2D topologies, i.e., they will be inactive in the same binding pocket. Interestingly, all of the molecules were identified as low-energy FABP4 binders in the SMD data, which was confirmed by the low F
max for all of the compounds, demonstrating the ability of the model to identify true negative compounds.
To exploit the new model in drug design, we generated a novel series of FABP4is and evaluated them with the proposed Fmax-based approach by using the computer-assisted scaffold hopping technique.
As shown in
Figure 10, we focused on the search for bioisosteric-replacements/scaffold hopping in the central core of a recently published novel FABP4i scaffold with a pyridazin-3-(2
H)-one central core [
46]. Our bioisosteric replacement analysis led to 500 novel molecules for each series. Among the best compounds, as scored by the generated field similarity analysis (
Tables S1 and S2), the 3-methoxy-6-phenylpyridazin-4-amine core was identified as a common one (compounds
18 and
22) when considering the results of both series (
Table 8). The newly identified core was then evaluated by using SMD-based calculations (
Figure 11). Both compounds,
18 and
22, were identified as promising FABP4is with calculated F
max values of 3313.93 and 3693.72 pN, respectively.
4. Conclusions
Inhibiting FABP4 is a viable and appealing therapeutic opportunity for treating metabolic disorders [
59,
60,
61,
62]. Furthermore, given the discovery of the protein’s role in cancer progression, the inhibition of FABP4 might offer a viable therapeutic option for cancer patients through the suppression or decrease of early-stage tumors and metastasis, and they have a possible use as biomarkers for cancer detection [
63,
64,
65,
66,
67,
68]. Nevertheless, no FABP4i has entered the clinical research phase so far. This is mainly due to several unavoidable adverse effects of FAPBis, including metabolic issues, in vivo toxicity, and rapidly acquired drug resistance [
69]. Believing that this family of transporter proteins holds promise as a valid therapeutic target, research must still try to pursue the common aim of bringing FAPB inhibitors into clinics.
Computational approaches were used to identify novel scaffolds for FABP4 inhibition. Our research group stayed widely active in this field by applying several structures and ligand-based computational tools in FABP4is research [
14,
22,
25,
26,
27,
46]. SMD simulation was never employed to study and rank small-ligand FABP4is.
SMD simulations can be used to evaluate the conformation, stability, and interactions of proteins with surrounding macromolecules (membrane, DNA, RNA, or other proteins). Moreover, SMD is a solid and practical approach for gaining insight into binding mechanisms and acquiring the relative binding energies between candidate drugs and targeted proteins by simply considering the mechanical components, such as ligands and target flexibility. In contrast to the majority of previously reported computational approaches, which focus on accurate binding energy calculations using MD simulations, SMD simulation has the potential to provide more effective ranking procedures while reducing computational expense when combined with existing methods for endpoint free energy calculations, such as molecular mechanics/Poisson–Boltzmann surface area, free energy perturbation, and thermodynamic integration. Thus, we reasoned that SMD simulation might play an essential role in FABP4is’ rational design.
For the first time, we established good agreement between SMD simulation-retrieved data and experimental inhibition or dissociation constants for a dataset of compounds targeting the FABP4 protein. Our study supports SMD simulation as a promising method for evaluating new small-molecule FABP4is. The key benefit of SMD simulation is that it is computationally less expensive than the MM/PB(GB)SA method and, in general, substantially more accurate than the docking approach and scoring functions. Moreover, this work proves SMD simulation is a valuable strategy for ranking novel FABP4is. It will consequently enlarge the arsenal of tools to assist medicinal chemists working in the field, as it demonstrates a practical approach for the future identification of FABP4is for clinical use.
In comparison to the MM/PB(GB)SA method, despite being highly accurate, one limit of the SMD-derived ranking is that it cannot directly predict absolute binding affinities. Another limitation is that other MD simulation-based experiments, such as the US method, could be more capable of calculating a ligand’s free energy of binding. US-method calculations were recently reported to be able to reliably estimate binding free energy for a complex of small molecules/proteins. Compared with SMD, the better results obtained with the US method come at the cost of computational resources. The total MD simulation time with the US method was recently reported to be as long as 120 ns of simulation [
70], which is remarkably high compared to the 50–150 ps required with our SMD methodology (16 h vs. 9–12 min with an AMD Ryzen Threadripper PRO 5975WX equipped with a GeForce RTX 3060 Ti graphics card).
Instead, the computational method presented here can be regarded as a tool to help medicinal chemists pursue molecular modifications and new synthetic directions in FABP4is research that would have been too risky with no computational validation support. It can also be used to readily identify compounds that would be unlikely to meet the desired affinity [
71]. This significantly lowers the risk of embarking on laborious synthetic protocols by anticipating if such a compound is/is not projected to accomplish the potency objectives (as informed by SMD ranking assessments), allowing researchers to focus on target molecules endowed with more promising characteristics as active FABP4is. Moreover, the SMD methodology was used to rank a newly designed library of FABP4is that was built from a biologically active heterocyclic framework recently identified through scaffold hopping replacement [
46]. Synthetic procedures are being investigated to produce compounds
18 and
22 (with the 3-methoxy-6-phenylpyridazin-4-amine core) identified in this work, resulting in potentially valid FABP4is; this is definitely worth further biological study.

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























